一种用于λ-Red重组系统基因敲除重组阳性克隆检测的引物及检测方法与流程
本发明属于生物技术工程领域,具体涉及通过λ-red重组系统对目标基因敲除后,对重组阳性克隆进行检测的引物及检测方法。
背景技术:
基于λ噬菌体red重组酶的同源重组系统目前广泛用于大肠杆菌基因工程研究。red重组系统由三种蛋白组成:exo蛋白是一种核酸外切酶,结合在双链dna的末端,从5'端向3'端降解dna,产生3'突出端;beta蛋白结合在单链dna上,介导互补单链dna退火;gam蛋白可与recbcd酶结合,抑制其降解外源dna的活性。red同源重组技术具有同源序列短(40~60bp)、重组效率高的特点。这种技术可在dna靶标分子的任意位点进行基因敲除、敲入、点突变等操作,无需使用限制性内切酶和连接酶。
四环素抗性基因tetra作为遗传筛选标记容易检测,而且具有条件致死特性,因此常作为替换基因用于取代需要敲除的基因或插入序列。red同源重组技术使难度较大的基因工程实验顺利进行,大大推动功能基因组研究的发展。四环素抗性tetra基因作为敲除目的基因的替换基因,其大小为1911bp;同源重组序列通常为目标基因两侧同源臂序列,以长度为40bp为例;用于鉴定基因是否重组的鉴定引物选择位置通常为同源重组序列外侧同源臂,以长度为18bp为例;因此用于同源重组的片段大小为1911+40+40=1991bp;重组阳性的pcr检测结果为1991+18+18=2027bp,相应的阴性对照大小为:原基因大小x+40+40+18+18=x+116bp。由于待敲除基因大小不一,如果待敲除的序列与四环素抗性tetra基因的片段大小相差较大,则阳性结果与阴性对照的片段相差较大容易识别;如果待敲除基因序列与四环素抗性tetra基因片段大小相差较小,则由于琼脂糖凝胶电泳的分辨率问题,会导致很难识别阳性结果与阴性对照,进而无法准确判断结果。
技术实现要素:
为克服现有检测方法的不足,本发明的目的是提供一种通用检测引物,其设计方法为在四环素tetra基因中间约1/3处设计一对互补的引物,与原有的检测重组阳性的鉴定引物进行组合使用,分别用于检测以四环素基因tetra为替换基因的λ-red重组系统基因敲除重组阳性克隆的3’端序列和5’端序列。这样设计检测引物使检测重组阳性克隆有了3种选择,可以完全避免因为待敲除的序列与四环素tetra基因相差较小而带来的难以判断阳性结果难题。而且用通用引物检测重组阳性克隆,可以更有效的证明四环素tetra基因插入位置的正确性。
本发明另一目的,提供一种以四环素基因tetra为替换基因的λ-red重组系统基因敲除重组阳性的检测方法。
为了实现上述目的本发明采用如下技术方案:
第一方面,提供一种以四环素抗性基因tetra为替换基因的λ-red重组系统基因敲除重组阳性克隆的检测方法,包含如下步骤:
(1)设计引物:
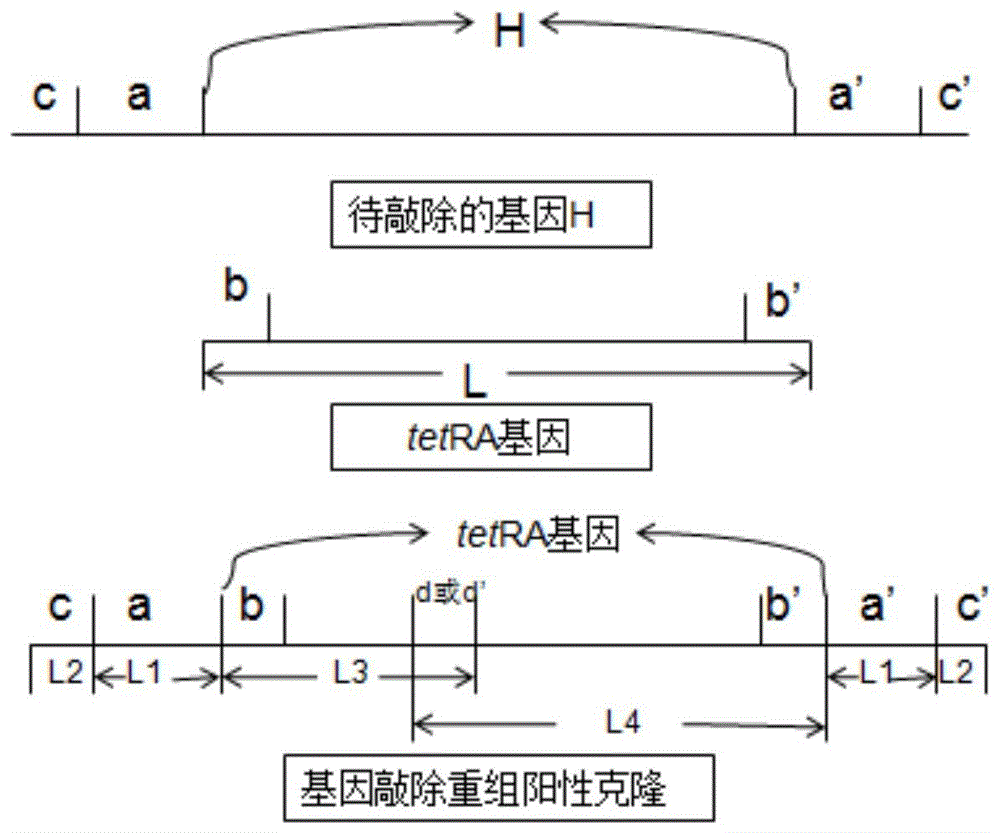
从ncbi数据库获得tetra基因序列和待敲除的基因h的序列,设计一对引物h-tetrar和h-tetraf用于扩增同源重组序列,每一条引物的5’端有35~60bp,分别为待敲除的基因h上下游同源序列a或a’片段,其序列长度均为l1,3’端分别为四环素抗性基因tetra两侧自带的引物序列b或b’片段,四环素抗性基因tetra的序列长度为l;设计用于鉴定基因是否重组的鉴定引物jc-hf和jc-hr,其序列分别位于待敲除的基因h上下游同源序列a或a’片段的外侧的c或c’片段,引物jc-hf和jc-hr的序列长度均为l2;设计两条互补的通用引物tyjc-tetraf和tyjc-tetrar,通用引物的序列为四环素抗性基因tetra内部的d片段,四环素抗性基因tetra上从序列b至d之间的序列长度为l3,四环素抗性基因tetra上从序列d’至b’之间的序列长度为l4;
(2)含tetra基因的dna片段的扩增;
(3)感受态细胞的制备;
(4)电转及目的菌株的筛选与鉴定;
用鉴定引物jc-hf和jc-hr检测筛选的目的菌株,目的片段长度理论值=l2+l1+l+l1+l2;用jc-hr和tyjc-tetraf检测筛选的目的菌株,目的片段长度理论值=l1+l2+l4,用jc-hf和tyjc-tetrar检测筛选的目的菌株,目的片段长度理论值=l1+l2+l3;
