一种单管实现连续区域的非特异性PCR扩增方法与流程
本发明实施例涉及基因工程技术领域,具体涉及一种单管实现连续区域的非特异性pcr扩增方法及单管实现多重pcr扩增方法。
背景技术:
随着技术的进步,高通量测序技术由lifesciences公司开发出,2005年春发表人类历史上第一篇高通量测序文章(genomesequencinginmicrofabricatedhigh-densitypicolitrereactors),展示了广阔的应用前景。通过高通量测序可以得到snp、cnv、indel、基因组结构变异、群体多态性等重要信息。
很多物种基因组较大,比如人基因组,有30亿个碱基,而在很多研究中,并不需要对全基因组进行测序,因此需要特定多靶区域测序的方法。这其中主要以探针捕获和多重pcr两种方法为主,首先安捷伦2009年第一次推出探针靶向捕获的技术,并于2010年推出第一个人外显子区域捕获产品。多重pcr技术应用于高通量测序最早见于lifetechnologies于2012年推出的ampliseq技术,该技术的推出,使得小而精的靶向区域捕获迅速的进入科研和临床应用。另外,联川生物在2014年推出了基于特殊引物设计(omega引物)的多重技术,将两轮pcr方式简化为一轮。padlockprimer最早由mnilsson等人于1994年提出,用于单点的snp检测效果明显;再后来,哈佛大学的georgem.church等人第一次将padlock结构的probe用于靶区域测序。随着技术的发展和进步,2016年,美国专利描述了利用互补结构解决了两扩增子之间的重叠区扩增问题,实现单管pcr可以进行连续区域的捕获,解决了传统多重pcr捕获实现连续区域需要分多管pcr的局限。
ampliseq技术虽然最早提出了多重捕获的技术方法,但对连续区域并未能实现单管内捕获,且ampliseq技术并未体现降低基因组非特异性扩增的技术;omega引物的多重技术也能实现多重pcr捕获效果,同时,其omega结构的引物,可以降低非特异性基因组扩增,且能将实验在一轮pcr内完成,但对于连续区域的捕获,仍然需要分两管或者多管以上。padlockprimer各个文章仍然用的是单条probe通过连接酶介导链接方法来实现单点以及小区域的靶向捕获,该方法需要的基因组投入量较大,且时间较长,一般需要2天左右时间。而在ampliseq技术基础上做了较大改进,采用重叠区域相邻两扩增子的引物序列有互补成为茎环结构,可以实现连续区域的pcr靶向捕获,但该方法并没有解决对重复区附近或者相似序列的非特异性扩增的问题。
综上所述,现有技术中,在基因测序的过程中,无法通过单管扩增实现对连续区域的基因捕获,并且具有明显降低特异性基因组扩增的效果。
技术实现要素:
为此,本发明实施例提供一种单管实现连续区域的非特异性pcr扩增方法,以解决现有技术中无法通过单管扩增实现对连续区域基因进行捕获的问题。
为了实现上述目的,本发明的实施方式提供如下技术方案:
一种单管实现连续区域的非特异性pcr扩增方法,其包括:根据扩增的靶序列设计plmmamp引物,其中,所述plmmamp引物包括模板杂交区核苷酸序列、靶区域延伸区核苷酸序列以及连接所述模板杂交区核苷酸序列和靶区延伸区核苷酸序列的中间链接区核苷酸序列,所述模板杂交区核苷酸序列为扩增子核苷酸序列一部分,所述靶区域延伸区核苷酸序列为扩增子核苷酸序列边界区的一部分,且所述模板杂交区核苷酸序列与所述靶区域延伸区核苷酸序列无重叠。
优选的,还包括多重pcr扩增方法,根据多个把序列设计多对所述plmmamp引物。
优选的,所述多重pcr扩增中,相邻所述靶序列的重叠区内,所述plmmamp引物的中间链接区的核苷酸序列相同。
优选的,所述模板杂交区核苷酸序列与所述中间链接区核苷酸序列采用无法用于扩增的非碱基修饰,如c3、c6基因修饰。
优选的,所述plmmamp引物3’端为靶区域延伸区核苷酸序列,5’端为模板杂交区核苷酸序列,靶区域延伸区核苷酸序列tm值的范围在40-50度之间,所述模板杂交区核苷酸序列tm值范围在65-75度之间。
优选的,所述中间链接区核苷酸序列的核苷酸碱基的数量至少为1个。
优选的,所述靶序列为微生物核苷酸序列、人染色体核苷酸序列、动物染色体核苷酸序列或植物染色体核苷酸序列。
优选的,所述模板杂交区核苷酸序列的碱基数量为12-25个;
