MOFs晶体材料及其制备、应用的制作方法
本申请涉及一种mofs晶体材料及其制备、应用,属于光催化领域。
背景技术:
乙炔是重要的工业原料,目前工业制备乙炔的方法主要是电石法和天然气法,这两种方法制备的乙炔都会含有二氧化碳杂质。
利用孔材料吸附分离乙炔和二氧化碳能减少能耗和环境污染。与传统的孔材料如沸石、碳分子筛及介孔氧化硅相比,mofs材料显示出更优越的分离效果。但是由于二氧化碳和乙炔具有非常相近的物化性质,目前发现的能分离这两种气体的mofs材料极其罕见,其中多数材料却又显示出乙炔吸附大于二氧化碳的吸附,相反二氧化碳吸附优于乙炔的却仅有少数几例。显然,直接从混合气中移除二氧化碳可以减少脱附乙炔的过程而更为节能且方便,但是已有的6例此类材料其设计不可控,并且273k,1bar条件下二氧化碳和乙炔的吸附量比值最大的为6.4,分离性较低。因此可控合成具有二氧化碳吸附优于乙炔的mofs材料并提高其分离效果成为材料化学领域的研究热点。
技术实现要素:
根据本申请的一个方面,提供了一种mofs晶体材料,该晶体材料对二氧化碳的吸附优于对乙炔的吸附,省去了脱附乙炔的过程从而更为节能且方便,并且对二氧化碳和乙炔具有很好的分离效果,且该材料的合成过程可以受控。
一种mofs晶体材料,具有式ⅰ所示的化学式:
[m5(l)2(bdc)4(oh)2]·nh2o
式ⅰ
其中,m为zn和/或cd;
l表示具有式ⅱ所示化学式的配体:
式ⅱ中的r选自亚烃基中的至少一种;
bdc为对苯二甲酸根。
本申请提供的mofs晶体材料,是一种用于气体存储和分离的孔材料,且在室温下具有光致变色性能。该晶体材料合成可控,具有良好的二氧化碳和乙炔混合气的分离效果。本申请中的l配体是一种紫精类鎓盐,具有光致变色性能,配体自身的电场梯度有利于二氧化碳的吸附,并且光致变色后的配体其吡啶环间的扭转角变小,有利于增大的孔隙率。同时辅助配体bdc的加入可以保持光致变色后mofs材料的电场梯度。这样光致变色后mof材料对二氧化碳的吸附提高,而对乙炔的吸附反而减少,因此两种气体的吸附值比增高了。
具体地,r可以为c1~6亚烷基,或者还可以为苄基,或者也可以为苯亚基,优选地,r为对苯亚基。
当r为对苯亚基时,配体l的结构如下所示:
n可以为任意数值,例如n可以为9。
所述的mofs晶体材料可以为块状的晶体或者也可以为晶体粉末。
可选地,mofs晶体材料属于三斜晶系的p-1空间群。
可选地,mofs晶体材料的晶体学数据为:
优选地,mofs晶体材料的晶胞参数为:
α=104.557(3)–105.557(3)°,β=103.252(3)–104.252(3)°,
γ=95.979(3)–96.979(3)°;
可选地,当m为zn时,所述mofs晶体材料的晶体学数据为:
可选地,当m为cd时,所述mofs晶体材料的晶体学数据为:
可选地,mofs晶体材料为光致变色材料。
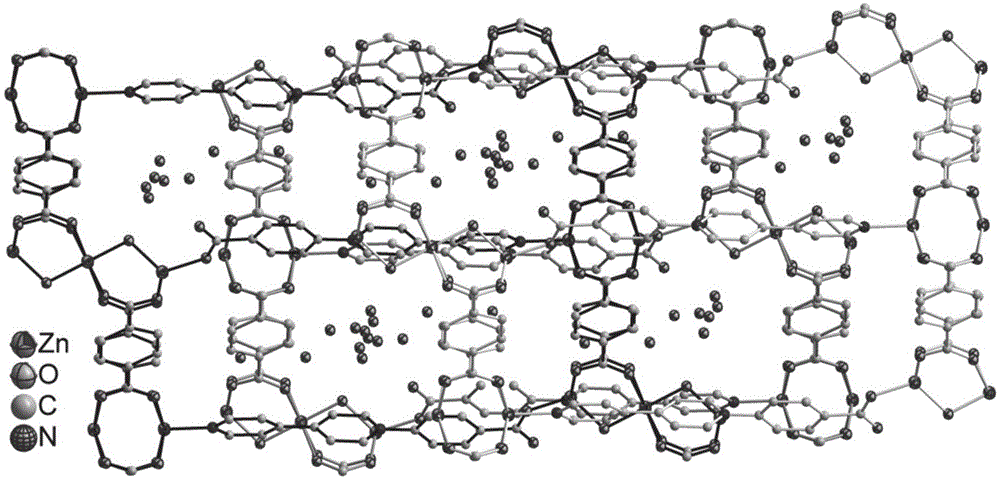
