一种锇配合物、制备方法及其应用与流程
本发明涉及过渡金属配合物
技术领域:
,尤其涉及一种锇配合物、制备方法及其应用。
背景技术:
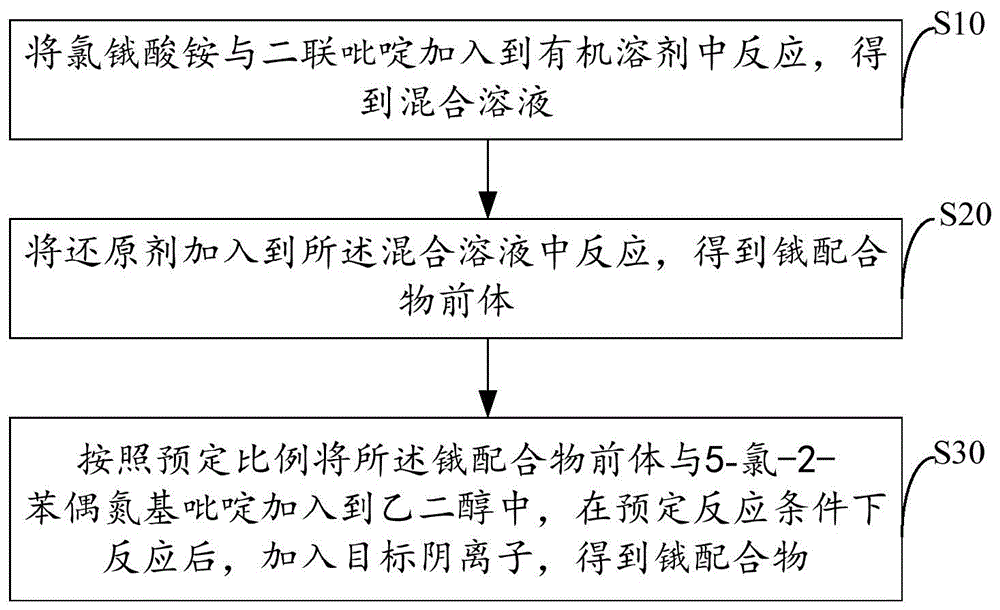
:过渡金属配合物一直是近几十年的研究热点之一,大量的含有d6、d8和d10电子组态的过渡金属配合物被成功合成和研究。在已经合成的金属配合物中,以铱和钌配合物数量最多、应用最为广泛。但是研究发现饿配合物具有更加红外的吸收。p.t.chou等人合成出一系列[os(ii)(n~n)2(pr)2](n~n=2-吡啶基三唑或2-吡啶基吡唑pr=pph2me或pphme2)配合物。研究表明,此配合物最低能吸收属于mlct和ilct混合跃迁,mlct跃迁贯穿于整个跃迁过程中,具有较长波长的紫外吸收。其中,较长波长的mlct吸收峰使配合物在测定共振拉曼光谱时的激发波长也相应延长,从而使其更适合生物样品的分析,增强组织穿透能力和生物相容性。在医药上,金属抗癌药物如含铂抗癌药物是临床上广泛使用的抗肿瘤药物,而钌、锇配合物将成为最有前途的抗癌药物之一,锇配合物分子结构具有很好的可塑性,容易在配体上引入其它分子活性基团,因而其性能也具有可塑性,合成新型的、具有优越稳定性和抗肿瘤活性的锇配合物在医药、材料领域具有重大意义。现有的技术中所制备的锇配合物稳定性不高,抗肿瘤活性低。因此,现有技术还有待于改进和发展。技术实现要素:鉴于上述现有技术的不足,本发明的目的在于提供一种锇配合物、制备方法及其应用,旨在解决现有锇配合物稳定性差、抗肿瘤活性低的问题。本发明为解决上述技术问题所采用的技术方案如下:一种锇配合物,其中,该配合物具有如下通式:其中:x为cl-、br-和i-中的任一种,m-为pf6-。一种锇配合物的制备方法,其中,包括步骤:将氯锇酸铵与二联吡啶加入到有机溶剂中反应,得到混合溶液;将还原剂加入到所述混合溶液中反应,得到锇配合物前体;按照预定比例将所述锇配合物前体与5-氯-2-苯偶氮基吡啶加入到乙二醇中,在预定反应条件下反应后,加入目标阴离子,得到锇配合物。所述的锇配合物的制备方法,其中,所述还原剂为连二亚硫酸钠或亚硫酸钠。所述的锇配合物的制备方法,其中,所述步骤将氯锇酸铵与二联吡啶加入到有机溶剂中反应,得到混合溶液,具体包括:将氯锇酸铵与二联吡啶加入到有机溶剂中,在惰性气体保护下回流反应第一预定时间,反应后得到混合溶液。所述的锇配合物的制备方法,其中,所述步骤将还原剂加入到所述混合溶液中反应,得到锇配合物前体,具体包括:将还原剂的水溶液加入到所述混合液中,放入4℃冰箱,过夜得到锇配合物前体。所述的锇配合物的制备方法,其中,所述预定比例为锇配合物前体与5-氯-2-苯偶氮基吡啶的摩尔比为1:1-1.2。所述的锇配合物的制备方法,其中,所述预定反应条件为在惰性气体保护下,设置温度120-140℃。所述的锇配合物的制备方法,其中,所述第一预定时间为4-8小时。一种拉曼探针,其中,所述拉曼探针采用锇配合物制备而成。一种锇配合物的应用,其中,用于制备以细胞核为作用靶点的抗肿瘤药。有益效果:本发明所提供的锇配合物具有极高的结构稳定性,几乎不发荧光,对肿瘤细胞的细胞毒性很强,对癌细胞也具有选择性,能够应用在肿瘤细胞的拉曼成像领域。附图说明图1是本发明实施实例所提供的锇配合物的制备方法流程图。图2是本发明实施例所提供的锇配合物的合成途径图。图3是本发明实施实例所提供的锇配合物的紫外和荧光光谱图。图4是本发明实施实例所提供的饿配合物的红外光谱图。图5是本发明实施实例提供的饿配合物在细胞内不同部位的含量分布柱状图。图6是本发明实施实例所提供的饿配合物在溶液中和细胞中的拉曼光谱图。图7是本发明实施实例所提供的饿配合物在肺癌细胞中的拉曼成像图,b是c-h峰,代表整个细胞区域,c、d分别是bpy辅助配体和主配体里n=n键的峰,e为b、c、d的叠加图。