一种用于测定细菌与真菌种类和丰度比较的人工合成外源性参照分子的制作方法
本发明涉及微生物检测和分析技术领域,尤其涉及一类用于测定细菌与真菌种类和丰度比较的人工合成的外源性参照分子。
背景技术:
皮肤表面存在着多种多样的的微生物,包括细菌、真菌及病毒等群体在其上生长,然而每一处皮肤的微生物组成都是不尽相同的,这些群体的组成取决于皮肤的特征,如皮脂腺密度、含水量、温度及宿主基因和外生环境因素,特殊的环境造就了特殊的微生物组成,这种有着特定生态架构的组成被称为皮肤微生物组。皮肤表面的微生物组曾经一度被认为是导致皮肤疾病的主要原因,然而随着现代科技的不断进展,越来越多的新观点、新看法不断涌现,从过去的皮肤微生物培养基到今天的实时定量聚合酶链式反应(qpcr)技术的广泛应用,人们对于皮肤微生物组有了更加全面而深刻的认识。
皮肤作为人类免疫的第一屏障,有着不可或缺的作用。人类皮肤作为外在屏障保护皮肤于许多环境威胁,并且处理大量的微生物包括自身微生物群及潜在的致病微生物。从一个微生物试图定植并侵袭皮肤的视角,研究者们强调了皮肤一线防卫屏障的重要性。皮肤自身存在的微生物群一般具有占位的保护作用,使致病菌及外籍菌无法定植于皮肤,例如革兰氏阳性的皮肤共生菌,其中包括乳酸菌、链球菌、葡萄球菌可产生杀菌素,表皮葡萄球菌可产生抗菌肽,对金黄色葡萄球菌、a组链球菌、大肠杆菌具有选择性杀伤作用,詹森菌属就有抑制足癣常见红色毛癣菌生长的作用。人类皮肤在正常状态下每平方厘米有大约100万细菌,上百种类型,传统的皮肤取材和活检标本在标准的实验条件下,仅有1%的细菌与真菌生长,更多的细菌和真菌的生长会被具有生长优势的微生物抑制,因此只能检验到部分的细菌或真菌群落,例如葡萄球菌及马拉色菌等。
在当前的应用广泛的宏基因组测序技术辅助下,人类皮肤的不同部位的微生物组成得到了深入的探索。然而,单一样本中的微生物的组成,只能在真菌或者细菌中进行分析。单一样本中细菌和真菌之间的比例及丰度差异无法进行量化比较。
同种属的微生物间的基因序列既有在进化过程中基本不变化或变异程度很小的保守序列区域,又有种类之间差异表达的可变序列区域。在有核生物中,核糖体rna(rrna)及对应的编码基因核糖体dna(rdna)成为很好的区域,同时包含两种序列区域,并且长度大小合适,适合现有技术框架下的序列分析。目前使用比较多的有5srrna/rdna、5.8srrna/rdna、16srrna/rdna、18srrna/rdna、23srrna/rdna、its(internaltranscribedspace,内转录间隔区)等区的序列。具体原理为根据这些保守区设计出通用物,从而扩增出采集的样本中的所有同一个属的微生物片段,对这些片段进行测序,就可以检测到两个保守的通用引物之间的可变区的基因序列,分析这些可变区的序列差异就可以实现对微生物的分类。检测不同种类微生物的测序条数(reads)就可以知道微生物间的相对丰度。
不同属别的微生物间的基因序列差异比较大,适合用以测序检测的保守区域及其序列也往往不同。而不同的基因序列以及扩增片段的长度制约扩增反应条件。因此目前比较普遍的做法是针对有相同通用引物的同一属别的微生物,设计扩增试剂盒,进行扩增及测序检测。通用引物不同的微生物,各自单独扩增和测序。而不同批次的扩增反应和测序存在批次间扩增效率以及其他影响因素的差异。这就造成微生物种类和丰度的检测只能在同属别之间进行,而同时定居在同一空间区域的不同属的微生物间则不能进行比较。
技术实现要素:
鉴于现有技术存在的缺陷,本发明提供一种用于测定细菌与真菌种类和丰度比较的人工合成外源性参照分子,可以在细菌及真菌的试剂盒中得到扩增,并参与每个扩增反应中的不同微生物之间种类和相对丰度的比较。以此外源性合成物的检测量为参照,可以进一步比较细菌与真菌的微生物之间的相对丰度。
为了实现上述目的,本发明采用以下技术方案。
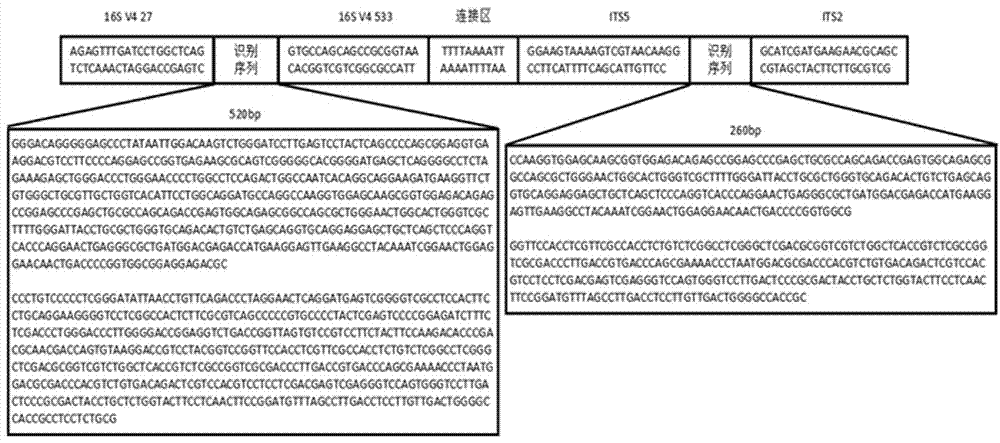
一种人工合成的外源性序列片段,其中是由细菌及真菌的保守序列及外源性序列组合而成的新序列。采取16sv427及16sv4533作为细菌引物序列区的前后序列,its5及its2作为真菌引物序列区的前后序列,连接区为随机序列。同时,相应的引物遵循引物设计原则,保证人工合成的外源性序列片段与样本中片段的有效扩增。
一种人工合成外源性参照序列由16s、its、特异识别区、连接序列区组成(如图1所示)其中。
1)16sv427区为16s上游(或下游)引物序列区。
