一种乳腺癌无创分子分型试剂盒及方法与流程
本发明提供了一种乳腺癌无创分子分型产品,具体地说,是一种基于血浆游离dna高通量测序技术的乳腺癌无创分子分型产品,本发明还是涉及该分子产品的构建方法,属于生物技术领域。
背景技术:
乳腺癌是女性最常见的浸润性癌症。根据世界卫生组织2015年报道,全球每年新增的乳腺癌病例约115万例,死亡病例约41万例,并且发病率逐年增加。乳腺癌也是中国女性最常见的癌症,每年中国乳腺癌新发数量和死亡数量分别占全世界的12.2%和9.6%。
乳腺癌的分子分型与乳腺癌的治疗和预后密切相关。目前乳腺癌分子分型方法主要采用2013年乳腺癌国际会议上规定的乳腺癌分类标准。即采用免疫组织化学方法,根据癌组织中雌激素受体(estrogenreceptor,er)、孕激素受体(progester-onereceptor,pr)和her-2的检测结果,将乳腺癌分为4种分子亚型:管腔a型(luminala)、管腔b型(luminalb)、her2富集型(her2-enriched)和基底细胞型(basal-like)。分子分型结果广泛用于临床治疗方案的确定、疗效和预后判断等不同亚型乳腺癌的生物学行为、治疗策略和预后存在明显差异。管型为激素受体表达阳性,预后良好;her-2型为激素受体无表达但her-2表达阳性,预后虽较导管型差但可通过特定的靶点治疗得到明显改善。基底细胞样型,即通常所说的三阴性乳腺癌,治疗效果和预后较差。
目前的分子分型方法都是基于有创的诊断方法,即通过针吸细胞学检查、切取活检等直接对人体组织有创伤的检查手段,目的是获取肿瘤细胞或肿瘤组织、转移淋巴结。这种有创的诊断方法增加了患者的痛苦及肿瘤播散的风险。此外由于肿瘤组织具有较大异质性,局部穿刺的活检分析有可能造成结果偏倚,针对活检组织的病理诊断也需要具有良好的病理学理论基础和实践经验。需要二次活检的患者需要确定肿瘤进展部位,对于已手术切后的患者后期则无法获取可用的组织标本来判断预后及转移情况。
细胞死亡或者快速分裂时,会释放游离的dna进入外周血,主要分布于血浆和血清中。在正常人中,游离dna主要来源于造血细胞。由于细胞坏死或者细胞更新速度变快,感染、缺血、肿瘤、自身免疫性疾病、肥胖和怀孕时,体内游离dna含量会增加。在肿瘤的发生发展过程中,肿瘤细胞也会释放dna至外周血中,即游离得肿瘤细胞dna(cellfreetumordna,ctdna)。cfdna和ctdna的发现及arms-pcr和高通量测序技术的发展为无创性检测带来了新的契机。ctdna可代替肿瘤组织广泛应用于肿瘤的早期筛查、辅助诊断、靶向药物选择和预后疗效监测。
所以肿瘤患者血浆游离dna主要来源于快速代谢的造血细胞和肿瘤细胞。即血浆游离dna是细胞死亡后,染色质经酶切片段化后形成dna片段,其半衰期很短,半小时就会被清除。其中被核小体固定的dna序列因核小体保护而降解速度较慢,而裸露的dna序列很快被降解,所以游离dna长度一般为核小体固定长度的整数倍。不同的肿瘤组织、不同类型的肿瘤细胞,其血浆游离dna的核小体足迹是存在差异的。可以通过高通量测序技术对血浆cfdna进行全基因组测序,获得核小体定位信息,通过定位信息对患者进行分子分型。
技术实现要素:
针对上述不足,本发明的第一个目的是提供一种使用简单,检测准确率高的试剂盒。
本发明的第二个目的是提供一种样本易得、取样流程简单、临床体验较好、且可流程化操作,非常适合临床应用的非疾病诊断目的的乳腺癌无创分子分型的方法。
为此,本发明提供的第一个技术方案是这样的:
一种乳腺癌无创分子分型的试剂盒,包括血浆游离dna提取试剂、文库构建试剂、测序试剂、测序芯片。
本发明提供的第二个技术方案是这样的:
一种非疾病诊断目的的乳腺癌无创分子分型的方法,基于对血浆游离dna高通量测序数据,通过生物信息学分析,获得血浆游离dna中全基因组范围内启动子区的核小体足迹差异信息,进而根据足迹信息的差异对乳腺癌患者实现管腔a型、管腔b型、her2富集型和基底细胞型四种类别的无创分子分型。
进一步的,上述的非疾病诊断目的的乳腺癌无创分子分型的方法,所述的核小体足迹差异是血浆游离dna全基因组范围内每个基因转录起始位点tsss区域的核小体分布与该基因其余区域的差异,与核小体解离的区域被快速降解掉导致差异产生的。
进一步的,上述的非疾病诊断目的的乳腺癌无创分子分型的方法,具体包括以下步骤:
(1)血浆游离dna的高通量测序
采用权利要求1所述的血浆游离dna提取试剂,提取血浆的游离dna,进行末端修复、接头连接、pcr扩增形成测序文库,进行高通量测序,每个样本至少检测6mreads的数据量;
(2)核小体印迹分析
通过ucsc的refseq数据库,获得目前人类蛋白编码基因的启动子区域,数据库注释信息计算统计每个基因的转录起始位点tsss上下游1kb区域的基因组位置,并将该区域定义为原始启动子区域;采用权利要求1测序的原始数据,利用bowtie软件对测序数据进行比对,去除比对数据中的重复序列,统计出每个蛋白编码基因的原始启动子区域的reads数,并利用rpkm方法对数据进行标准化,原始启动子区域覆盖度能够提示核小体印迹,覆盖度越低核小体结合数目较少,反之核小体数目较多;
(3)核小体足迹差异分析
根据核小体足迹差异,利用kruskal-wallis非参数单因子方差分析筛选出管腔a型、管腔b型、her2富集型和基底细胞型组中存在覆盖度差异的基因启动子区域,通过秩和检验同一个基因的4组覆盖度数值进行两两比较,并利用holm法对p值进行校正,筛选出fdr值小于0.1的基因启动子区域;
(4)聚类分析
利用cluster软件对差异表达的基因启动子覆盖度数据进行标准化,采用等级聚类法根据样本间的相关性对标准化后的数据进行聚类分析,并用r语言pheatmap包对数据进行可视化展示;根据tsss区核小体的印迹差异,样本类型聚类为管腔a型、管腔b型、her2富集型和三阴型4类。
进一步的,上述的非疾病诊断目的的乳腺癌无创分子分型的方法,步骤(1)所述的高通量测序是根据常规的血浆游离dna基因组高通量测序方法进行,最终获得大于6mreads的数据量。
进一步的,上述的非疾病诊断目的的乳腺癌无创分子分型的方法,步骤(1)所述的高通量测序的检测平台为iontorrentproton测序平台。
更进一步的,上述的非疾病诊断目的的乳腺癌无创分子分型的方法,所述的血浆游离dna的高通量测序具体方法为:
步骤一)外周血血浆的制备
采用两步离心法进行血浆分离
步骤二)血浆游离dna提取
使用血浆游离dna提取试剂盒提取血浆游离dna,提取完成后,使用qubit3.0进行浓度测定;
步骤三)文库构建及定量
使用ionplusfragmentlibrarykit试剂盒对dna进行末端修复,磁珠纯化后使用dna连接酶在dna两端添加上测序接头,连接产物进行磁珠纯化后进行扩增,磁珠纯化后即完成文库构建,进行浓度测定;根据浓度将每个文库稀释到100pm,使用abi7500进行qpcr定量后,每10个文库等量进行混合,使其终浓度为100pm;
步骤四)高通量测序
使用ionpitmhi-qtmot2200kit对混合文库进行乳液pcr,使文库与测序isp结合成isp模板并得到扩增,随后用dynabeadstmmyonetmstreptavidinc1进行isp模板富集,使用ionpitmhi-qtmsequencingkit初始化测序仪,将isp模板与质控模板、测序引物进行混合退火后,loading到pi芯片上,在iontorrentprotontm测序仪选择预先设置好的plan(程序),进行测序反应;