具体实施方式为使本发明的目的、技术方案及优点更加清楚、明确,以下参照附图并举实施例对本发明进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。本发明提供一种锇配合物,该配合物具有如下式所示的结构:其中,x为cl-、br-和i-中的任一种,m-为pf6-。上述锇配合物的结构中锇与配体之间通过锇-氮键相连,增强了母体与配体之间的稳定性。同时上述结构中的锇是正二价,因此使得配合物的结构更加稳定,适于制备拉曼探针。请参阅图1,本发明还提供了一种锇配合物的制备方法,包括步骤:s10、将氯锇酸铵与二联吡啶加入到有机溶剂中反应,得到混合溶液;具体来说,先将氯锇酸铵与二连吡啶溶解在有机溶剂中,制备得到混合溶液,其中所述有机溶剂可以是dmf、乙二醇等。s20、将还原剂加入到所述混合溶液中反应,得到锇配合物前体;具体来说,将步骤s10中得到的混合物在惰性气体保护条件下回流反应,回流反应结束后,向混合溶液中加入一定量的还原剂,搅拌均匀,放入4℃冰箱,静置过夜,抽滤后得到黑色沉淀,用水和乙醚对所述黑色沉淀进行洗涤,得到了锇配合物前体。所述还原剂为连二亚硫酸钠、亚硫酸铵等。该步骤中具体的反应机理为:2,2-联吡啶配体上电子云密度较高的氮原子提供电子与氯锇酸铵以金属配体键(即锇-氮键)形成七配位的中间体,氯离子离去,再重复这一过程,是一个典型的类似sn2亲核取代的双分子缔合机理,其中,正三价的锇再被连二亚硫酸钠还原形成更稳定的二价锇,最终形成锇配合物前体[os(bpy)2cl2]。s30、按照预定比例将所述锇配合物前体与5-氯-2-苯偶氮基吡啶加入到乙二醇中,在预定反应条件下反应后,加入阴离子盐,得到锇配合物。具体来说,按照一定的比例称取锇配合物前体和5-氯-2-苯偶氮基吡啶(主配体)加入到乙二醇中,设置反应温度120℃,在氩气保护下加热回流预定时间,反应结束后将溶液温度降至室温,向其中加入六氟磷酸铵的水溶液,析出黑色固体即锇配合物。其中,锇配合物前体:5-氯-2-苯偶氮基吡啶的摩尔比为1:1-1.2,适当增加主配体的量可以原料纯度不够所带来偏差的问题。该步骤中具体的反应机理为:主配体(5-氯-2-苯偶氮基吡啶)上电子云密度较高的氮原子提供电子与锇前体的空位形成锇-氮键,产生七配位的中间体,氯离去,另一个氮原子再重复这一过程,两个锇-氮键与右侧配体成环,再与六氟磷酸根成盐,从而形成目标锇配合物,也是基于sn2亲核取代的双分子缔合机理。本发明还提供了一种拉曼探针,所述拉曼探针是由上述的锇配合物制备得到。该分子探针光稳定好,自身几乎不发荧光。本发明还提供了一种锇配合物的应用,用于制备以细胞核为作用靶点的抗肿瘤药。下面通过具体的实施例对本发明所提供的锇配合物的制备方法进行进一步的解释说明。称取0.439g的氯锇酸铵固体和0.328g的二联吡啶粉末加入到15ml的dmf溶液中,在氮气保护下回流反应5h,冷却至室温后,往混合溶液中加入含有6.8g连二亚硫酸钠的水溶液,混合均匀后放入冰箱,静置过夜后,抽滤,黑色沉淀用水和乙醚洗涤数次,得到锇配合物前体。然后按计量摩尔比(1:1)称取[os(bpy)2cl2]约57.35mg和主配体(5-氯-2-苯偶氮基吡啶)约21.77mg反应,加入10ml乙二醇,设置温度为120℃,氩气保护,加热回流8h,待溶液冷却后,加入nh4pf6水溶液,析出黑色固体,真空抽滤后,得深黑色固体。具体合成途径见图2。