2)16sv4533区为相应的16s下游(或上游)引物序列。
3)its5区为its上游(或下游)引物序列区。
4)its2区为相应的its下游(或上游)引物序列。
5)特异识别区为人类apoe基因随机序列,设计者明确知道该序列的排列信息,与待检测样本中的基因组dna序列无同源性,存在明显特异区别,16s间识别序列长度为520bp,its间识别序列长度为260bp。
6)连接序列区,设计10bp的与待测序列不同的随机序列。
与现有技术相比,本发明设计了一种人工合成的序列,可以成为外源性参照序列(图1)。该序列可以同时含有细菌及真菌的的通用引物序列区域,并且含有可以人为识别的特异序列。将此外源性人工合成的基因分子(rna或dna)片段,以已知并且特定的量加入到样本核酸样本中。将样本核酸均等分为两份后,分别进行后续特定的扩增。本外源性参照序列可以在细菌及真菌的试剂盒中得到扩增,并参与每个扩增反应中的不同微生物之间种类和相对丰度的比较。以此外源性合成物的检测量为参照,可以进一步比较细菌与真菌的微生物之间的相对丰度。
附图说明
图1是人工合成外源性片段的碱基组成图。目的是为分析同一样本中细菌与真菌的相对比例,人工合成的外源性参照核酸序列。该序列中连接区左侧设置有用于模拟细菌的16sv4区通用序列,二者间插入人类载脂蛋白apolipoproteine(apoe)mrna序列片段中520bp。连接区为人工设置的用于识别外源性掺入片段数量的特异序列ttttaaaatt。连接区右侧为用于模拟真菌its区通用序列,二者间插入人类载脂蛋白apolipoproteine(apoe)mrna序列片段260bp。
图2是根据设计序列的片段,设计细菌16sv4区及its区前后引物。
图3是经过对各个样本的16srrna测序后所得测序结果,反映出各个样本内细菌含量的相对丰度。用不同颜色代表不同的真菌的属种,色条越长代表该属种在样本中的含量越高。
图4是经过对各个样本的its测序后所得测序结果,反映出各个样本内真菌含量的相对丰度。用不同颜色代表不同的真菌的属种,色条越长代表该属种在样本中的含量越高。
图5各个样本中相对丰度在前十位的细菌及真菌,因为细菌与真菌间不能使用同样的通用引物扩增,分子生物学反应以及后续分析必须分开进行,物种之间的相对丰度或相互影响的数据无法获得。a图为健康个体皮屑的细菌与真菌的相对丰度,列表中为细菌中丰度前十位及真菌前十位的菌株,b图为临床确诊的浅表真菌感染个体的皮屑细菌与真菌的相对丰度,列表中为细菌中丰度前十位及真菌前十位的菌株。蓝色字体为细菌组成,红色字体为真菌组成。
图6是人工合成的外源性分子做标准化后,得到样本内细菌和真菌的相对丰度及比例。为了实现同一样本中真菌与细菌的相对丰度比例,每个反应中均加入浓度为0.001ng/ul,体积为1ul的外源性参照序列(图1),扩增之后,均以外源性参照为基准,进行数据标准化,使得不同属别的物种之间也可以相互比较。将设计完成的人工合成的片段(图1),以浓度为0.001ng/ul,体积为1ul加入到待测样本中,进行测序分析,分别获得细菌及真菌的相对丰度,同时也能获得人工合成片段的相对丰度。按照其进行标准化,获得单一样本中,细菌和真菌的丰度比例。a图为统计健康皮肤皮屑中细菌及真菌相对丰度在前10位的菌种,通过人工合成片段的标准化后,整合而成的相对丰度。b图为统计临床确诊的浅表真菌感染的皮屑中细菌及真菌相对丰度在前10位的菌种,通过人工合成参照序列(图1)的标准化后,整合而成的相对丰度。蓝色字体为细菌组成,红色字体为真菌组成。
图7是人工合成外源性片段的碱基组成测定图。腺嘌呤(a)205个,胸腺嘧啶(t)133个,胞嘧啶(c)218个,鸟嘌呤(g)315个,全长871个碱基,%gc含量为61.19。
图8是用16s及its前后引物扩增目的片段所得胶图。可见细菌目的片段大小为510bp左右,真菌目的片段大小为280bp左右。
具体实施方式
下面结合附图和实施例详细描述本发明,以下所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员,在不脱离本发明方法的前提下,还可以做出若干改进和补充,这些改进和补充也应视为本发明的保护范围。
实施例。
