多环不饱和缩酮化合物及其合成方法
1.本发明属于药物化学及有机合成领域,具体涉及芳香聚酮类天然产物abx-a、abx-c、 abx-e、babx以及多环不饱和缩酮化合物、以及其合成方法和应用。
背景技术:
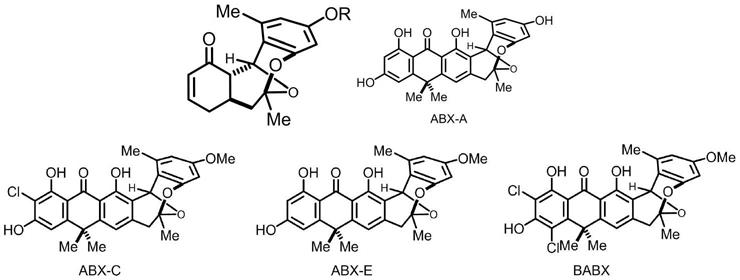
2.anthrabenzoxocinones类属于芳香聚酮类天然产物。abx-a则是由suda等人从紫黑链霉菌a24566分离得到。abx-c、abx-e则是由瞿旭东等人通过组合生物学的方法分离得到;而babx 是由singh等人在2005年从土壤链霉菌ma6657中分离得到。据初步的研究表明, anthrabenzoxocinones类属于芳香聚酮类天然产物均对于耐甲氧西林金黄色葡萄球菌和枯草芽孢杆菌表现出良好的活性。据初步的研究表明,anthrabenzoxocinones类属于芳香聚酮类天然产物均对于耐甲氧西林金黄色葡萄球菌和耐万古霉素粪肠球菌表现出良好的活性。例如, abx-a对于包括耐甲氧西林金黄色葡萄球菌在内革兰氏阳性菌表现出良好的生物活性,其mic 分别为3.13μg/ml。例如,abx-c对于包括耐甲氧西林金黄色葡萄球菌和枯草芽孢杆菌表现出良好的生物活性,其mic分别为0.89μg/ml、1.94μg/ml。多环不饱和缩酮化合物结构通常具有良好的生物活性,如,abx-a、abx-c、abx-e、babx对于包括耐甲氧西林金黄色葡萄球菌在内革兰氏阳性菌均表现出良好的生物活性。
3.结构如下:
[0004][0005]
至目前尚未见有关于anthrabenzoxocinones类天然产物的全合成的相关报道。
技术实现要素:
[0006]
本发明首次创新提出了芳香聚酮类天然产物anthrabenzoxocinones及其类似物的合成方法,包括abx-a、abx-c、abx-e、babx、多环不饱和缩酮化合物及其合成方法和应用,从而实现了芳香聚酮类天然产物及其类似物的合成,克服了现有技术存在的问题。本发明合成方法简洁高效,不仅在步骤上简洁高效有利于天然产物的大量制备,而且还有利于天然产物衍生物的合成,这将为后续药物化学研究奠定一定的基础。
[0007]
本发明提出了一种芳香聚酮类化合物天然产物abx-a的合成方法,以式1化合物为起始原料和式2化合物发生分子间狄尔斯-阿尔德反应得到片段式3化合物,式4化合物酚羟基分别用溴化苄和氯甲基加醚试剂保护制备片段式6化合物;将片段式3化合物与式6化合
物发生亲核加成反应以及分子内杂狄尔斯-阿尔德反应制备片段式7化合物;式7化合物通过氧化生成式12化合物,所述片段式12化合物和芳基醛式11化合物发生钛酸四异丙酯介导的光催化的狄尔斯-阿尔德反应得到片段式13化合物,随后经氧化芳构化得到片段式15化合物,再经脱保护及在苯环上选择性氯代等得到所述芳香聚酮类化合物abx-a。
[0008]
所述合成方法的具体路线如下:
[0009][0010]
其中,r1为叔丁基二甲基硅基、三甲基硅基、三异丙基硅基、叔丁基二苯基硅基;r2为c1-c10 烷基、苄基、对甲氧基苯甲基、甲氧基甲基、叔丁基二甲基硅基、三甲基硅基、三异丙基硅基、叔丁基二苯基硅基;r3为c1-c10烷基、苄基、对甲氧基苯甲基、甲氧基甲基,r4为c1-c10 烷基、苄基、对甲氧基苯甲基、甲氧基甲基,r5为c1-c10烷基、叔丁基二甲基硅基、三甲基硅基、三异丙基硅基、叔丁基二苯基硅基,r6为c1-c10烷基,x为卤素。
[0011]
其中,从式1化合物至化合物式15化合物的合成路线及步骤,如下所示:
[0012]