如果分别用jc-hf和jc-hr、jc-hr和tyjc-tetraf或jc-hf和tyjc-tetrar3对引物检测筛选的目的菌株获得的pcr扩增的片段长度与目的片段长度理论值接近,表明tetra基因打靶片段已经成功的将待敲除的基因h敲除。
优选地,上述检测方法中的步骤(1)所述的两条互补的通用引物tyjc-tetraf和tyjc-tetrar在四环素抗性基因tetra离5’端约1/3处,其序列如seqidno:1和seqidno:2所示。
本发明的技术方案的原理图如图1所示。
与现有技术相比,本发明的优势在于:
本发明设计的检测引物使检测阳性克隆是有了3种选择,可以完全避免因为待敲除的序列与四环素tetra基因相差较小而带来的难以判断阳性结果难题。而且用通用引物检测阳性克隆,可以更有效的证明四环素tetra基因插入位置的正确性。
附图说明
图1为本发明技术方案的原理图示意图;
图2用于tar基因重组的pcr产物切胶回收电泳检测;
m:dl2000dna标样;1:四环素抗性基因胶回收产物。
图3tar基因敲除株菌落pcr鉴定图;
m:dl2000dna标样;泳道1是对照组tar基因,大小为1662bp,泳道2是引物jc-tarr和jc-tarf进行pcr检测结果,与目的片段理论值2070bp接近;泳道3是引物jc-tar-r和tyjc-tetraf进行pcr扩增的片段,与目的片段理论值1469bp接近;泳道4是引物tyjc-tetrar和jc-tar-f进行pcr扩增的片段,与目的片段理论值576bp接近。
图4在大肠杆菌基因组中相邻的两个基因cheb和cher的位置示意图;
图5用于cheb和cher基因重组的pcr产物切胶回收电泳检测;
m:dl2000dna标样;1:四环素抗性基因胶回收产物。
图6cheb和cher基因敲除株菌落pcr鉴定图;
m:dl2000dna标样;泳道1是以mg1655原始菌株对照检测,大小为2029bp,泳道2、3、4是引物jc-cherf和jc-chebr进行pcr检测阳性克隆,大小为2070bp;泳道5为引物jc-chebr和tyjc-tetraf进行pcr检测mg1655原始菌株的对照组,没有条带,和预期相符;泳道6、7、8是引物jc-chebr和tyjc-tetraf进行pcr检测阳性克隆,大小为1469bp;泳道9为引物tyjc-tetrar和jc-cherf进行pcr检测mg1655原始菌株的对照组,没有条带,和预期相符;泳道10、11、12是引物tyjc-tetrar和jc-cherf进行pcr检测阳性克隆,大小为576bp。
具体实施方式
通过以下详细说明结合附图可以进一步理解本发明的特点和优点。所提供的实施例仅是对本发明方法的说明,而不以任何方式限制本发明揭示的其余内容。
以下实施例中,从ncbi数据库获得tetra基因序列,双下划线部分为tetra基因的18bp的序列,用于四环素同源重组的外源dna的pcr扩增引物的3’端。单下划线为通用检测引物,与用于鉴定基因是否重组的鉴定引物搭配使用。
通用引物序列如下,两个引物序列互补:
tyjc-tetraf:5’acaatgtaggctgctcta3’(用于检测3’序列)
tyjc-tetrar:5’tagagcagcctacattgt3’(用于检测5’序列)
【实施例1】利用通用检测引物检测敲除大肠杆菌阿斯帕特酸(天冬氨酸)受体tar基因的阳性克隆
(1)设计引物:
用于同源重组的引物为tar-tetrar和tar-tetraf;设计原则是5'端为tar外侧基因的同源区(未加下划线),3'端为四环素抗性基因tetra的同源序列(加下划线),重组体的检测所用引物为jc-tarf及jc-tarr。这些引物的序列如表1所示,由生工生物工程(上海)股份有限公司合成。
表1实施例1所用的引物
(2)含tetra基因的dna片段的扩增:
以含有四环素抗性基因的大肠杆菌dh10bac的dna为模板,tar-tetrar和tar-tetraf为引物进行pcr扩增,获得两端分别与tar基因上、下游同源,中间为四环素抗性基因tetra的dna片段。将该片段进行琼脂糖凝胶回收,以用于下一步电转化,如图2所示。