更进一步的,上述的基于血浆游离dna高通量测序技术对乳腺癌无创分子的分型方法,两步离心法进行血浆分离具体包括:
(1)在4℃,1600g离心10min将血浆与白细胞、血小板、红细胞进行分离,将血浆转移到2ml离心管中;
(2)4℃,16000g离心10min去除残余细胞,将上清转移至2ml离心管中,即得到外周血血浆。
更进一步的,上述的基于血浆游离dna高通量测序技术对乳腺癌无创分子的分型方法,所述的程序flow数为300,每个样本数据量大于6m。
与现有技术相比,本发明提供的技术方案具有如下优点:
1、本发明提供的技术方案通过对血浆游离dna高通量测序、生物信息学分析及机器学习,实现对乳腺癌患者管腔a型(luminala)、管腔b型(luminalb)、her2富集型(her2-enriched)和基底细胞型(basal-like)无创精准分型,便于医护人员选择乳腺癌患者临床治疗的方式。
2、本发明提供的技术方案克服已有的乳腺癌分子分型产品采用有创性方法的局限性,特别是对乳腺癌分子分型的材料需要有创性的穿刺取样,其临床体验不佳、并且具有无法解决肿瘤异质性、穿刺禁忌证、造成肿瘤传播转移风险等缺点;本发明提供的技术方法的原材料是3-5ml外周血,其样本易得、取样流程简单、临床体验较好、且可流程化操作,非常适合临床应用。
3、本发明所述的检测样本为外周血血浆,较佳地为用edta抗凝管采集新鲜的外周全血标本经离心后获得的血浆标本,亦可为2年内-70℃以下保存的血浆标本。
4、本发明提供的技术方案通过对血浆游离dna高通量测序数据的生物信息学分析,获得血浆游离dna中全基因组范围内启动子区的核小体足迹信息,进而根据足迹信息的差异对乳腺癌患者实现管腔a型、管腔b型、her2富集型和基底细胞型四种类别的无创分子分型;其检测材料为乳腺癌患者外周血,属于无创检测范畴,可用于乳腺癌化疗、内分泌治疗、靶向治疗或者手术方案的选择依据。
5、本发明提供的技术方案通过分析乳腺癌患者血浆游离dna中全基因组范围内每个基因转录起始位点(tsss)区域与该基因其余区域的核小体分布差异信息对乳腺癌患者进行无创分子分型,可为选择合适的治疗方案提供依据。
附图说明
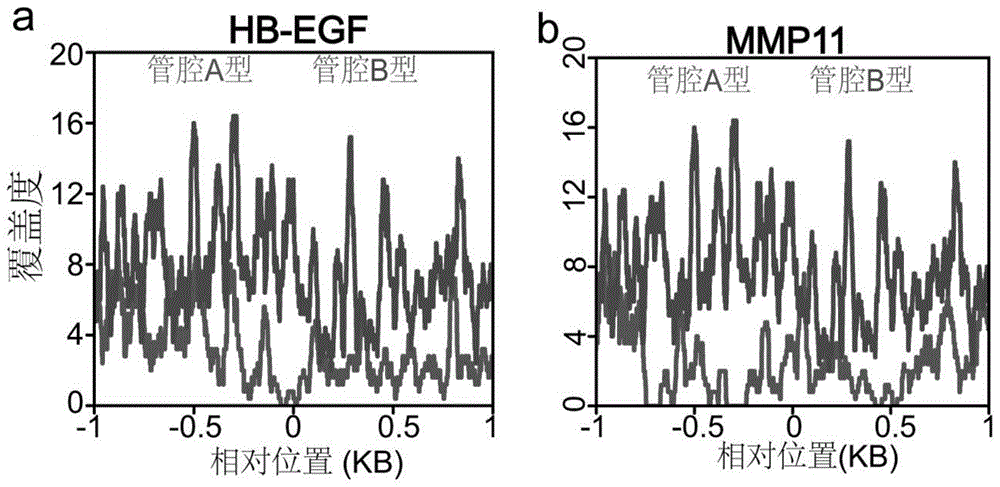
图1是hbegf基因和mmp11基因的tss区域覆盖度;
图2是基于47例治疗前乳腺癌患者血浆游离dna全基因组核小体足迹差异的聚类结果。
具体实施方式
下面以具体实施例对本发明作进一步说明,但本发明并不局限于这些实施例。
本发明提供的试剂均为本领域常规试剂,均可通过市售获得。
实施例1
本发明提供的所述的乳腺癌无创分子分型产品包括血浆游离dna提取试剂(血浆游离dna提取试剂为magabio公司的dna提取试剂盒)、文库构建试剂(lifetechnologies公司的ionplusfragmentlibrarykit试剂盒)、测序试剂、测序芯片。其中文库构建试剂盒包括末端修复、接头连接、pcr扩增试剂。
需要说明的是,上述的血浆游离dna提取试剂和文库构建试剂和测序试剂也可以采用其它厂家的,比如:
iontorrentproton测序平台:使用的测序试剂ionpitmhi-qtmot2200kit货号:a26434和ionpitmhi-qtmsequencing200kit货号:a26433,测序芯片ionpitmchipkitv3货号:a26771。
illuminamiseq测序平台:使用的测序试剂miseqreagentkitv2(300-cycles)货号:ms-102-2002。