所述靶区域延伸区核苷酸序列的碱基数量为15-40个。
优选的,所述pcr扩增采用具有双链5’-3’外切活性的dna聚合酶。
本发明还提供单管实现连续区域的非特异性pcr扩增方法获得的pcr产物在测序中的应用,其中,所述中间链接区核苷酸序列采用高通量测序平台通用序列。
根据本发明的实施方式,本发明实施例的一种单管实现连续区域的非特异性pcr扩增方法具有如下优点:
本发明中,plmmamp(pad-lockmediatedmutiplexamplification)技术结合padlockprimer技术特点,利用pcr上下游引物均采用padlock结构的引物,其中3’端作为靶区延伸区,tm(meltingtemperature)设置较低,5’端作为杂交区,tm值设置较高;对于有重叠区临近扩增子,设计时,5’端的模板杂交区在重叠区之外,这样重叠区的扩增就无法进行,原先连续区域多重捕获需要分多管的,可以在单管内完成多重pcr捕获,极大的降低了劳动成本和耗用成本。另外一方面,本发明的plmmamp引物由于具有杂交区和延伸区双区功能,因此,能够很好的避开重复区的非特异性扩增现象,降低非特异性扩增产物。
附图说明
为了更清楚地说明本发明的实施方式或现有技术中的技术方案,下面将对实施方式或现有技术描述中所需要使用的附图作简单地介绍。显而易见地,下面描述中的附图仅仅是示例性的,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据提供的附图引伸获得其它的实施附图。
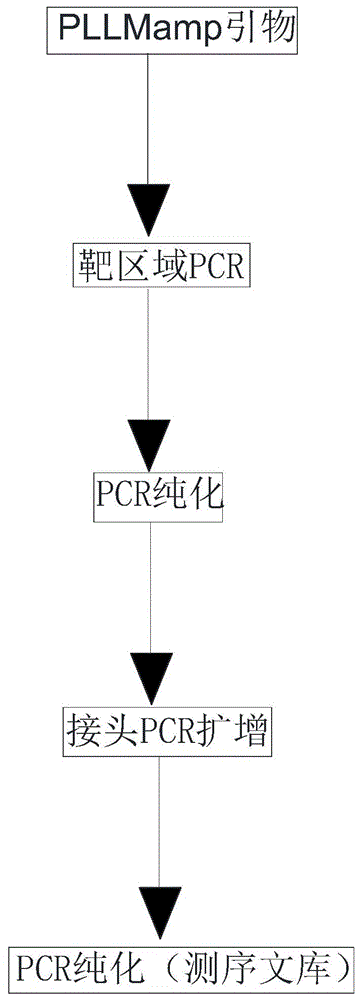
图1为本发明的实施例提供的利用本发明的plmmamp引物pcr扩增测序的技术流程图;
图2为本发明的实施例1的常规pcr引物和plmmamp引物pcr扩增产物的电泳图,其中,lane1:plmmamp引物pcr产物电泳条带;lane2:常规引物pcr产物电泳条带;lanem:dna标准marker。
图3为本发明的实施例2常规pcr引物和plmmamp引物pcr扩增产物的电泳图,lane1:常规引物pcr产物电泳条带;lane2:plmmamp引物pcr产物电泳条带;lanem:dna标准marker。
图4为本发明的实施例3常规pcr引物和plmmamp引物pcr扩增产物的电泳图;lane1-1,1-2,1-3:常规设计brca引物pcr产物电泳条带;lane2-1,2-2,2-3:plmmamp设计引物pcr产物电泳条带;lanem:dna标准marker。
具体实施方式
以下由特定的具体实施例说明本发明的实施方式,熟悉此技术的人士可由本说明书所揭露的内容轻易地了解本发明的其他优点及功效,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
本发明实施例中,所谓靶序列为在pcr扩增过程中的目标基因扩增序列。
本发明实施提供一种单管实现连续区域的非特异性pcr扩增方法,plmmamp引物包括:模板杂交区核苷酸序列,即probe、靶区域延伸区核苷酸序列,即primer,以及中间链接区核苷酸序列。其中模板杂交区核苷酸序列的长度为15-40个碱基,tm值在65-75度之间;模板延伸区核苷酸序列长度为12-25个碱基,tm值在40-50度之间,用于模板区域的延伸。其中,模板杂交区核苷酸序列位于扩增子核苷酸序列内部,靶区域延伸区核苷酸序列位于扩增子核苷酸序列边界。模板杂交区核苷酸序列与靶区延伸区核苷酸序列不能处于相互重叠状态,需有1个或者以上碱基的空域,利于聚合酶延伸。模板杂交区核苷酸序列与靶区延伸区核苷酸序列之间有一段中间链接序列,用于将两端区域连接,中间链接区核苷酸序列可以是高通量ngs测序测序平台的通用序列。该通用序列选自以下序列a(seq.no17)-f(seq.no22)中的任一序列,或者任一序列中的部分序列。a(seq.no17):tcgtcggcagcgtcagatgtgtataagagacag;b(seq.no18):acactctttccctacacgacgctcttccgatct;c(seq.no19):aatgatacggcgaccaccgagatctacactctttccctacacgacgctcttccgatct;d(seq.no20):gtctcgtgggctcggagatgtgtataagagacag;e(seq.no21):gtgactggagttcagacgtgtgctcttccgatct;f(seq.no22):gtgactggagttccttggcacccgagaattcca。