1.首先采取16sv427及16sv4533作为细菌引物序列区的前后序列,its5及its2作为真菌引物序列区的前后序列,连接区为随机序列。按照此合成思路,合成外源性参照核酸(图1),同时根据细菌16s及真菌its序列设计相应引物(图2),引物设计过程中遵循引物设计原则:引物应用核酸系列保守区内设计并具有特异性,产物不能形成二级结构,长度不超过38bp,gc含量在40%-60%之间,碱基随机分布,引物自身不能有连续4个碱基的互补,引物之间不能有连续4个碱基的互补,引物5’端可以修饰,引物3′端不可修饰,引物3′端要避开密码子的第3位等。
2.扩增片段长度的确定:扩增区域为16sv1-v4区,其中前引物5'-agagtttgatcctggctcag-3'的在16srrnagene的起始位点为8,即8f,后引物5'-ttaccgcggctgctggcac-3'在16srrnagene的终止位点为533,即533r,所以理论上的扩增区域为533-8=525bp,而且从扩增的胶图(图8)来看,目的片段的长度也超过500bp。而且,its区的扩增片段通过胶图(图8)确定其长度超过250bp。采用520bp作为16s特异识别区的片段长度及260bp作为its特异识别区片段长度,才能够有效扩增出片段,实现后续的测序分析及比较分析。
3.皮屑采用qiagendna提取试剂盒提取dna,进行0.8%琼脂糖凝胶电泳进行分子大小判断(图8),利用紫外分光光度计对dna进行定量。
4.样本核酸充分混匀后均等分开,将浓度为0.001ng/ul,体积为1ul的外源性参照核酸(图1)加入待测样本核酸。使用omegae.z.n.a.®soildnakit进行pcr扩增。
核糖体rna含有多个保守区和高度可变区,通常利用保守区域设计引物来扩增rrna基因的单个或多个可变区,然后测序分析微生物多样性。前后引物序列如表所示(图2)。前引物中的barcode是一个7个碱基的寡核苷酸序列,用来区分同一文库中的不同样品。pcr采用nebq5dna高保真聚合酶,体系见表1.,程序见表2.扩增结果进行2%琼脂糖凝胶电泳(图8),切取目的片段,细菌目的片段大小为510bp左右,真菌目的片段大小为280bp左右,然后用axygen凝胶回收试剂盒回收目的片段。
5.文库构建是利用illumina公司的truseqnanodnaltlibraryprepkit进行建库。
5.1进行的末端修复过程是利用试剂盒中的endrepairmix2将dna5’端突出的碱基切除,3’端缺失的碱基补齐,同时在5’端加上一个磷酸基团,具体步骤如下。
(1)取30ng混好的dna片段补水至60μl,加入40μlendrepairmix2。
(2)用枪吹打混匀并置于pcr仪(abi,2720)上30℃孵育30min。
(3)利用beckmanampurexpbeads纯化末端修复体系,最终用17.5μlresuspensionbuffer洗脱。
5.23’端加a,在此过程中dna的3’端会单独加上一个a碱基,目的是防止dna片段的自连,同时保证dna与3’端有一个突出t碱基的测序接头相连,具体步骤如下。
(1)向片段选择后的dna中加入12.5μla-tailingmix。
(2)用枪吹打混匀,并置于pcr仪上孵育,程序如下:37℃,30min;70℃,5min;4℃,5min;4℃,∞。
5.3加在扩增前引物前的7个特异性碱基,作为有特异性标签的接头,目的是让dna最终杂交到flowcell上,具体步骤如下。
(1)在已加a的体系中,加入2.5μlresuspensionbuffer,2.5μlligationmix和2.5μldnaadapterindex。
(2)用枪吹打混匀,并置于pcr仪上,30℃孵育10min。
(3)加入5μlstopligationbuffer。
(4)利用beckmanampurexpbeads纯化已加接头的体系。
5.4通过pcr扩增已经加上接头的dna片段,然后利用beckmanampurexpbeads纯化pcr体系。
5.5通过2%琼脂糖凝胶电泳来对文库做最终的片段选择与纯化。
6.文库质检与定量:取1μl文库,在agilentbioanalyzer机器上用agilenthighsensitivitydnakit对文库做2100质检,合格的文库应该有单一的峰,无接头。利用quant-itpicogreendsdnaassaykit在promegaquantifluor上对文库进行定量,合格的文库计算后浓度应在2nm以上。