(3)感受态细胞的制备:
将含有pkd46质粒的mg1655菌株在含有20μg/mlamp的lb培养基中30℃下过夜培养,之后以1:100的比例接种到含20μg/mlamp的lb中30℃下震荡培养,当od600达到0.6时加入0.2%的阿拉伯糖进行诱导,4小时后4℃下收集细胞,用1/2体积预冷的无菌去离子水洗2次,并浓缩为原体积的1/40,作为感受态细胞备用。
(4)电转及目的菌株的筛选与鉴定
将胶回收的dna片段(100ng)与80μl感受态细胞在电转杯中混合,冰浴5-10min后,用2200v电击,电击后迅速加入1000μllb培养液,30℃下震荡培养30min后在20μg/ml的四环素平板上涂板,37℃下过夜培养,平板上长出的菌落为抗四环素的菌落,对其进一步在四环素平板上划线纯化,然后用检测引物检测结果,如图3所示。泳道1是对照组tar基因,大小为1662bp,泳道2是引物jc-tarr和jc-tarf进行pcr扩增的片段,大于2000bp,与目的片段理论值2070bp接近;泳道3是引物jc-tar-r和tyjc-tetraf进行pcr扩增的片段,在1000到2000bp之间,靠近2000bp,与目的片段理论值1469bp接近;泳道4是引物tyjc-tetrar和jc-tar-f进行pcr扩增的片段,与目的片段理论值576bp接近。表明tetra基因打靶片段已经成功的将tar基因敲除。
【实施例2】利用通用检测引物检测同时敲除大肠杆菌甲基修饰系统cheb(甲基化酶)和cher(甲基脂酶)基因的阳性克隆
(1)设计引物:
如图4所示,cheb和cher在大肠杆菌基因组中是相邻的两个基因,cheb基因序列号为:nc_000913.3(1967452..1968501,complement);cher基因序列号为:nc_000913.3(1968504..1969364,complement)。因此同时敲除这两个基因时目标序列大小为1913bp,用于同源重组的引物为cheb-tetrar和cher-tetraf;设计原则是5'端为cheb和cher外侧基因的同源区(未加下划线),3'端为四环素抗性基因tetra的同源序列(加下划线),重组体的检测所用引物为jc-cherf及jc-chebr。这些引物的序列如表2所示,由由生工生物工程(上海)股份有限公司合成。
表2实施例2所用的引物
(2)含tetra基因的dna片段的扩增:
以含有四环素抗性基因的大肠杆菌dh10bac的dna为模板,cheb-tetrar和cher-tetraf为引物进行pcr扩增,获得两端分别与cheb和cher基因上、下游同源,中间为四环素抗性基因tetra的dna片段。将该片段进行琼脂糖凝胶回收,以用于下一步电转化,如图5所示。
(3)感受态细胞的制备:
将含有pkd46质粒的mg1655菌株在含有20μg/mlamp的lb培养基中30℃下过夜培养,之后以1:100的比例接种到含20μg/mlamp的lb中30℃下震荡培养,当od600达到0.6时加入0.2%的阿拉伯糖进行诱导,4小时后4℃下收集细胞,用1/2体积预冷的无菌去离子水洗2次,并浓缩为原体积的1/40,作为感受态细胞备用。
(4)电转及目的菌株的筛选与鉴定
将胶回收的dna片段(100ng)与80μl感受态细胞在电转杯中混合,冰浴5-10min后,用2200v电击,电击后迅速加入1000μllb培养液,30℃下震荡培养30min后在20μg/ml的四环素平板上涂板,37℃下过夜培养,平板上长出的菌落为抗四环素的菌落,对其进一步在四环素平板上划线纯化,然后用检测引物检测结果,如图6所示。泳道1是以mg1655原始菌株对照检测,大小为2029bp,泳道2、3、4是引物jc-cherf和jc-chebr进行pcr检测阳性克隆,大小为2070bp;泳道5为引物jc-chebr和tyjc-tetraf进行pcr检测mg1655原始菌株的对照组,没有条带,和预期相符;泳道6、7、8是引物jc-chebr和tyjc-tetraf进行pcr检测阳性克隆,大小为1469bp;泳道9为引物tyjc-tetrar和jc-cherf进行pcr检测mg1655原始菌株的对照组,没有条带,和预期相符;泳道10、11、12是引物tyjc-tetrar和jc-cherf进行pcr检测阳性克隆,大小为576bp。