一种2,4-二氯-3-氰基-5-氟苯甲酸的制备方法与流程
本发明涉及药物中间体的合成方法技术领域,具体指一种2,4-二氯-3-氰基-5-氟苯甲酸的制备方法。
背景技术:
2,4-二氯-3-氰基-5-氟苯甲酸是制备新型氟喹诺酮药物非那沙星和普拉沙星的关键中间体,具有很好的市场前景。
现有2,4-二氯-3-氰基-5-氟苯甲酸的合成方法主要有三种:
(1)2,6-二氯-3-氰基-5-氟溴苯通过制备烷烃格氏试剂,与2,6-二氯-3-氰基-5-氟溴苯进行卤素-金属交换反应制备2,6-二氯-3-氰基-5-氟溴苯的格氏试剂,并进一步与二氧化碳反应,经后处理得目标产物2,4-二氯-3-氰基-5-氟苯甲酸;该路线操作工艺较为复杂,溴化反应和格式反应均产生大量杂质。
(2)以2,4-二氯-5-氟苯甲酸为原料,经硝化得2,4-二氯-3-硝基-5-氟苯甲酸,后经还原得2,4-二氯-3-氨基-5-氟苯甲酸,再经重氮化反应及桑德迈尔反应得2,4-二氯-3-氰基-5-氟苯甲酸;该方法硝化反应一步产生大量酸化废水,并且桑德迈尔反应收率不高且需用到当量的氰化亚铜以及三倍以上当量的剧毒物质氰化钠,在工业化生产中将带来极大的安全隐患。
(3)5-氟-1,3-二甲苯在路易斯酸催化下与氯气反应得2,4-二氯-5-氟-1,3-二甲苯,后与氯气经自由基反应得2,4-二氯-5-氟-3-二氯甲基-1-三氯甲基苯,再水解得2,4-二氯-5-氟-3-甲酰基-苯甲酸,继续与羟胺反应得2,4-二氯-5-氟-3-n-羟基亚胺基-苯甲酸,最后经脱水反应得终产物2,4-二氯-3-氰基-5-氟苯甲酸;该路线方法步骤长,操作工艺较为复杂,且总收率较低(低于30%)。
因此,对于2,4-二氯-3-氰基-5-氟苯甲酸的制备方法,有待改进。
技术实现要素:
本发明所要解决的技术问题是针对现有技术的现状,提供一种收率及纯度高、操作简单、成本低、对环境友好、有利于实现工业化生产的2,4-二氯-3-氰基-5-氟苯甲酸的制备方法。
本发明解决上述技术问题所采用的技术方案为:一种2,4-二氯-3-氰基-5-氟苯甲酸的制备方法,其特征在于包括以下步骤:
(1)以取代氰基氟苯为原料,在强碱存在下,低温环境中与二氧化碳反应,反应完毕后用水淬灭;
(2)水相用稀酸调节ph至酸性,然后用萃取剂萃取有机相,静置分层,将有机相浓缩得到所述的2,4-二氯-3-氰基-5-氟苯甲酸;
其中,r为氢原子或卤素原子;
优选地,所述的强碱选自正丁基锂、仲丁基锂、叔丁基锂、异丁基锂、三甲基硅甲基锂,双(三甲基硅烷基)氨基钠、双(三甲基硅烷基)氨基钾、叔丁醇钾。
进一步优选,所述的强碱为正丁基锂或仲丁基锂。
在上述方案中,当所述强碱为正丁基锂,且r为氢原子时,反应体系中还需要加入有机配体;当所述强碱为仲丁基锂或r为卤素原子时,反应体系中不需要加入有机配体。采用这样的反应方式,是为了确保r原子能顺利完成置换。