a)式1化合物和式2化合物混合均匀,100℃下反应,得到狄尔斯-阿尔德反应产物式3 化合物;
[0013]
b)式4化合物在丙酮作为溶剂、室温的条件下加入碳酸钾,室温反应,再加入4-甲基苯磺酰氯,加热回流,冷却至室温,加入氯甲基甲醚,室温反应,得到式5化合物;
[0014]
c)式5化合物在四氢呋喃作为溶剂、0℃的条件下加入钠氢,0℃反应,再加入溴化苄, 50℃反应,得到式6化合物;
[0015]
d)式6化合物在四氢呋喃作为溶剂、0℃的条件下加入正丁基锂,0℃反应,再加入式3 化合物,0℃反应,加入2n盐酸,调ph为酸性,50℃反应,得到式7化合物;
[0016]
e)式8化合物在二氯甲烷作为溶剂、0℃的条件下加入三溴化硼,室温反应,得到脱甲基的式9化合物;
[0017]
f)式9化合物在乙腈作为溶剂、0℃的条件下加入n,n-二异丙基乙胺,再加入氯甲基甲醚,室温反应,得到式10化合物;
[0018]
g)式10化合物在四氢呋喃作为溶剂、0℃的条件下加入碳酸钾,再加入碘甲烷,室温反应,滤除碳酸钾固体,再加入6n盐酸,50℃反应,经简单后处理,得到粗产物,粗产物溶于n,n-二甲基甲酰胺,加入咪唑、4-二甲氨基吡啶、三异丙基氯硅烷,室温反应,得到式11化合物;
[0019]
h)式7化合物在二氯甲烷作为溶剂、0℃的条件下加入戴斯马丁氧化剂,室温反应,得到式12化合物;
[0020]
i)式12化合物在无水二氧六环作为溶剂、室温的条件下加入式11化合物,再加入钛酸四异丙酯,用紫外光照射,得到式13化合物;
[0021]
j)式13化合物在二氯甲烷作为溶剂、室温的条件下加入戴斯马丁氧化剂,室温反应,得到式14化合物;
[0022]
k)式14化合物在甲苯作为溶剂、室温的条件下加入2,3-二氯-5,6-二氰基-1,4-苯醌, 80℃反应,得到所述式15化合物。
[0023][0024]
其中,r1为叔丁基二甲基硅基、三甲基硅基、三异丙基硅基、叔丁基二苯基硅基;r2为c1-c10 烷基、苄基、对甲氧基苯甲基、甲氧基甲基、叔丁基二甲基硅基、三甲基硅基、三异丙基硅基、叔丁基二苯基硅基;r3为c1-c10烷基、苄基、对甲氧基苯甲基、甲氧基甲基;r4为c1-c10 烷基、苄基、对甲氧基苯甲基、甲氧基甲基;r5为c1-c10烷基、叔丁基二甲基硅基、三甲基硅基、三异丙基硅基、叔丁基二苯基硅基,r6为c1-c10烷基,x为卤素。
[0025]
其中,从式15化合物至化合物abx-a的合成路线及步骤,为:
[0026][0027]
其中,r2为c1-c10烷基、苄基、对甲氧基苯甲基、甲氧基甲基、叔丁基二甲基硅基、三甲基硅基、三异丙基硅基、叔丁基二苯基硅基;r5为c1-c10烷基、叔丁基二甲基硅基、三甲基硅基、三异丙基硅基、叔丁基二苯基硅基;r6为c1-c10烷基。
[0028]
所述步骤为:
[0029]
l)所述式15化合物在四氢呋喃作为溶剂、室温的条件下加入碘化镁,70℃反应,得到式 16化合物;
[0030]
m)式16化合物在四氢呋喃作为溶剂、0℃的条件下加入四丁基氟化胺,室温反应,得到式17化合物;
[0031]
n)式17化合物在甲醇作为溶剂、室温的条件下加入氯化钯,氢气氛围下室温反应,得到所述式abx-a化合物。
[0032]
在一具体实施方案中,所述合成路线包括如下步骤:
[0033]
a)式1化合物和式2化合物混合均匀后100℃反应5小时,即可得到狄尔斯-阿尔德反应产物式3化合物。其中,其物质的量的比为式1化合物:式2化合物=1:1;
[0034]