样本说明:收集临床免疫组化确认的乳腺癌患者外周血47例,其中管腔a型12例、管腔b型26例、her2富集型3例和三阴型6例。具体如表1所示。
实施例乳腺癌无创分子分型
步骤1:血浆分离:使用两步离心法进行血浆分离(1)4℃,1600g离心10min将血浆与白细胞、血小板、红细胞进行分离,将血浆转移到2ml离心管中;(2)4℃,16000g离心10min去除残余细胞,将上清转移至2ml离心管中,即得到外周血血浆。
步骤2:血浆游离dna提取:使用血浆游离dna提取试剂盒(magabio公司)提取血浆游离dna,严格按说明书进行操作,提取完成后,使用qubit3.0(lifetechnologies)进行浓度测定。
步骤3:文库构建及定量:使用ionplusfragmentlibrarykit(lifetechnologies公司)试剂盒对dna进行末端修复,磁珠纯化后(该方法为本领域常规磁珠纯化方法)使用dna连接酶在dna两端添加上测序接头(所述的测序接头接头试剂为ionxpresstmbarcodeadapters1-96kit),连接产物进行磁珠纯化后进行扩增,磁珠纯化后即完成文库构建,使用qubit3.0进行浓度测定。根据浓度将每个文库稀释到100pm,使用abi7500(thermofisher)进行qpcr定量后,每10个文库等量进行混合,使其终浓度为100pm。
步骤4:高通量测序:使用ionpitmhi-qtmot2200kit(lifetechnologies)对混合文库进行乳液pcr,使文库与测序isp结合成isp模板并得到扩增,随后用dynabeadstmmyonetmstreptavidinc1(invitrogen)进行isp模板富集。使用ionpitmhi-qtmsequencingkit(lifetechnologies)初始化测序仪,将isp模板与质控模板、测序引物进行混合退火后,loading到pi芯片上,在iontorrentprotontm测序仪选择预先设置好的程序flow数为300,每个样本数据量大于6m),进行测序反应。测序结果如表1所示。
表147例乳腺癌血浆标本信息及测序reads数、平均读长信息
步骤5:生物信息学分析
(1)核小体印迹分析
通过ucsc的refseq数据库,获得目前人类蛋白编码基因的启动子区域,我们根据数据库注释信息计算统计每个基因的转录起始位点(tsss)上下游1kb区域的基因组位置,并将该区域定义为原始启动子区域。我们获得测序的原始数据后,利用bowtie软件对测序数据进行比对,去除比对数据中的重复序列,统计出每个蛋白编码基因的原始启动子区域的reads数,并利用rpkm方法对数据进行标准化。
以原始覆盖度差异的肝素结合性表皮生长因子(heparin-bindingepidermalgrowthfactor-likegrowthfactor,hb-egf)基因为例,其管腔b型患者在原始启动子的覆盖度低于其管腔a型患者的覆盖度,如图1a所示。hb-egf基因是表皮生长因子(epidermalgrowthfactor,egf)家族的成员。hb-egf基因可以直接激活egfr和erbb4,或间接激活erbb2和erbb3组成的异源二聚体受体。
以基质金属蛋白酶-11(matrixmetalloproteinase-11,mmp-11)基因为例,同样其在管腔b型患者在原始启动子的覆盖度低于其在管腔a型患者的覆盖度,如图1b所示。mmp11基因在多种肿瘤中过度表达,参与了肿瘤细胞的浸润及转移过程,与肿瘤的发生和发展密切相关。
(2)核小体印迹差异分析