从图中泳道1和泳道2、3、4可以看出,由于敲除的基因大小和替代基因大小相差较小,如果用原始的检测引物做pcr检测,琼脂糖凝胶电泳的分辨率无法辨认阳性菌株和原始菌株的区别,因此也无法断定基因是否敲除;如果配合通用检测引物则可以明确判断目标基因cheb和cher被四环素抗性基因tetra替换成功。
序列表
<110>武汉生物工程学院
<120>一种用于λ-red重组系统基因敲除重组阳性克隆检测的引物及检测方法
<160>10
<170>siposequencelisting1.0
<210>1
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>1
acaatgtaggctgctcta18
<210>2
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>2
tagagcagcctacattgt18
<210>3
<211>58
<212>dna
<213>人工序列(artificialsequence)
<400>3
ctccccatcaggcggcaatgaccgcgttagtaaatactcgctaagcacttgtctcctg58
<210>4
<211>58
<212>dna
<213>人工序列(artificialsequence)
<400>4
tgccgataacgagatcaacttgttttcaggaaggtgccttttaagacccactttcaca58
<210>5
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>5
taaagtttcccccctcct18
<210>6
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>6
tacgattaaacatcaacg18
<210>7
<211>58
<212>dna
<213>人工序列(artificialsequence)
<400>7
aaaaatttaagttctttatccgccatttcacactcctgatctaagcacttgtctcctg58
<210>8
<211>58
<212>dna
<213>人工序列(artificialsequence)
<400>8
aattgcgccagtggtatcctgaagtgattgagaaggcgctttaagacccactttcaca58
<210>9
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>9
catgagtcggtgcagtta18
<210>10
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>10
gaaaagtcatccacaacc18
- 还没有人留言评论。精彩留言会获得点赞!
- 用CRISPR/Cas9技术易于筛选获得目的基因敲除细胞系的方法及产品与制造工艺
- 一种Candida amazonensis的FLP/FRT基因敲除方法与制造工艺
- 玉米低镁条件下特异性转运镁离子基因ZmMGT10及其应用的制造方法与工艺
- CRISPR‑Cas9特异性敲除猪SLA‑3基因的方法及用于特异性靶向SLA‑3基因的sgRNA与制造工艺
- 利用CRISPR/Cas9敲除人PD‑1基因构建靶向CD19CAR‑T细胞的方法与制造工艺
- 一种构建基因替代小鼠研究野生型和突变体非肌性肌球蛋白重链II功能的方法与制造工艺
- 构建在海马体区域特异性敲除IKKα基因的小鼠模型的方法及打靶载体和试剂盒与制造工艺
- 猪ApoE基因敲除打靶载体及其构建方法和应用与制造工艺
- 一种血小板减少症的斑马鱼模型的制造方法与工艺
- 基于CRISPR技术敲除FUT8基因的方法与制造工艺