b)式4化合物在丙酮作为溶剂,室温下加入碳酸钾,室温反应30分钟,再加入4-甲基苯磺酰氯,加热回流24h,冷却至室温,加入氯甲基甲醚,室温反应6小时后,即可得到式5 化合物;其中,每毫摩尔(mmol)式4化合物对应适用溶剂的用量范围为1ml到10ml;其物质的量的比为式4化合物:碳酸钾:4-甲基苯磺酰氯:氯甲基甲醚=1:3:1.05:2;
[0035]
c)式5化合物在四氢呋喃作为溶剂,0℃下加入钠氢,0℃反应30分钟,再加入溴化苄,50℃反应4小时,即可得到式6化合物;其中,每毫摩尔(mmol)式5化合物对应适用溶剂的用量范围为1ml到10ml;其物质的量的比为式5化合物:钠氢:溴化苄=1:3:2;
[0036]
d)式6化合物在四氢呋喃作为溶剂,0℃下加入正丁基锂,0℃反应30分钟,再加入式3化合物,0℃反应2小时,加入2n盐酸,调ph为酸性,50℃反应10小时即可得到式7化合物;其中,每毫摩尔(mmol)式6化合物对应适用溶剂的用量范围为1ml到10ml;其物质的量的比为式6化合物:正丁基锂:式3化合物=1:2:2;
[0037]
e)式8化合物在二氯甲烷作为溶剂,0℃下加入三溴化硼,室温反应12小时,即可得到脱甲基的式9化合物;其中,每毫摩尔(mmol)式8化合物对应溶剂的用量适用范围为1ml到 10ml;其物质的量的比为式8化合物:三溴化硼=1:5;
[0038]
f)式9化合物在乙腈作为溶剂,0℃下加入n,n-二异丙基乙胺,再加入氯甲基甲醚,室温反应12小时,即可得到式10化合物;其中,每毫摩尔(mmol)式9化合物对应溶剂的用量适用范围为1ml到10ml;其物质的量的比为式9化合物:n,n-二异丙基乙胺:氯甲基甲醚=1:1:1;
[0039]
g)式10化合物在四氢呋喃作为溶剂,0℃下加入碳酸钾,再加入碘甲烷,室温反应4小时,滤除碳酸钾固体,再加入6n盐酸,50℃反应3小时,经简单后处理,得到粗产物,粗产物溶于n,n-二甲基甲酰胺,加入咪唑、4-二甲氨基吡啶、三异丙基氯硅烷,室温反应12小时,即可得到式11化合物;其中,每毫摩尔(mmol)式10化合物对应适用溶剂的用量范围为1ml到10ml;其物质的量的比为式10化合物:碳酸钾:碘甲烷:6n盐酸:咪唑:4
-ꢀ
二甲氨基吡啶:三异丙基氯硅烷=1:1.5:1.5:6:1.3:0.1:1.3;
[0040]
h)式7化合物在二氯甲烷作为溶剂,0℃下加入戴斯马丁氧化剂,室温反应2小时,即可得到式12化合物;其中,每毫摩尔(mmol)式7化合物对应适用溶剂的用量范围为1ml到10ml;其物质的量的比为式7化合物:戴斯马丁氧化剂=1:2;
[0041]
i)式12化合物在无水二氧六环作为溶剂,室温加入式11化合物,再加入钛酸四异丙酯,用 300纳米紫外光照射4小时,即可得到式13化合物;其中,每毫摩尔(mmol)式12化合物对应适用溶剂的用量范围为1ml到10ml;其物质的量的比为式12化合物:式11化合物:钛酸四异丙酯=1:3:9;
[0042]
j)式13化合物在二氯甲烷作为溶剂,室温加入戴斯马丁氧化剂,室温反应2小时,即可得到式14化合物;其中,每毫摩尔(mmol)式13化合物对应适用溶剂的用量范围为1ml到10ml;其物质的量的比为式13化合物:戴斯马丁氧化剂=1:3;
[0043]
k)式14化合物在甲苯作为溶剂,室温加入2,3-二氯-5,6-二氰基-1,4-苯醌,80℃反应2小时,即可得到式15化合物;其中,每毫摩尔(mmol)式14化合物对应适用溶剂的用量范围为1ml到10ml;其物质的量的比为式14化合物:2,3-二氯-5,6-二氰基-1,4-苯醌=1:3;
[0044]