在dna扩增上,只有模板杂交区核苷酸序列,靶区域延伸区核苷酸序列同时与被扩增模模板,即靶序列结合,才能进行pcr扩增。
对于连续区域的情况,相邻两个扩增子各自的交叉引物区设计时,模板杂交区核苷酸序列probe,由于在各自扩增子内部,并不在相邻扩增子内,这样使得相邻扩增子的重叠区无法有效扩增,这样可实现连续区域的在单个管中扩增。进一步的,在模板杂交区核苷酸序列probe与中间链接区核苷酸序列可采用无法用于扩增的非碱基修饰如c3、c6等基团修饰。
如图1所示,本发明实施例还提供一种多重pcr法,其包括使用plmmamp设计的多重pcr组合plmmamp引物对,对dna模板进行扩增,其中在pcr扩增过程中,采用具有双链5’-3’外切活性的dna聚合酶。
本发明实施例的pcr扩增方法流程如下:第一轮pcr反应:0.2mlpcr按照如下体系配置反应:kapa2xhi-fipcrmix15-25μl,1ulplatinumtaq,primermix(每条引物50nm-1um)2-16μl,基因组dna10-300ng,共30-50μl。扩增程序设置:95℃3min;95℃15s,60℃4min,15-30个循环,72℃4min,10℃保存;
pcr产物纯化:向pcr反应液/酶促反应液加入1.0-1.5倍ampurexpbeads混匀,用强力磁铁或磁力架吸附磁珠,用移液器小心吸取上清,抛弃上清,留磁珠;加入100μl70%乙醇洗涤后蒸干。
第二轮pcr反应:采用0.2mlpcr管,在超净台里按照如下体系配置反应:phusionhigh-fidelitypcrmix15μl,f通用引物(10um)1μl,rindex引物(10um)1μl,去离子水13μl,总共30μl,直接加到上步的回收管内。
f通用seq.no1:
aatgatacggcgaccaccgagatctacactctttccctacacgacgctcttccg
r_indexseq.no2:
caagcagaagacggcatacgagatgtgactggagttccttggcacccgagaattcca
扩增程序:95℃1min;循环95℃15s,60℃15s,72℃15s,6-8个循环。
pcr反应结束后加入0.6-1.2倍ampurexpbeads混匀,用强力磁铁或磁力架吸附磁珠,用移液器小心吸取上清,抛弃上清,留磁珠;加入100μl70%乙醇洗涤后蒸干,用20μl去离子水溶解dna,该pcr产物可利用illumina测序平台进行测序。
本发明实施例的plmmamp引物模板杂交区核苷酸序列,靶区域延伸区核苷酸序列进行分区设计,将两者分别位于不同的基因组位置,避开基因组的严重重叠区,有益于降低模板的非特异性扩增;本发明实施例plmmamp引物的模板杂交区核苷酸序列,靶区域延伸区核苷酸序列分区设计,模板杂交区核苷酸序列位于各自扩增子内部,对于相邻扩增子,有益于降低重叠区的扩增,实现连续区域单管内多重pcr扩增;本发明实施例的plmmamp引物由于靶区域延伸区核苷酸序列相对常规引物短一些,因此能节省测序数据,引物越短,引物浪费的数量越少。
本发明实施例采用plmmamp设计类型引物为上下游引物共同作pcr,模板杂交区核苷酸序列,靶区域延伸区核苷酸序列分区设计;plmmamp引物中间链接区核苷酸序列可以是基因组非同源的任意序列,一般设计为测序设备通用序列;相邻重叠区的plmmamp引物的中间链接区的核苷酸序列相同。
实施例1
本发明实施例采用常规pcr引物与本发明实施例的plmmamp引物,扩增基因组一段序列,即靶序列,其前后有重复区,以验证本发明实施例的plmmamp引物的良好扩增性。
1、扩增区域为人染色体:chr7:2022500-2022799中间的一段序列,如seq.no3所示,有下划线字母区为靶序列,非划线字母区是两侧核苷酸序列seq.no3:
gaaattacccatatattcctattttattaagagtttttaatcaaggctgagtgttggattttgtcaaacgcccccatgaagagggtattctataaattgatatcctaaaatcaagctgacaggctggtcctggaataaatcttactgcatatgatgtatttcaacctgctgttgaatttcactcgtgacatgttaacaatgacactgatacacgtatgcgcgtgcatgcacacacacacacgtgcacacacgcacacacacccacagacgcgcgcgcgcgcgcgcgcgcgcacacacaca
2、常规引物:
a:常规设计
f(seq.no4):
tccttggcacccgagaattccagctgagtgttggattttgtcaaac
r(seq.no5):
cctacacgacgctcttccgatctgtgtgcacgtgtgtgtgtgtg
b:本发明实施例的plmmamp引物设计,有下划线字母表示核酸序列为中间链接区核苷酸序列:
f(seq.no6):
atgaagagggtattctataaattgatatcctaaaatcaagctccttggcacccgagaattccagttggattttgtcaaac
r(seq.no7):
cacgcgcatacgtgtatcagtgtccctacacgacgctcttccgatctgtgtgcacgtgtgtg
3、引物pcr:对每对pcr引物,进行如下pcr反应,采用0.2mlpcr管,每对引物在超净台里按照如下体系配置反应:kapa2xhi-fipcrmix15μl;platinumtaq1ul;primerf(1um)2μl;primerr(1um)2μl;基因组dna10ng;h2o总共30μl。
扩增程序设置:95℃3min;95℃15s,60℃20s,72℃1min,2-4步骤25个循环。72℃4min,10℃保存。
pcr结束后,所有pcr产物进行电泳检测,如图2所示,本发明实施例的plmmamp引物pcr扩增产物的电泳条带11单一,常规设计引物pcr产物的电泳条带21呈弥散状。这是由于靶区域两侧全是基因组重复区,因此,常规pcr引物扩增时,会产生很多非特异性产物,但靶区域的大写字母部分是特异性核苷酸序列区域,没有高度重复,plmmamp引物利用模板杂交区核苷酸序列位于高度特异区特点,降低了非pcr特异性扩增,所扩增的产物较为单一,无非特异性产物。
实施例2
本实施例采用2对常规引物与本发明实施例的plmmamp引物进行多重扩增,扩增连续序列,其有重叠区,验证plmmamp引物可以有效的避免重叠区非特异性扩增。
1、扩增区域为人染色体:下划线字母核苷酸序列部分区域为重叠区,seq.no8
cctcatccctttcagggaagaggttgtgggtagggggactcccctgcctggtagcctcaaagcttcttagttcatccatttccgacaattccaggctccacagtggcaggggatcttgtgctgatcgtcctcaaaatcaaactgatagtcataggttagctgaaaagagaagagggcagaatcaatgctag
2、引物设计
a:常规设计:
001f(seq.no9):
cctacacgacgctcttccgatctcctcatccctttcagggaaga
001r(seq.no10):
tccttggcacccgagaattccattgaggacgatcagcacaagatc
001f(seq.no11):
cctacacgacgctcttccgatctagttcatccatttccgacaattcc
001r(seq.no12):
tccttggcacccgagaattccactagcattgattctgccctcttct
b:plmmamp引物,划线部分字母区表示核苷酸序列为中间链接区核苷酸序列
001f(seq.no13):
gaagaggttgtgggtagggggactcctacacgacgctcttccgatctcctcatccctttcag
001r(seq.no14):
agatcccctgccactgtggagctccttggcacccgagaattccattgaggacgatcagc
001f(seq.no15):
gctccacagtggcaggggatctccttggcacccgagaattccaatttccgacaattcc
001r(seq.no16):