核小体缺失区域即在tss位点较邻近区域明显降低的区域,为高表达基因,反之则为低表达基因。首先通过kruskal-wallis非参数单因子方差分析筛选出管腔a型、管腔b型、her2富集型和基底细胞型组中存在覆盖度差异的基因启动子区域,通过秩和检验同一个基因的4组覆盖度数值进行两两比较,并利用holm法对p值进行校正,筛选出fdr值小于0.1的基因启动子区域。
(3)聚类分析
利用cluster软件对差异表达的基因启动子覆盖度数据进行标准化,采用等级聚类法根据样本间的相关性对标准化后的数据进行聚类分析。并用r语言pheatmap包对数据进行可视化展示。
基于47例乳腺癌的血浆游离dna的高通量检测进行tsss区域的覆盖度差异分析,参阅表2,通过两两比较,发现管腔a型与管腔b型的差异tsss区域295个,三阴型与管腔a型的差异tsss区域104个,三阴型与管腔b型的差异tsss区域116个,her2富集型与管腔a型的差异tsss区域2个,her2富集型与管腔b型的差异tsss区域21个,her2富集型和三阴型的差异tsss区域个数为0,这可能与her2富集型的样本例数较少有关。通过利用差异表达基因进行无监督聚类,结果发现基于474个基因能够将样本聚成4种类别,管腔a型、管腔b型、her2富集型和三阴型均能各自聚类为1个分支。各分支间覆盖度差异基因的模式显著差异。
表2.差异覆盖度分析tsss区域及对应的基因列表
管腔a型即免疫组织化学检测er阳性或pr阳性,her-2阴性,ki67低表达。此类型是乳腺癌最常见的分子亚型,发病率为44.5%-69.0%。管腔a型型对内分泌治疗敏感,有效率高达40%,而且er水平与内分泌治疗的敏感性呈正相关。因为her-2水平为阴性,不适合进行分子靶向治疗。
管腔b型即免疫组织化学检测er阳性或pr阳性,而her2也阳性,多见于高龄乳腺癌患者。此亚型也属于内分泌治疗敏感的肿瘤,但由于her2阳性,对他莫昔芬的疗效较管腔a型差,对芳香化酶抑制剂的效果较好。
her2富集型即免疫组织化学检测er、pr阴性,her2阳性,ki-67多为高表达。在原发乳腺癌患者中,有20%-30%存在her2过表达,且研究显示预后较差。多数为晚期病例,容易出现腋窝淋巴结转移。此型对her2靶向药物相对敏感,目前广泛采用赫赛汀联合化疗的治疗方案。
基底细胞型即免疫组化检测er、pr、her-2均为阴性,但不等同于三阴性乳腺癌,有研究显示,基底细胞型乳腺癌患者中有5%-45%er为阳性,14%her2阳性,而三阴性乳腺癌中只有80%-90%属于基底细胞型乳腺癌。其发病年龄较其他亚型低,在乳腺癌中占18.9%-28.8%。基底细胞型一直以来被认为是乳腺癌预后最差的1个亚型,无病生存期和总生存期较其他亚型短,容易出现肺、脑等远处转移。对目前的乳腺癌治疗方案均不敏感,化疗是唯一的全身治疗途径。一般临床预后较差。而在分子靶向治疗方面,有可能寻找到新的治疗靶点。
乳腺癌的分子分型是后续临床治疗方案选择的主要依据。目前乳腺癌分子分型广泛使用标准是2013年乳腺癌国际会议上规定的乳腺癌分类标准,基于免疫组化分子分型结果。但免疫组化采用的材料为穿刺组织,是有创性的,临床体验不佳。此外,根据不同免疫组化分子分型结果采取相应治疗方案的大规模临床应用发现,仍有部分患者的治疗效果并不明显,或者反而加强了副作用,因此临床上需要更精细的分子分型。本发明通过血浆游离dna高通量测序数据的生物信息学分析,可以实现对乳腺癌的无创分子分型。而且该方法是对全基因组范围内进行扫描,可比传统的免疫组化获得更多的信息,有望通过后期进一步分析得到更精细的分子分型,可获得更个体化的治疗方案。总之,本发明开创了乳腺癌无创分子分型的新方法,有助于乳腺癌的精准治疗。
- 还没有人留言评论。精彩留言会获得点赞!