l)式15化合物在四氢呋喃作为溶剂,室温加入碘化镁,70℃反应4小时,即可得到式16化合物;其中,每毫摩尔(mmol)式15化合物对应适用溶剂的用量范围为1ml到10ml;其物质的量的比为式15化合物:碘化镁=1:5;
[0045]
m)式16化合物在四氢呋喃作为溶剂,0℃加入四丁基氟化胺,室温反应2小时,即可得到式 17化合物;其中,每毫摩尔(mmol)式16化合物对应适用溶剂的用量范围为1ml到10ml;其物质的量的比为式16化合物:四丁基氟化胺=1:2;
[0046]
n)式17化合物在甲醇作为溶剂,室温加入氯化钯,氢气氛围下室温反应10小时,即可得到式abx-a天然产物;其中,每毫摩尔(mmol)式17化合物对应适用溶剂的用量范围为1ml 到10ml;其物质的量的比为式17化合物:氯化钯=1:0.5。
[0047]
本发明还提出了按以上所述方法合成得到的芳香聚酮类化合物abx-a,其结构式如下:
[0048][0049]
本发明还提出了所述芳香聚酮类化合物abx-a在制备化合物abx-c,abx-e,babx中的应用。
[0050]
本发明还提出了式12所示的化合物,其结构式如下所示:
[0051][0052]
其中,所述式12化合物中,r2为c1-c10烷基、苄基、对甲氧基苯甲基、甲氧基甲基、叔丁基二甲基硅基、三甲基硅基、三异丙基硅基、叔丁基二苯基硅基。
[0053]
优选地,所述式12化合物中,r2为苄基、对甲氧基苯甲基。
[0054]
本发明还提出了所述式12化合物的合成方法,所述方法按前述步骤a)-步骤h)即合成得到式12化合物。
[0055]
本发明还提出了式15所示的化合物,其结构式如下所示:
[0056][0057]
其中,r2为c1-c10烷基、苄基、对甲氧基苯甲基、甲氧基甲基、叔丁基二甲基硅基、
三甲基硅基、三异丙基硅基、叔丁基二苯基硅基;r5为c1-c10烷基、叔丁基二甲基硅基、三甲基硅基、三异丙基硅基、叔丁基二苯基硅基;r6为c1-c10烷基。
[0058]
本发明还提出了式15所示化合物的合成方法,所述方法包括如上所述的步骤a)-步骤k)。
[0059]
本发明还提出一种芳香聚酮类化合物天然产物abx-e的合成方法,以式1化合物和式2 化合物发生狄尔斯-阿尔德反应得到片段式3化合物,片段式3化合物与式6化合物发生亲核加成反应及杂狄尔斯-阿尔德反应制备片段式7化合物;片段式12化合物和式11化合物发生钛酸四异丙酯介导的光催化的狄尔斯-阿尔德反应得到片段式13化合物,随后经氧化芳构化得到片段式15化合物,再经脱保护及在苯环上选择性氯代,得到所述芳香聚酮类化合物abx-e。
[0060]
其中,按前述反应路线及步骤a)至步骤k)合成得到所述式15化合物。
[0061]
接着,按以下反应路线,所述式15化合物甲基化再经脱保护,得到所述式21芳香聚酮类化合物abx-e;即,式15化合物脱r2保护基得到式18化合物,式18化合物经甲基化得式19 化合物,式19化合物脱保护基得到式20化合物,式20化合物脱保护基得式21化合物;
[0062][0063]
其中,r1为叔丁基二甲基硅基、三甲基硅基、三异丙基硅基、叔丁基二苯基硅基;r2为c1-c10 烷基、苄基、对甲氧基苯甲基、甲氧基甲基、叔丁基二甲基硅基、三甲基硅基、三异丙基硅基、叔丁基二苯基硅基;r3为c1-c10烷基、苄基、对甲氧基苯甲基、甲氧基甲基;r4为c1-c10 烷基、苄基、对甲氧基苯甲基、甲氧基甲基;r5为c1-c10烷基、叔丁基二甲基硅基、三甲基硅基、三异丙基硅基、叔丁基二苯基硅基;r6为c1-c10烷基;x为卤素。
