一种手性脲类配体及其制备方法与流程
本发明涉及脲类配体技术领域,具体涉及一种手性脲类配体及其制备方法。
背景技术:
不对称催化与合成是当前有机化学领域的热点。在不对称催化反应中,手性配体的选择是很重要的,往往直接影响到手性识别的效果。手性脲类催化剂是一种非常高效的催化剂,它具有较弱的
技术实现要素:
本发明提供一种在不对称反应中具有高对映选择性和高反应活性的手性脲类配体及其制备方法。
本发明采用的技术方案是:一种手性脲类配体,其结构如下所示:
式中:r1为氢、甲基、乙基和苄基中的一种,r2为甲基、乙基、异丙基和苯基中的一种,r3为氢和三氟甲基中的一种,x为氧和硫中的一种。
一种手性脲类配体的制备方法,包括以下步骤:
步骤1:将手性胺与异氰酸酯或异硫氰酸酯按摩尔比为1:1~1.2的比例溶于有机溶剂中,充分混合;
步骤2:在温度为-5℃~35℃条件下反应1~18h后,柱层析纯化后即得所需手性脲类配体。
进一步的,所述手性胺的制备方法如下:
s1:在不大于零度条件下带有保护基的羧酸甲酯溶液中加入烷烃类格式试剂,带有保护基的羧酸甲酯和烷烃类格式试剂的摩尔比为1:4~6;室温条件下反应10~18h后得到具有保护基保护的氨基叔醇;
s2:将步骤s1得到的氨基叔醇中加入三乙胺和甲基磺酰氯,溶于溶剂中在室温条件下反应2~3h,得到甲磺酸酯;其中氨基叔醇、甲基磺酰氯和三乙胺摩尔比为1:1.2:2~1:1.5:4;
s3:将步骤s2中得到甲磺酸酯溶液中加入三甲基硅叠氮,在85℃~110℃条件下,反应10~12h后得到叠氮化合物;其中甲磺酸酯和三甲基硅叠氮摩尔比为1:1.2~1:1.5;
s4:将步骤s3得到叠氮化合物溶液中加入四氢铝锂,反应10~12h后脱保护即得所需手性胺,其中叠氮化合物和四氢铝锂摩尔比为1:3~4。
进一步的,所述步骤s1中的保护基为苄基。
进一步的,所述烷烃类格式试剂为甲基溴化镁、乙基溴化镁、异丙基溴化镁和苯基溴化镁中的一种。
进一步的,所述步骤2中柱层析纯化后还包括脱保护,脱保护过程如下:
将步骤2得到的手性脲类配体溶到无水乙醇中,加入钯碳和冰醋酸,加氢还原即得所需手性脲类配体;其中手性脲类配体、钯碳和冰醋酸的摩尔比为1:0.1:0.2~1:0.2:0.2。
本发明的有益效果是:
(1)本发明得到的手性脲类配体,分子结构中为非简单直链,分子结构中具有软性骨架;
(2)本发明得到的手性脲类配体,可利用本身脲类结构的多重氢键作用活化亲电试剂或亲核试剂,提高其催化性能;
(3)本发明得到的手性脲类配体,在不对称反应中具有高对映选择性以及高的反应活性,制备方法简单,步骤少,且原料便宜易得。
附图说明
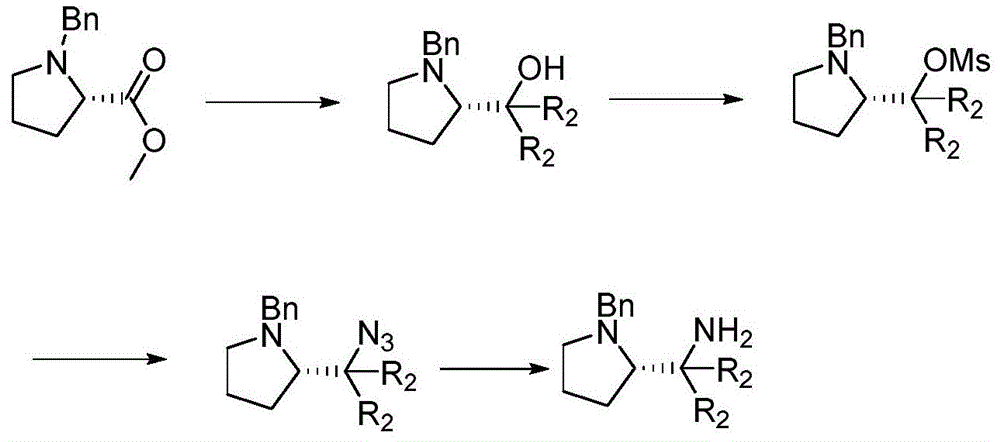
图1为手性胺a合成流程示意图。
图2为以手性胺a为中间体合成的脲类配体的流程示意图。
图3为本发明实施例1中n-bn-(l)-脯氨酸甲酯的1hnmr图。
图4为本发明实施例1中n-bn-(l)-氨基醇的1hnmr图。
图5为本发明实施例1中n-bn-(l)-甲磺酸酯的1hnmr图。
图6为本发明实施例1中n-bn-(l)-叠氮的1hnmr图。
图7为本发明实施例1中保护基为苄基时手性胺的1hnmr图。
图8为本发明实施例1中r1为苄基时手性脲类配体的1hnmr图。
图9为本发明实施例1中r1为氢时手性脲类配体的1hnmr图。
图10为本发明实施例1中r1为乙基时手性脲类配体的1hnmr图。
图11为本发明实施例1中r1为乙基时手性脲类配体的三维分子结构示意图。
具体实施方式
下面结合附图和具体实施例对本发明做进一步说明。
一种手性脲类配体,其结构如下所示:
式中:r1为氢(h)、甲基(me)、乙基(et)和苄基(bn)中的一种,r2为甲基(me)、乙基(et)、异丙基(i-pr)和苯基(bz)中的一种,r3为氢(h)和三氟甲基(cf3)中的一种,x为氧(o)和硫(s)中的一种。