优选地,所述有机配体选自二异丙胺、五甲基二乙二胺、六甲基二硅氮烷。
优选地,步骤(1)中,所述低温为-50℃~-100℃。
优选地,步骤(1)中,需要将取代氰基氟苯、强碱均溶于有机溶剂中进行反应,所述有机溶剂选自四氢呋喃、甲苯、2-甲基四氢呋喃。
在上述各方案中,步骤(1)中,所述取代氰基氟苯在强碱中保持时间不少于30min后,再向反应体系中通入二氧化碳,反应时间不少于30min后,先升温至室温,放出反应体系中的二氧化碳和氮气,再用冰水进行淬灭反应。只有采用这样的反应方式,才能获得纯度及收率高的产物。
优选地,步骤(2)中,所述的稀酸为2n~6n的稀盐酸,调节ph至1~3。
优选地,步骤(2)中,所述的萃取剂选自乙酸乙酯、乙醚、异丁醇。
与现有技术相比,本发明的优点在于:本发明以取代氰基氟苯为原料,在强碱条件下,在低温下与二氧化碳反应,然后用萃取剂萃取有机相,水相用稀盐酸调节ph至酸性,静置分层,将有机相浓缩得到所述取代苯甲酸类化合物,步骤少,分离纯化方法简单,不需要采用操作繁琐、成本较高的柱层析方法纯化产物和其它特殊设备,可操作性强,且工艺温和、安全;本发明的产率及纯度高,产率可达85%以上,纯度可达99%以上,且本发明的制备方法选择性好,重复性好,有利于实现工业化生产。
附图说明
图1为本发明实施例1的2,4-二氯-3-氰基-5-氟苯甲酸hplc图谱;
图2为本发明实施例1的2,4-二氯-3-氰基-5-氟苯甲酸质图谱;
图3为本发明实施例2的2,4-二氯-3-氰基-5-氟苯甲酸hplc图谱;
图4为本发明实施例2的2,4-二氯-3-氰基-5-氟苯甲酸nmr图谱。
具体实施方式
以下结合附图实施例对本发明作进一步详细描述。
实施例1:
本实施例中2,4-二氯-3-氰基-5-氟苯甲酸的制备方法包括以下步骤:
合成路线为:
(1)在干燥的反应装置中,加入100ml四氢呋喃和20.2g二异丙胺,通氮气至液面以下,降温至-80~-70℃,滴加80ml的2.5m正丁基锂的正己烷溶液,滴毕,保温半小时;
(2)将19g2,6-二氯-3-氟苯腈溶于40ml四氢呋喃中,保持温度在-80~-70℃,滴入步骤(1)的溶液中,滴毕,保温一小时;
(3)保持温度为-80~-60℃,向步骤(2)的反应体系中缓慢通入二氧化碳气体,反应30min至反应完全,逐渐升到室温,放出二氧化碳和氮气,用冰水淬灭反应;
(4)用6n的稀盐酸调节步骤(3)反应体系的ph至1~3,加入乙酸乙酯200ml,静置分层,分出的水层弃除,分出的有机层再用水洗,静止分层后得到的有机层在60℃以下减压蒸干,得到淡黄色粉末固体产物。
本实施例的合成路线为:
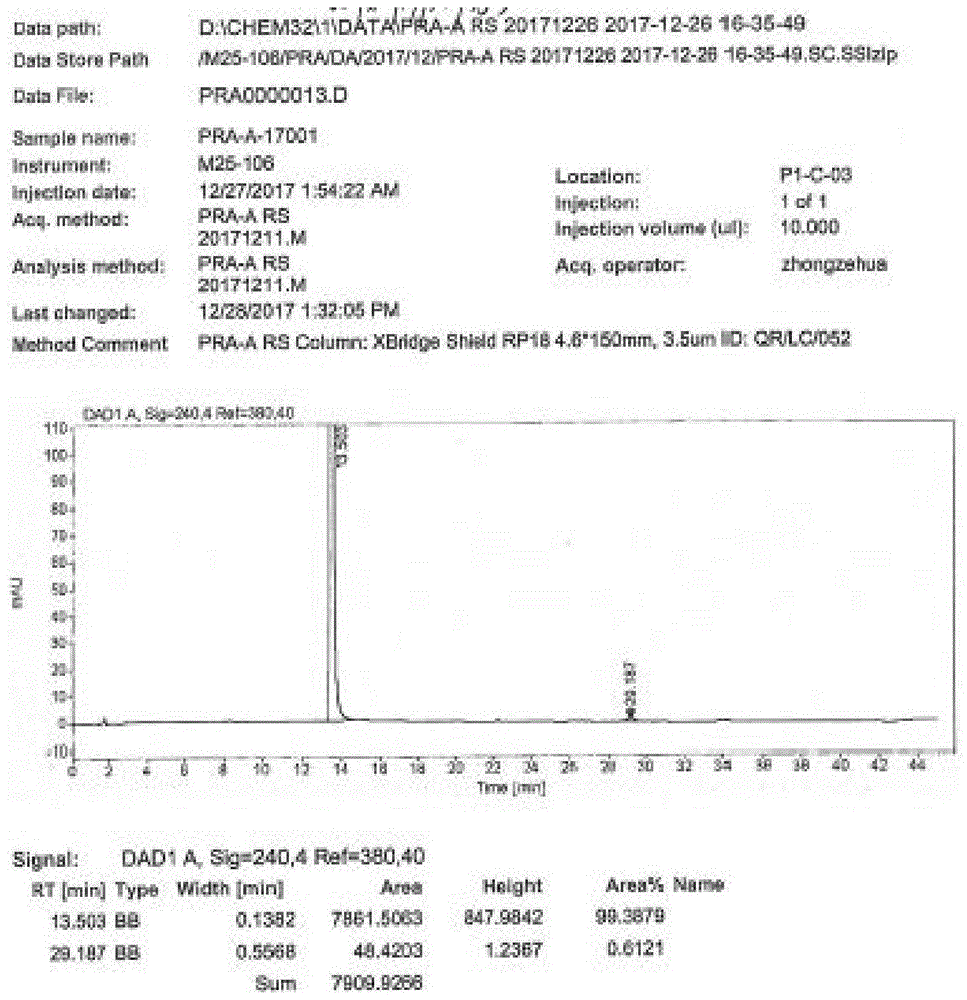