cctcttctcttttcagctaacctatgactatcagcctacacgacgctcttccgatctctagcattgattctgc
3、引物pcr扩增
将常规pcr引物按照每条终浓度50nm按照1:1混合,2对plmmamp引物按照每条终浓度50nm按照1:1混合,形成各自的primermix,对每primermix,进行如下pcr反应;
采用0.2mlpcr管,每对引物在超净台里按照如下体系配置反应:kapa2xhi-fipcrmix25μl,primermix16μl,基因组dna300ng,h2o总共50μl。
扩增程序设置:95℃3min;95℃15s,60℃4min,2-3步骤20个循环。95℃15s,72℃1min,4-5步骤6个循环。72℃4min,10℃保存。
pcr结束后,所有产物进行电泳检测,如图3所示,扩增产物的电泳图,其中lane1为常规pcr引物扩增条带,lane2为本发明实施例plmmamp引物扩增条带。本发明实施例的plmmamp引物在pcr扩增过程中,在重叠区k的扩增产物非常少,常规设计引物pcr产物在重叠区m特别明显。这是由于本发明实施例的plmmamp引物使得重叠区的扩增效率极大降低甚至无法扩增。
实施例3
本发明实施例采用常规引物与本发明实施例的plmmamp引物对brcai/ii两个基因进行多重pcr扩增,本发明实施例的plmmamp引物可以有效进行连续区域的多重扩增。
扩增区域为人brcai,brcaii两个基因全外显子区域;对其进行引物设计;a:常规引物122对;b:plmmamp引物122对。
引物pcr:将122常规引物按照每条终浓度200nm按照1:1混合,122对plmmamp引物按照每条终浓度200nm按照1:1混合,形成各自的primermix。对每primermix,进行如下pcr反应;每个重复3个样本。
采用0.2mlpcr管,每对引物在超净台里按照如下体系配置反应:5xnebq5pcrmix10μl;primermix12μl;基因组dna50ng;h2o总共50μl。
扩增程序设置:95℃3min;95℃15s,60℃4min,2-3步骤13个循环。95℃15s,72℃1min,4-5步骤6个循环。72℃4min,10℃保存。pcr结束后,所有pcr扩增产物进行电泳检测。
如图4所示,本发明实施例的plmmamp引物可以很好的进行超多重pcr扩增。在重叠区的扩增非常少,常规引物pcr扩增产物在重叠区,非特异性产物扩增特别明显。本发明实施例的plmmamp引物使得重叠区的扩增效率极大降低甚至无法扩增。
实施例4
本发明实施例利用plmmamp引物设计brcai/ii两个基因来说明如何进行多重扩增后高通量ngs测序。
第一轮引物pcr:按照预先测试的引物浓度将122对plmmamp引物混合,形成各自的primermix对每primermix,进行如下pcr反应;重复2个样本。采用0.2mlpcr管,每对引物在超净台里按照如下体系配置反应:2xphusionhigh-fidelitypcrmix15μl;primermix8μl;基因组dna10ng,300ngh2o总共30μl。
扩增程序设置:95℃3min;95℃15s,60℃4min,2-3步骤13个循环。95℃15s,72℃1min,4-5步骤6个循环。72℃4min,10℃保存。
本发明实施例中向30ulpcr反应液中加入1.1倍pcr体积ampurexpbeads;用移液器50ul量程上下吹打10-15次,以使pcr扩增产物与ampurexpbeads充分混匀,室温静置1-2分钟。
用强力磁铁或磁力架吸附磁珠,直至溶液澄清为止。用移液器小心吸取上清,抛弃上清,留磁珠。加入100ul75%乙醇,用磁力架反复在不同的两面来回吸附磁珠以充分悬浮磁珠便洗涤。用磁铁或磁力架吸附磁珠,2分钟,直至溶液澄清为止。用移液器小心去除上清,避免吸到磁珠。室温放置,直至乙醇挥发干净。本步骤磁珠亦可放于50度烘箱5分钟左右。
第二轮pcr:第二轮pcr主要是引入illumina测序相应接头与barcode。
a、按照下列的反应体系准备反应混合物,不同的样品请选择不同的barcode引物。2xphusionhigh-fidelitypcrmix15μl;h2o13ul;f通用引物1ul;index_r_xx1ul,总体积:30ul。将30ulmix直接加入到对应的样本磁珠中充分混合,然后进行如下pcr反应。