本申请提供的mofs晶体材料,具有以下结构:zn或cd离子被两种配体桥联形成具有三重交叠的立体框架结构,该结构中具有一维孔洞,孔洞内填充有客体分子,如水或者其他溶剂分子。
具体的,如图1所示,m为zn离子,r为对苯亚基,bdc为对苯二甲酸根。l配体中有两个自由端,一个为端部吡啶环中的n所提供的自由端,另一个为羧酸根所提供的自由端,该两个自由端分别与位于两种小环状结构中的zn离子相连,从而形成封闭的大环结构。其中一种小环结构是由2个对苯二甲酸各提供一个羧酸根与2个zn离子围合形成的八边形结构,两套八边形结构共用该2个zn离子。另一种小环结构是由2个对苯二甲酸各提供另外一个羧酸根与3个zn离子围合形成“8”字形的结构,在该“8”字形的结构中,双环的连接点处为1个zn离子,另外2个zn离子各处于双环的环架上。l配体端部吡啶环中的n与八边形结构中的zn连接,l配体羧酸根中的o与“8”字形的结构中的处于双环的环架上zn连接。
图1示出了3套该大环结构交叠所形成的立体框架结构。在该大环结构中,框架的一边按照八边形结构、对苯二甲酸、“8”字形的结构、对苯二甲酸、八边形结构的顺序排列,框架的另一边按照“8”字形的结构、对苯二甲酸、八边形结构、对苯二甲酸、“8”字形的结构的顺序反向排列从而组成一个大环,大环中部的“8”字形的结构和八边形结构之间还连接有l配体。
不同的大环结构之间则反向交替地交叠在一起,如图1所示,相邻的第一大环的一边按照八边形结构、对苯二甲酸、“8”字形的结构、对苯二甲酸、八边形结构的顺序排列;相邻的第二大环的一边则是按照“8”字形的结构、对苯二甲酸、八边形结构、对苯二甲酸、“8”字形的结构的顺序反向排列;相邻的第三大环的一边则又重复第一大环的一边按照八边形结构、对苯二甲酸、“8”字形的结构、对苯二甲酸、八边形结构的顺序排列,从而形成三重交叠的立体框架结构。在大环结构和大环结构之间的空隙为一维孔洞,该空洞中填充有氧离子。
根据本申请的另一方面,还提供了一种上述任一种mofs晶体材料的制备方法,至少包括步骤:
a)获得配体l:本配体的合成方法是基于已知的方法(参考文献helveticachimicaacta,2005,88,3200)改良的。
b)将含有配体l、m源、对苯二甲酸盐和水的原料,调节ph至8~9后,置于密闭容器中,于80~120℃晶化2~3天,即可得到所述的mofs晶体材料。
该mofs晶体材料为光致变色前的材料,具有储存气体的作用。例如,可以存储二氧化碳和乙炔混合气。
具体地,在步骤(a)中,配体l的制备方法可以为现有技术中任意合适的方法,本申请不做严格限定。以配体l中的r为对苯亚基为例,优选地,首先将二硝基氯苯和4,4’-联吡啶按1:1的比例加入100ml的圆底烧瓶,加入约20~40ml的丙酮,60℃条件下回流反应一天。过滤,得到的固体粉末用乙醚洗涤3次,得到中间体;然后将所得中间体和对氨基苯基酸按1:1.5的比例加入100ml的圆底烧瓶,加入约20~40ml的乙醇,90℃条件下回流反应一天。冷却,加入适量的三乙胺并搅拌直至产生米黄色沉淀,过滤,得到的固体粉末用乙醚洗涤3次,得到配体l。