[0064]
优选地,r2为苄基;r5为三异丙基硅基;r6为甲基。
[0065]
在一个具体实施方案中,反应路线如下所示,所述式15化合物中的r6为甲基,r5为三异丙基硅基tips;所述式15化合物甲基化再经脱保护,得到所述式21芳香聚酮类化合物abx-e:
[0066]
[0067]
其中,r2为c1-c10烷基、苄基、对甲氧基苯甲基、甲氧基甲基、叔丁基二甲基硅基、三甲基硅基、三异丙基硅基、叔丁基二苯基硅基。
[0068]
优选地,所述式15化合物,r2为c1-c10烷基、苄基、对甲氧基苯甲基、甲氧基甲基、叔丁基二甲基硅基、三甲基硅基、三异丙基硅基、叔丁基二苯基硅基;r5为c1-c10烷基、叔丁基二甲基硅基、三甲基硅基、三异丙基硅基、叔丁基二苯基硅基;r6为c1-c10烷基;经酸性条件或钯金属催化下脱出r2保护基得到式18化合物:
[0069][0070]
本发明还提出按上述合成方法制备得到的芳香聚酮类化合物天然产物abx-e,即式21化合物,其结构如下:
[0071][0072]
本发明还提出所述芳香聚酮类化合物abx-e在制备化合物abx-c,babx中的应用
[0073]
本发明还提出了一种芳香聚酮类化合物abx-c、babx的合成方法,式21化合物abx-e在四氢呋喃为溶剂、加入正丁基锂、0℃条件下反应,再加入次氯酸叔丁酯,0℃反应,得到所述化合物abx-c,babx;
[0074]
其中,所述合成反应路线如下:
[0075][0076]
其中,所述溶剂四氢呋喃的用量为,对应于每毫摩尔mmol式21化合物abx-e的溶剂用量为1ml-10ml;其物质的量的比为式21化合物abx-e:正丁基锂:次氯酸叔丁酯=1:3:2。
[0077]
所述式21芳香聚酮类化合物abx-e是由式15化合物再经脱保护及在苯环上选择性氯代反应得到,包括如下步骤:
[0078][0079]
其中,r2为c1-c10烷基、苄基、对甲氧基苯甲基、甲氧基甲基、叔丁基二甲基硅基、三甲基硅基、三异丙基硅基、叔丁基二苯基硅基;
[0080]
r5为叔丁基二甲基硅基、三甲基硅基、三异丙基硅基、叔丁基二苯基硅基;
[0081]
r6为c1-c10烷基。
[0082]
式15化合物经脱保护及在苯环上选择性氯代得到式21芳香聚酮类化合物abx-e。其中,式15化合物经酸性条件或者钯金属催化下脱出r2保护基,得到式18化合物。
[0083][0084]
其中,r2为c1-c10烷基、苄基、对甲氧基苯甲基、甲氧基甲基、叔丁基二甲基硅基、三甲基硅基、三异丙基硅基、叔丁基二苯基硅基;
[0085]
r5为c1-c10烷基、叔丁基二甲基硅基、三甲基硅基、三异丙基硅基、叔丁基二苯基硅基;
[0086]
r6为c1-c10烷基。
[0087]
然后,式18化合物在氯仿为溶剂,加入1,8-双二甲氨基萘,再加入三甲基氧鎓四氟硼酸,室温反应,得到上甲基的产物式19化合物;式19化合物在四氢呋喃为溶剂,加入碘化镁,80℃反应,得到脱甲基的产物式20化合物;式20化合物在四氢呋喃为溶剂,加入四丁基氟化胺,室温反应,得到脱硅基的天然产物abx-e。
[0088]
所述式15化合物通过下述方式合成得到,包括步骤:以式1化合物和式2化合物发生狄尔斯-阿尔德反应得到式3化合物,所述式3化合物与式6化合物发生亲核加成反应及杂狄尔斯-阿尔德反应得到式7化合物;所述式7化合物再经氧化得到式12化合物;所述式12化合物和式11化合物发生钛酸四异丙酯介导的光催化的狄尔斯-阿尔德反应得到式13化合物,随后经氧化芳构化得到式15化合物。
[0089][0090]