b、pcr反应条件:度1min;98度20s;60度20s;72度30s;2-3步骤6个循环。72度2min,10度保存。
pcr结束后,进行pcr产物回收,具体步骤如下:pcr产物中加0.8倍体积磁珠(30ul体系加24ul)用移液器量程上下吹打,以使回收产物与ampurexpbeads充分混匀。室温静置2分钟。用强力磁铁或磁力架吸附磁珠,2分钟,直至溶液澄清为止。用移液器小心吸取上清,抛弃上清,留磁珠。加入100ul75%乙醇,用磁力架反复在不同的两面来回吸附磁珠以充分悬浮磁珠便洗涤,用移液器小心去除上清。室温放置,直至乙醇挥发干净。6加入20-40μl去离子水,充分悬浮磁珠,室温静置2min以洗脱dna。将磁珠用磁铁吸附,所得到上清dna溶液吸至一新的1.5/0.5/0.2ml离心管/96孔pcr管。
本步骤的pcr产物可直接用于后续高通量测序,测序统计结果如表1所示:
表1
从表1可看,本发明实施例的plmmamp引物用于超过多重扩增后高通量ngs测序,覆盖率和均一性(目标区深度>20%平均深度比例)均超过95%,达到100%。
虽然,上文中已经用一般性说明及具体实施例对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
sequencelisting
<110>上海何因生物科技有限公司上海优甲医疗科技有限公司深圳人体密码基因有限公司石家庄博瑞迪生物技术有限公司
<120>一种单管实现连续区域的非特异性pcr扩增方法
<130>20181204
<160>22
<170>patentinversion3.5
<210>1
<211>54
<212>dna
<213>人工序列(artificialsequence)
<400>1
aatgatacggcgaccaccgagatctacactctttccctacacgacgctcttccg54
<210>2
<211>57
<212>dna
<213>人工序列(artificialsequence)
<400>2
caagcagaagacggcatacgagatgtgactggagttccttggcacccgagaattcca57
<210>3
<211>300
<212>dna
<213>人工序列(artificialsequence)
<400>3
gaaattacccatatattcctattttattaagagtttttaatcaaggctgagtgttggatt60
ttgtcaaacgcccccatgaagagggtattctataaattgatatcctaaaatcaagctgac120
aggctggtcctggaataaatcttactgcatatgatgtatttcaacctgctgttgaatttc180
actcgtgacatgttaacaatgacactgatacacgtatgcgcgtgcatgcacacacacaca240
cgtgcacacacgcacacacacccacagacgcgcgcgcgcgcgcgcgcgcgcacacacaca300
<210>4
<211>46
<212>dna
<213>人工序列(artificialsequence)
<400>4
tccttggcacccgagaattccagctgagtgttggattttgtcaaac46
<210>5
<211>44
<212>dna
<213>人工序列(artificialsequence)
<400>5
cctacacgacgctcttccgatctgtgtgcacgtgtgtgtgtgtg44
<210>6
<211>80
<212>dna
<213>人工序列(artificialsequence)
<400>6
atgaagagggtattctataaattgatatcctaaaatcaagctccttggcacccgagaatt60
ccagttggattttgtcaaac80
<210>7
<211>62
<212>dna
<213>人工序列(artificialsequence)
<400>7
cacgcgcatacgtgtatcagtgtccctacacgacgctcttccgatctgtgtgcacgtgtg60
tg62
<210>8
<211>191
<212>dna