对深黑色固体进行分析:1、esi(m/z):359.65[m-2pf6]2+/2.1hnmr(400mhz,dmso-d6)δ9.23(d,j=2.2hz,1h),9.05(d,j=8.2hz,1h),8.95(d,j=8.2hz,1h),8.67(d,j=8.1hz,1h),8.55(d,j=8.0hz,1h),8.30(t,j=7.9hz,1h),8.21(dd,j=13.8,6.6hz,2h),7.93(dd,j=11.3,4.5hz,1h),7.79(d,j=6.3hz,1h),7.72(d,j=5.3hz,1h),7.70–7.58(m,4h),7.58–7.53(m,1h),7.43(dt,j=10.9,6.3hz,3h),7.25(t,j=7.3hz,1h),7.18(t,j=7.5hz,2h),6.82(d,j=7.7hz,2h).2、对产物进行紫外/荧光/红外光谱实验:运用紫外光谱和荧光光谱对锇配合物的光学性质进行初步表征,确定其吸收峰的区域和荧光发射峰的位置,从图3可以看出,配合物在460-470nm处有强吸收峰,用460nm为激发波长测定其荧光光谱,发现其发射峰特别微弱,几乎没有荧光发射。运用kbr压片法测定锇配合物粉末的红外光谱,如图4所示,其中约1500cm-1处为c=c键的伸缩振动峰,1620cm-1为c=n键的伸缩振动峰。3、细胞毒性实验:通过mtt实验,探究锇配合物对肝癌细胞株(hep-g2)、肺癌细胞株(a549)和相应的正常细胞株(lo2,mrc-5)的细胞毒性。表1是锇配合物和顺铂对各种细胞株给药48h后的ic50值。ic50代表抑制一半细胞增殖所需要的浓度,与毒性负相关,表1中可以得出锇配合物对hep-g2肝癌细胞和a549肺癌细胞的毒性高于顺铂,并且其对两组正常细胞的的ic50值远远高于癌细胞的ic50,表明配合物对癌细胞具有选择性,但对正常细胞基本没有毒性,这将有望成为一种高效低毒的靶向药物。表1hep-g2a549mrc-5lo2os6.3±0.713.4±1.7102.4±2.775.2±3.9cis-pt9.5±1.710.2±1.813.5±1.513.2±1.34、细胞摄取实验:收集孵育配合物(10μm,8h)之后的肺癌a549细胞,分成2份,利用细胞核提取试剂提取一份细胞的细胞核,剩余部分为细胞质,总共分为整体、细胞核和细胞质3份样品(分装于1.5ml离心管中),用浓硝酸消化2天之后,加超纯水稀释至硝酸浓度为5%的澄清溶液,用滤膜过滤之后,取每份溶液10ml作为最终测试样品,并同时用锇标准溶液,联用抗坏血酸和硫脲作为抗氧化剂(防止金属锇被氧化,避免误差),配置浓度梯度为0,10,20...1000ppb的标准溶液,测试时先用标准液测定标准曲线(r2=0.99997),再依次测定细胞样品,经过计算得出配合物在细胞核的含量约为89.9ng/cell,细胞质约为9.1ng/cell,如图5所示,证明配合物大部分聚集在细胞核中。5、拉曼成像实验:先对锇配合物的粉末与溶液进行测试,用532nm激发得出拉曼光谱如图6所示,再对孵育锇配合物(100μm,4h)的肺癌a549细胞进行拉曼成像测试,在532nm激光激发下,选取一个细胞区域进行扫描,通过后期的mapping处理,得出直观的拉曼图像,如图7所示,进一步表明配合物能够聚集在细胞核中。综上所述,本发明所提供的一种锇配合物、制备方法及其应用。所述方法包括步骤,将氯锇酸铵与二联吡啶加入到有机溶剂中反应,得到混合溶液;将还原剂加入到所述混合溶液中反应,得到锇配合物前体;按照预定比例将所述锇配合物前体与5-氯-2-苯偶氮基吡啶加入到乙二醇中,在预定反应条件下反应后,加入目标阴离子,得到锇配合物。本发明所提供的锇配合物具有极高的结构稳定性,几乎不发荧光,对肿瘤细胞的细胞毒性很强,对癌细胞也具有选择性,能够应用在肿瘤细胞的拉曼成像领域。应当理解的是,本发明的应用不限于上述的举例,对本领域普通技术人员来说,可以根据上述说明加以改进或变换,所有这些改进和变换都应属于本发明所附权利要求的保护范围。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1