上述手性脲类配体是以简单的氨基酸作为原料,经过简单修饰制得非直链状构型手性配体化合物。
其中关键中间体为手性胺合成示意图如图1所示。
其中r2为甲基(me)、乙基(et)、异丙基(i-pr)和苯基(bz)中的一种。
即:
当r2为甲基时,结构如a所示。具体制备过程如下:
s1:在零度条件下带有保护基的羧酸甲酯溶于有机溶剂中,加入甲基溴化镁格式试剂,带有苄基的羧酸甲酯和烷烃类格式试剂的摩尔比为1:4~6;室温条件下反应10~18h后得到具有保护基保护的氨基叔醇,进行柱层析分离提纯;其中带有保护基的羧酸甲酯为苄基保护的脯氨酸甲酯;
s2:将步骤s1得到的苄基保护的氨基叔醇在零度条件下,加入三乙胺和甲基磺酰氯,在二氯甲烷中室温条件下搅拌2~3h,得到甲磺酸酯;苄基保护的胺基叔醇、甲基磺酰氯和三乙胺摩尔比为1:1.2:2~1:1.5:4
s3:将步骤s2得到的甲磺酸酯简单纯化后,溶于dmf中,加入三甲基硅叠氮,在85℃~110℃条件下,反应10~12h后得到叠氮化合物;其中甲磺酸酯和三甲基硅叠氮摩尔比为1:1.2~1:1.5;
s4:将步骤s3得到叠氮化合物溶于无水四氢呋喃中,在无水环境下加入四氢铝锂,反应10~12h后脱保护即得所需手性胺a,其中叠氮化合物和四氢铝锂摩尔比为1:3~4。
其中,b和c的反应如上所述。
具体的手性脲类配体较多,以合成硫尿配体a-03为例,其中手性胺为上述a,其中r1为et,r2为me,x为s,r3为cf3,具体反应如图2所示。
一种手性脲类配体的制备方法,包括以下步骤:
步骤1:将手性胺a溶于无水二氯甲烷中,无氧环境下,加入3,5-二(三氟甲基)异硫氰酸酯,室温条件下反应即可得到产物a-01,结构如图2所示。a和异硫氰酸酯的摩尔比为1.1~1:1.4,反应温度为5℃~45℃,反应时间为2~4h。
将产物a-01纯化后,溶到无水乙醇中,加入钯碳和冰醋酸,加氢还原即得a-02;其中苄基保护的手性脲类配体、钯碳和冰醋酸的摩尔比为1:0.1:0.2~1:0.2:0.2。反应温度为15℃~45℃,反应时间为1~18h。
将产物a-02纯化后,溶于无水乙腈中,加入碳酸钾和溴乙烷,回流,即得a-03;其中a-02、碳酸钾和溴乙烷摩尔比为1:3:2~1:6:4,反应温度为85℃~110℃,反应时间为1~18h。
下面结合具体实施例对本发明做进一步说明。
a-03结构如上所示,其三维分子结构示意图如图11所示,晶体结构参数如表1-表6所示。
表1.a-03的晶体数据和组织细化
表2.a-03的分数原子坐标(×104)和等效各向同性位移参数
表3.a-03的各向异性位移参数
表4.a-03的键长
表5.a-03的键角
表6.a-03的氢原子坐标
其反应过程如下:
(1)l-脯氨酸甲酯的合成
l-脯氨酸甲酯的结构式如下:
合成过程为:在装有温度计,机械搅拌的500ml三颈瓶中,将l-脯氨酸(1eq)溶到无水甲醇中,室温下,逐滴加入二氯亚砜(1.1eq),室温搅拌10-12h,反应完成后,直接旋掉体系多余溶剂和二氯亚砜,加入一定量的二氯甲烷,用饱和碳酸钠水溶液洗有机相,分离有机相,蒸干即得产品,收率:95%,
(2)n-bn-(l)-脯氨酸甲酯的合成
n-bn-(l)-脯氨酸甲酯的结构式如下:
合成过程为:将l-脯氨酸甲酯(1eq)溶到二氯甲烷中,零度下,加入三乙胺(2.4eq),体系产生大量固体,滤掉体系固体,零度下逐滴加入苄溴(1.2eq),体系在室温下反应10-16小时,待反应完成后,加入一定量的0.5n盐酸溶液洗有机相,静置分液,分出有机相,再用饱和碳酸氢钠水溶液洗有机相,最后用饱和食盐水洗,分液干燥,蒸干即得到产品,柱层析得产品,收率:98%;1hnmr(400mhz,dmso-d6)δ7.34–7.28(m,4h),7.26–7.22(m,1h),3.89(d,j=12.8hz,1h),3.65(s,3h),3.57(d,j=12.8hz,1h),3.28–3.20(m,1h),3.10–2.99(m,1h),2.46–2.31(m,1h),2.20–2.04(m,1h),2.02–1.85(m,2h),1.83–1.73(m,1h);