具体地,首先设计合成具有光致变色性能的配体l;m源、配体l、对苯二甲酸盐配料并加入水中混合均匀,用碱液调节ph值至8.0~9.0,装入水热罐中,放于烘箱中调至80~120℃晶化温度晶化2~3天,反应结束后取出冷却至室温,得到mofs晶体材料。
可选得,步骤b)中的m源选自硝酸锌、氯化锌、硝酸镉、氯化镉的至少一种;
步骤b)中采用氢氧化钠将所述含有配体l、m源、对苯二甲酸盐和水的原料的ph值调节至8~9。
上述任一项所述的mofs晶体材料经光照射后得到的光致变色后材料。
优选地,采用波长为200~450nm氙灯照射0.5~2h。
本申请还提供了上述任一种mofs晶体材料、上述光致变色后材料在储存和/或分离二氧化碳和乙炔中的应用。
具体的,利用制备好的mofs晶体材料吸附二氧化碳和乙炔混合气以将混合气储存在mofs晶体中,然后将该晶体置于氙灯下照射一段时间,例如1个小时,即可得到光致变色后的晶体,光致变色前后的mofs晶体对二氧化碳和乙炔的吸附量的比值发生了变化,从而将二氧化碳和乙炔分离。
本申请能产生的有益效果包括:
1)本申请所提供的mofs晶体材料,在273k、1bar条件下,对二氧化碳和乙炔的吸附量比值在光照后为7.1,因此该材料具有很好的分离效果。
2)本申请所提供的mofs晶体材料,对二氧化碳的吸附大于对乙炔的吸附,减少了脱附乙炔的过程,更为节能且方便。
3)本申请所提供的制备mofs晶体材料的方法,使得mofs晶体材料的合成过程受控。
附图说明
图1为试样cⅰ-1#中的[zn5(l)2(bdc)4(oh)2]n·9nh2o的立体结构示意图;
图2为试样cⅰ-1#中的[zn5(l)2(bdc)4(oh)2]n·9nh2o的粉末x-射线衍射图谱;
图3为试样cⅰ-1#中的[zn5(l)2(bdc)4(oh)2]n·9nh2o的对二氧化碳和乙炔的在光照前后的吸附图谱。
具体实施方式
下面结合实施例详述本申请,但本申请并不局限于这些实施例。
如无特别说明,本申请的实施例中的原料和催化剂均通过商业途径购买。
实施例1配体l的制备
制备过程:首先将二硝基氯苯和4,4’-联吡啶按1:1的比例加入100ml的圆底烧瓶,加入约30ml的丙酮,60摄氏度条件下回流反应一天。过滤,得到的固体粉末用乙醚洗涤3次,得到中间体;然后将所得中间体和对氨基苯基酸按1:1.5的比例加入100ml的圆底烧瓶,加入约30ml的乙醇,90摄氏度条件下回流反应一天。冷却,加入适量的三乙胺并搅拌直至产生米黄色沉淀,过滤,得到的固体粉末用乙醚洗涤3次,得到配体。将此样品记作:l-1#,在l-1#中,r为对苯亚基。
制备过程:将卤代烃酸和4,4’-联吡啶按1:1的比例加入100ml的圆底烧瓶,加入约35ml的乙腈,70摄氏度条件下回流反应一天。过滤,得到的固体粉末用乙醚洗涤3次,得到配体,将此样品记作:l-2#,在l-2#中,r为亚烷基。