<213>人工序列(artificialsequence)
<400>8
cctcatccctttcagggaagaggttgtgggtagggggactcccctgcctggtagcctcaa60
agcttcttagttcatccatttccgacaattccaggctccacagtggcaggggatcttgtg120
ctgatcgtcctcaaaatcaaactgatagtcataggttagctgaaaagagaagagggcaga180
atcaatgctag191
<210>9
<211>44
<212>dna
<213>人工序列(artificialsequence)
<400>9
cctacacgacgctcttccgatctcctcatccctttcagggaaga44
<210>10
<211>45
<212>dna
<213>人工序列(artificialsequence)
<400>10
tccttggcacccgagaattccattgaggacgatcagcacaagatc45
<210>11
<211>47
<212>dna
<213>人工序列(artificialsequence)
<400>11
cctacacgacgctcttccgatctagttcatccatttccgacaattcc47
<210>12
<211>46
<212>dna
<213>人工序列(artificialsequence)
<400>12
tccttggcacccgagaattccactagcattgattctgccctcttct46
<210>13
<211>62
<212>dna
<213>人工序列(artificialsequence)
<400>13
gaagaggttgtgggtagggggactcctacacgacgctcttccgatctcctcatccctttc60
ag62
<210>14
<211>59
<212>dna
<213>人工序列(artificialsequence)
<400>14
agatcccctgccactgtggagctccttggcacccgagaattccattgaggacgatcagc59
<210>15
<211>58
<212>dna
<213>人工序列(artificialsequence)
<400>15
gctccacagtggcaggggatctccttggcacccgagaattccaatttccgacaattcc58
<210>16
<211>73
<212>dna
<213>人工序列(artificialsequence)
<400>16
cctcttctcttttcagctaacctatgactatcagcctacacgacgctcttccgatctcta60
gcattgattctgc73
<210>17
<211>33
<212>dna
<213>人工序列(artificialsequence)
<400>17
tcgtcggcagcgtcagatgtgtataagagacag33
<210>18
<211>33
<212>dna
<213>人工序列(artificialsequence)
<400>18
acactctttccctacacgacgctcttccgatct33
<210>19
<211>58
<212>dna
<213>人工序列(artificialsequence)
<400>19
aatgatacggcgaccaccgagatctacactctttccctacacgacgctcttccgatct58
<210>20
<211>34
<212>dna
<213>人工序列(artificialsequence)
<400>20
gtctcgtgggctcggagatgtgtataagagacag34
<210>21
<211>34
<212>dna
<213>人工序列(artificialsequence)
<400>21
gtgactggagttcagacgtgtgctcttccgatct34
<210>22
<211>33
<212>dna
<213>人工序列(artificialsequence)
<400>22
gtgactggagttccttggcacccgagaattcca33
- 还没有人留言评论。精彩留言会获得点赞!