化合物n-bn-(l)-脯氨酸甲酯的1hnmr图见图3。
(3)n-bn-(l)-氨基醇的合成
n-bn-(l)-氨基醇的结构式如下:
合成过程为:将n-bn-(l)-脯氨酸甲酯溶到无水四氢呋喃中,氩气环境下,室温下,滴加乙基溴化镁(4.5eq),加毕后,体系在室温下反应,至原料消失产物生成后,用饱和氯化铵水溶液淬灭,用乙酸乙酯萃取,干燥旋干,柱层析得产品,收率:76%;1hnmr(400mhz,chloroform-d)δ7.40–7.27(m,4h),7.27–7.19(m,1h),4.02(d,j=13.8hz,1h),3.61(d,j=13.9hz,1h),2.93–2.86(m,1h),2.86–2.71(m,1h),2.52–2.38(m,1h),1.88–1.78(m,2h),1.73–1.55(m,5h),1.43–1.34(m,1h),0.89(q,j=7.2hz,6h);
化合物n-bn-(l)-氨基醇的1hnmr图见图4。
(4)n-bn-(l)-甲磺酸酯的合成
n-bn-(l)-甲磺酸酯的结构式如下:
合成过程为:将n-bn-(l)-氨基醇溶到无水二氯甲烷中,氩气环境下,低温下,依次加入三乙胺(2eq)和甲基黄酰氯(1.2eq),加毕后,体系在室温下反应,至原料消失产物生成后,用饱和食盐水溶液洗有机相,用二氯甲烷萃取,干燥旋干,柱层析得产品,收率:76%;1hnmr(400mhz,dmso-d6)δ7.47–7.27(m,4h),7.25–7.18(m,1h),4.07(d,j=13.8hz,1h),3.56(d,j=14.0hz,1h),3.32(s,3h),3.26–3.19(m,1h),2.79–2.72(m,1h),2.40–2.29(m,1h),2.02–1.92(m,3h),1.90–1.75(m,2h),1.68–1.60(m,3h),0.97(t,j=4.1hz,6h);
化合物n-bn-(l)-甲磺酸酯的1hnmr图见图5。
(5)n-bn-(l)-叠氮化合物的合成
n-bn-(l)-叠氮化合物的结构式如下:
合成过程为:将n-bn-(l)-甲磺酸酯溶到dmf中,氩气环境下,室温下加入三甲基硅叠氮,加毕后,体系升温85-110℃下反应,至原料消失产物生成后,加入一倍量蒸馏水,用乙酸乙酯萃取,干燥旋干,柱层析得产品,收率:70%;
1hnmr(400mhz,dmso-d6)δ7.35–7.28(m,4h),7.22–7.14(m,1h),3.92(d,j=15.0hz,1h),3.69(dd,j=10.3,4.2hz,1h),3.43(d,j=15.0hz,1h),2.41–2.31(m,1h),1.90–1.86(m,1h),1.84–1.75(m,2h),1.71–1.64(m,1h),1.55–1.43(m,2h),0.94(t,j=7.6hz,3h),0.86(t,j=7.4hz,3h);
化合物n-bn-(l)-叠氮的1hnmr图见图6。
(6)n-bn-(l)-手性伯胺化合物b的合成
n-bn-(l)-手性伯胺化合物的结构式如下:
合成过程为:将n-bn-(l)-叠氮溶到无水四氢呋喃中,氩气环境下,0℃下分批加入四氢铝锂,加毕后,体系室温下反应,至原料消失产物生成后,加入湿十水硫酸钠萃灭反应,用乙酸乙酯萃取,干燥旋干得产品,收率:82%;
(7)n-bn-(l)-手性伯胺化合物a的合成
n-bn-(l)-手性伯胺化合物a的结构式如下:
合成过程为:将n-bn-(l)-叠氮溶到无水四氢呋喃中,氩气环境下,0℃下分批加入四氢铝锂,加毕后,体系室温下反应,至原料消失产物生成后,加入湿十水硫酸钠萃灭反应,用乙酸乙酯萃取,干燥旋干得产品,收率:80%;1hnmr(400mhz,dmso-d6)δ7.47–7.24(m,5h),4.14(d,j=14.0hz,1h),3.64–3.55(m,1h),3.48(d,j=14.1hz,1h),2.83–2.71(m,1h),2.60–2.54(m,1h),2.27–2.19(m,1h),1.78–1.71(m,2h),1.70-1.66(m,1h)1.38–1.22(m,2h),1.01(s,3h),0.98(s,3h);
化合物a的1hnmr图见图7。
(8)n-bn-(l)-手性伯胺化合物c和化合物d的合成
n-bn-(l)-手性伯胺化合物c和化合物d的结构式如下:
合成过程同化合物a和b相似,在此不做重述;
化合物a-01合成:
化合物a-01的结构式如下:
合成过程为:将n-bn-(l)-手性伯胺化合物a溶到无水二氯甲烷中,在室温下以下加入3,5-双(三氟甲基)苯基异硫氰酯,室温搅拌3-4h,蒸除多余溶剂,柱层析得到化合物a-01,收率:25%,1hnmr(400mhz,chloroform-d)δ13.68(s,1h),7.55(s,1h),7.46(s,2h),7.19–7.09(m,3h),7.08–7.01(m,2h),6.22(s,1h),4.13(d,j=12.2hz,1h),3.53(d,j=12.3hz,1h),2.94–2.85(m,2h),2.77–2.67(m,1h),2.12–2.04(m,1h),2.00–1.96(m,1h),1.84(s,3h),1.74–1.57(m,2h),1.34(s,3h);
化合物a-01的1hnmr图见图8。
化合物a-02合成:
化合物a-02的结构式如下:
合成过程为:将化合物a-01溶到无水甲醇中,在室温下以下加入无水钯碳和冰醋酸,室温下加氢还原24h,蒸除多余溶剂,调节体系ph至10以上,二氯萃取,干燥后柱层析得到化合物a-02,收率:55%,1hnmr(400mhz,chloroform-d)δ7.94(s,1h),7.46–7.39(m,1h),7.38–7.36(m,2h),7.28(s,1h),4.88(d,j=12.9hz,1h),4.34(d,j=13.0hz,1h),4.25–4.17(m,1h),3.60-3.58(m,1h),3.26(s,1h),2.20–2.09(m,1h),2.00–1.93(m,1h),1.89–1.83(m,1h),1.26(s,3h),0.86(s,3h);
化合物a-02的1hnmr图见图9。
化合物a-03合成:
化合物a-03的结构式如下:
合成过程为:将化合物a-02溶到无水乙腈中,加入无水碳酸钾和溴乙烷,体系升温24h,蒸除多余溶剂,加水二氯甲烷萃取,干燥后柱层析得到化合物a-03,收率:75%,1hnmr(400mhz,chloroform-d)δ14.15(s,1h),8.13–7.91(m,2h),7.61(s,1h),6.10(s,1h),3.16–3.07(m,1h),3.04–2.89(m,1h),2.75–2.67(m,1h),2.65–2.56(m,2h),2.04–1.89(m,2h),1.87–1.77(m,2h),1.61(s,3h),1.29(s,3h),1.10(t,j=7.2hz,3h);
化合物a-03的1hnmr图见图10。
本发明以简单的氨基酸为原料,通过对其进行修饰,然后与偶联剂、碱等物质发生反应,得到非链状构型多重氢键手性脲类配体化合物。该手性脲类配体结构中为非简单直链,分子结构中具有软性骨架,以及含有氢、氮等杂原子的识别基因,拥有脲类结构具有多重氢键作用;在不对称反应中实现高对映选择性以及高的反应活性,其制备方法简单、步骤少且原料便宜易得。
- 还没有人留言评论。精彩留言会获得点赞!