制备过程:将对卤甲基苯甲酸和4,4’-联吡啶按1:1的比例加入100ml的圆底烧瓶,加入约35ml的乙腈,70摄氏度条件下回流反应一天。过滤,得到的固体粉末用乙醚洗涤3次,得到配体,将此样品记作:l-3#,在l-3#中,r为苄基。
实施例2mofs晶体材料的制备
将六水合硝酸锌(1189mg),l-1#配体(138mg)和对苯二甲酸钠(210mg)配料,并加入10毫升的水搅拌均匀后,用浓度为0.2m的氢氧化钠水溶液调节ph值至8.0,装入20毫升的聚四氟乙烯水热罐,放入烘箱中加热至100℃,保温2天后,取出自然冷却至室温,得到化学式为[zn5(l)2(bdc)4(oh)2]·nh2o的mofs晶体,记作cⅰ-1#。
将六水合硝酸锌(1189mg),l-2#配体(138mg)和对苯二甲酸钠(210mg)配料,并加入10毫升的水搅拌均匀后,用浓度为0.2m的氢氧化钠水溶液调节ph值至9.0,装入20毫升的聚四氟乙烯水热罐,放入烘箱中加热至80℃,保温2天后,取出自然冷却至室温,得到化学式为[zn5(l)2(bdc)4(oh)2]·nh2o的mofs晶体,记作cⅰ-2#。
将六水合硝酸锌(1189mg),l-3#配体(138mg)和对苯二甲酸钠(210mg)配料,并加入10毫升的水搅拌均匀后,用浓度为0.2m的氢氧化钠水溶液调节ph值至8.0,装入20毫升的聚四氟乙烯水热罐,放入烘箱中加热至90℃,保温2天后,取出自然冷却至室温,得到化学式为[zn5(l)2(bdc)4(oh)2]·nh2o的mofs晶体,记作cⅰ-3#。
将四水合硝酸镉(1234mg),l-1#配体(138mg)和对苯二甲酸钠(210mg)配料,并加入10毫升的水搅拌均匀后,用浓度为0.2m的氢氧化钠水溶液调节ph值至9.0,装入20毫升的聚四氟乙烯水热罐,放入烘箱中加热至110℃,保温3天后,取出自然冷却至室温,得到化学式为[cd5(l)2(bdc)4(oh)2]·nh2o的mofs晶体,记作cⅱ-1#。
将四水合硝酸镉(1234mg),l-2#配体(138mg)和对苯二甲酸钠(210mg)配料,并加入10毫升的水搅拌均匀后,用浓度为0.2m的氢氧化钠水溶液调节ph值至9.0,装入20毫升的聚四氟乙烯水热罐,放入烘箱中加热至80℃–120℃,保温3天后,取出自然冷却至室温,得到化学式为[cd5(l)2(bdc)4(oh)2]·nh2o的mofs晶体,记作cⅱ-2#。
将四水合硝酸镉(1234mg),l-3#配体(138mg)和对苯二甲酸钠(210mg)配料,并加入10毫升的水搅拌均匀后,用浓度为0.2m的氢氧化钠水溶液调节ph值至8.0,装入20毫升的聚四氟乙烯水热罐,放入烘箱中加热至120℃,保温3天后,取出自然冷却至室温,得到化学式为[cd5(l)2(bdc)4(oh)2]·nh2o的mofs晶体,记作cⅱ-3#。
实施例3样品的结构表征
样品cⅰ-1#~cⅰ-3#、cⅱ-1#~cⅱ-3#研磨后的x-射线粉末衍射物相分析(xrd)在rigaku公司的miniflexⅱ型x射线衍射仪上进行,cu靶,kα辐射源(λ=0.154184nm)。结果表明,实施例2所制备的样品均为高纯度和高结晶度的样品。