7.测序:对合格的文库,在miseq机器上利用miseqreagentkitv3(600cycles)进行2×300bp的双端测序。首先将需要上机的文库(index不可重复)梯度稀释到2nm,然后按所需数据量比例混样。混好的文库经0.1nnaoh变性成单链进行上机测序。所上文库量的多少可根据实际情况控制在15-18pm之间。上机后,选择细菌参考数据库为greengenes,真菌参考数据库为unite进行注释。
8.分析流程。
8.1首先对高通量测序的原始下机数据根据序列质量进行初步筛查。
8.2通过质量初筛的序列按照引物和barcode信息,识别分配入对应样本,并去除嵌合体等疑问序列。
8.3对获得的序列进行otu归并划分,每个otu的代表序列用于分类地位鉴定以及系统发育学分析。
8.4根据otu在不同样本中的丰度分布,评估每个样本的多样性水平,并通过稀疏曲线反映测序深度是否达标。
8.5对各样本(组)在不同分类水平的具体组成进行分析(并检验组间是否具有统计学差异)。
8.6通过多种多变量统计学分析工具,进一步衡量不同样本(组)间的菌群结构差异及与差异相关的物种。
8.7根据物种在各样本中的组成分布,构建互作关联网络。
8.8根据16srrna基因测序结果(图3),及its基因测序结果(图4),获得样本中外源性人工合成序列(图1)及各种微生物物种的在样本中的相对丰度。基于每个物种的定量或相对定量分析结果(图5),以外源性参照核酸序列(图1)的检测量为基准进行标准化;物种间标准化后的数据可以进一步相互比较,已得到物种间的相对种类和丰度的数据(图6)。
在未考虑外源性人工合成参照核酸序列(图1)的因素时,单一样本中的细菌分类及含量与真菌的分类及含量,可以通过测序获得其相对丰度,表格中列举的是细菌和真菌中相对丰度在前十位的菌株(图5)。然而,当用外源性合成参考序核酸序列(图1)在每个样本中含量纳为标准时,各种菌株的相对丰度参照其进行标准化,得到单一样本中细菌与真菌整体的相对丰度及比例(图6)。
sequencelisting
<110>中国医科大学附属第一医院
<120>一种用于测定细菌与真菌种类和丰度比较的人工合成外源性参照分子
<130>1
<160>1
<170>patentinversion3.3
<210>1
<211>871
<212>dna
<213>人工序列
<400>1
agagtttgatcctggctcaggggacagggggagccctataattggacaagtctgggatcc60
ttgagtcctactcagccccagcggaggtgaaggacgtccttccccaggagccggtgagaa120
gcgcagtcgggggcacggggatgagctcaggggcctctagaaagagctgggaccctggga180
acccctggcctccagactggccaatcacaggcaggaagatgaaggttctgtgggctgcgt240
tgctggtcacattcctggcaggatgccaggccaaggtggagcaagcggtggagacagagc300
cggagcccgagctgcgccagcagaccgagtggcagagcggccagcgctgggaactggcac360
tgggtcgcttttgggattacctgcgctgggtgcagacactgtctgagcaggtgcaggagg420
agctgctcagctcccaggtcacccaggaactgagggcgctgatggacgagaccatgaagg480
agttgaaggcctacaaatcggaactggaggaacaactgaccccggtggcggaggagacgc540
gtgccagcagccgcggtaattttaaaattggaagtaaaagtcgtaacaaggccaaggtgg600
agcaagcggtggagacagagccggagcccgagctgcgccagcagaccgagtggcagagcg660
gccagcgctgggaactggcactgggtcgcttttgggattacctgcgctgggtgcagacac720
tgtctgagcaggtgcaggaggagctgctcagctcccaggtcacccaggaactgagggcgc780
tgatggacgagaccatgaaggagttgaaggcctacaaatcggaactggaggaacaactga840
ccccggtggcggcatcgatgaagaacgcagc871
- 还没有人留言评论。精彩留言会获得点赞!