如图2所示,质谱:esi-ms(m/z):231.6([m-h]-),本实施例中的淡黄色粉末固体产物即为目标产物2,4-二氯-3-氰基-5-氟苯甲酸;如图1所示,2,4-二氯-3-氰基-5-氟苯甲酸的纯度为99.4%,所得淡黄色粉末固体产物19.9g,收率为85%。
实施例2:
本实施例中2,4-二氯-3-氰基-5-氟苯甲酸的制备方法包括以下步骤:
(1)在干燥的反应装置中,加入100ml2-甲基四氢呋喃及13.5g、50mmol的2,6-二氯-3-氰基-5-氟溴苯,通氮气至液面以下,降温至-80~-70℃;
(2)向步骤(1)的溶液中缓慢滴加1.3m的仲丁基锂的四氢呋喃溶液,(58ml,75mmol),滴加完毕后,-80~-70℃下搅拌反应1小时;
(3)向步骤(2)的反应体系中通入二氧化碳气体半小时,在-78℃下反应半小时,反应完毕后逐渐升到室温,放出二氧化碳和氮气,用冰水淬灭反应;
(4)用2n的稀盐酸调节步骤(3)反应体系的ph至1~3,分出水层弃除,有机层再用2n氢氧化钠调节ph至11~12;分出有机层弃除,向水层中加入异丁醇200ml,用6n的盐酸调节ph至1~3;分出水层弃除,有机层再用水洗,静止分层,得有机层,60℃以下减压蒸干得淡黄色粉末固体产物。
如图4所示,nmr:1hnmr(dmso-d6,d2o)δ:8.17(d,j=9.1hz,1h);13cnmr(dmso-d6)δ:112.76,116.52,122.98,127.52,131.31,132.75,155.97,163.85;本实施例中的淡黄色粉末固体产物即为目标产物2,4-二氯-3-氰基-5-氟苯甲酸;如图3所示,2,4-二氯-3-氰基-5-氟苯甲酸的纯度为99.7%,所得淡黄色粉末固体产物10.37g,收率为88.6%。
- 还没有人留言评论。精彩留言会获得点赞!
- 用对氯甲苯生产2,4-二氯甲苯的方法
- 用对氯甲苯制备2,4-二氯甲苯的方法
- 一种含有3,5-二氯水杨醛缩4-硝基邻氨基酚Schiff碱与吡啶的锌配合物及其制备方法和应用
- 一种含有3,5-二氯水杨醛缩4-硝基邻氨基酚Schiff碱与吡啶的镍配合物及其制备方法和应用
- 一种含有3,5-二氯水杨醛缩4-氯邻氨基酚Schiff碱与吡啶的镍配合物及其制备方法和应用
- 一种合成4,6-二氯-2-(丙硫基)-5-氨基嘧啶的方法
- 一种2,9-二氯-4,7-二苯基-1,10菲啰啉的制作方法
- 一种含有3,5-二氯水杨醛缩4-硝基邻氨基酚Schiff碱与吡啶的锰配合物及其制备方法和应用
- 一种邻氯甲苯氯化制备2,6-二氯甲苯的方法
- 一种新型1-(2,4-二氯-10,11-二氢-5H-二苯并[a,d]环庚烯-5-基)咪唑的合成方法