样品cⅰ-1#~cⅰ-3#、cⅱ-1#~cⅱ-3#的x-射线单晶衍射在mercuryccd型单晶衍射仪上进行,mo靶,kα辐射源(λ=0.07107nm),测试温度293k。并通过shelxtl97对其进行结构解析。以cⅰ-1#为典型代表,通过将单晶衍射结构数据cif文件导入软件mercury3.5计算得到的xrd衍射理论图谱与实验测得的xrd衍射图谱比较如图2所示,可以看出,通过单晶数据拟合得到的xrd衍射图谱与其实验测得的xrd衍射图谱高度一致,证明所得样品为高纯度和高结晶度的样品。
x-射线粉末衍射和多晶衍射结果表明:
cⅰ-1#~cⅰ-3#、cⅱ-1#~cⅱ-3#(化学式[m5(l)2(bdc)4(oh)2]·nh2o)均属于三斜晶系的p1空间群。且形成金属离子(zn离子或者cd离子)被两种配体桥联形成具有三重交叠的立体框架结构,该结构中具有一维孔洞,孔洞内填充有氧离子。其中,以cⅰ-1#为典型代表,具体结构如图1所示。
具体地,cⅰ-1#~cⅰ-3#的晶胞参数为
实施例4光致变色后材料的制备
分别将cⅰ-1#~cⅰ-3#、cⅱ-1#~cⅱ-3#的置于波长为200~450nm氙灯下照射1h,得到光致变色后的材料样品,分别记为cⅰ-1#-l~cⅰ-3#-l、cⅱ-1#-l~cⅱ-3#-l。
实施例5二氧化碳和乙炔吸附比值测试实验
分别将100~150毫克cⅰ-1#~cⅰ-3#、cⅱ-1#~cⅱ-3#、cⅰ-1#-l~cⅰ-3#-l、cⅱ-1#-l~cⅱ-3#-l的晶体或粉末在80℃左右的温度下抽真空活化约16小时后,用asap2020的吸附系统测试273k条件下晶体化合物光致变色前后的二氧化碳和乙炔的等温吸附、脱附性能。以压力为横坐标,所测吸附性能为纵坐标作图,得到不同气体在273k、1bar条件下的吸附值,判断mofs晶体材料对二氧化碳和乙炔气体的存储能力、以及对这两种气体的分离能力。
结果表明:光致变色前的样品cⅰ-1#~cⅰ-3#、cⅱ-1#~cⅱ-3#和光致变色后的样品cⅰ-1#-l~cⅰ-3#-l、cⅱ-1#-l~cⅱ-3#-l均可以较好的吸附/分离二氧化碳和乙炔。其中,光致变色后的样品对二氧化碳和乙炔的分离性能优于光致变色前的样品。
以cⅰ-1#和cⅰ-1#-l为典型代表,图3为[zn5(l)2(bdc)4(oh)2]·nh2o的吸附图谱,图3表明:cⅰ-1#在光致变色后对二氧化碳和乙炔气体具有较好的分离性能。如表1所示:cⅰ-1#晶体材料对二氧化碳和乙炔的吸附量比值从变色前的5.0提升到变色后的7.1。
表1在273k、1bar条件下,cⅰ-1#多晶材料变色前后对二氧化碳和乙炔的吸附量
以上所述,仅是本申请的几个实施例,并非对本申请做任何形式的限制,虽然本申请以较佳实施例揭示如上,然而并非用以限制本申请,任何熟悉本专业的技术人员,在不脱离本申请技术方案的范围内,利用上述揭示的技术内容做出些许的变动或修饰均等同于等效实施案例,均属于技术方案范围内。
- 还没有人留言评论。精彩留言会获得点赞!