所述反应路线中,r1为叔丁基二甲基硅基、三甲基硅基、三异丙基硅基、叔丁基二苯基硅基;r2为c1-c10烷基、苄基、对甲氧基苯甲基、甲氧基甲基、叔丁基二甲基硅基、三甲基硅基、三异丙基硅基、叔丁基二苯基硅基;r3为c1-c10烷基、苄基、对甲氧基苯甲基、甲氧基甲基;r4为c1-c10烷基、苄基、对甲氧基苯甲基、甲氧基甲基;r5为c1-c10烷基、叔丁基二甲基硅基、三甲基硅基、三异丙基硅基、叔丁基二苯基硅基;r6为c1-c10烷基;x为卤素。
[0091]
本发明还提出了按所述方法合成制备得到的芳香聚酮类化合物abx-c、babx,其结构如下。
[0092][0093]
本发明还提出一种式12所示的多环不饱和缩酮化合物,其结构如下:
[0094]
[0095]
所述式12中,r2为c1-c10烷基、苄基、对甲氧基苯甲基、甲氧基甲基、叔丁基二甲基硅基、三甲基硅基、三异丙基硅基、叔丁基二苯基硅基。
[0096]
本发明还提出一种所述式12所示多环不饱和缩酮化合物的合成方法,包括如下合成路线中的步骤e或步骤d~e或步骤a/c~d~e或步骤b~c/a~d~e。即该合成方法的步骤包括如下:
[0097]
由式7化合物经氧化得到所述式12化合物;
[0098]
或,由式3化合物与式6化合物发生亲核加成反应及杂狄尔斯-阿尔德反应得到式7化合物;所述式7化合物再经氧化得到式12化合物;
[0099]
或,由式1化合物和式2化合物发生狄尔斯-阿尔德反应得到式3化合物;所述式3化合物与式6化合物发生亲核加成反应及杂狄尔斯-阿尔德反应得到式7化合物;所述式7化合物再经氧化得到式12化合物;
[0100]
或,由式1化合物和式2化合物发生狄尔斯-阿尔德反应得到式3化合物;由式4化合物经化合物5生成式6化合物;所述式3化合物与所述式6化合物发生亲核加成反应及杂狄尔斯
ꢀ-
阿尔德反应得到式7化合物;所述式7化合物再经氧化得到式12化合物。
[0101][0102]
其中,r1为叔丁基二甲基硅基、三甲基硅基、三异丙基硅基、叔丁基二苯基硅基;r2为c1-c10 烷基、苄基、对甲氧基苯甲基、甲氧基甲基、叔丁基二甲基硅基、三甲基硅基、三异丙基硅基、叔丁基二苯基硅基;x为卤素。
[0103]
其中,所述式1化合物和式2化合物混合均匀后100℃反应5小时,即可得到狄尔斯-阿尔德反应产物式3化合物;其中,其物质的量的比为式1化合物:式2化合物=1:1。
[0104]
式4化合物在丙酮作为溶剂,室温下加入碳酸钾,室温反应30分钟,再加入4-甲基苯磺酰氯,加热回流24h,冷却至室温,加入氯甲基甲醚,室温反应6小时后,得到式5化合物;其中,每毫摩尔mmol式4化合物对应适用溶剂的用量范围为1ml到10ml;其物质的量的比为式4化合物:碳酸钾:4-甲基苯磺酰氯:氯甲基甲醚=1:3:1.05:2。
[0105]
式5化合物在四氢呋喃作为溶剂,0℃下加入钠氢,0℃反应30分钟,再加入溴化苄,50℃反应4小时,得到式6化合物;其中,每毫摩尔(mmol)式5化合物对应适用溶剂的用量范围为1ml到10ml;其物质的量的比为式5化合物:钠氢:溴化苄=1:3:2。
[0106]
式6化合物在四氢呋喃作为溶剂,0℃下加入正丁基锂,0℃反应30分钟,再加入式3化合物,0℃反应2小时,加入2n盐酸,调ph为酸性,50℃反应10小时,得到式7化合物;其中,每毫摩尔mmol式6化合物对应适用溶剂的用量范围为1ml到10ml;其物质的量的比为式 6化合物:正丁基锂:式3化合物=1:2:2。
[0107]
式7化合物在二氯甲烷作为溶剂,0℃下加入戴斯马丁氧化剂,室温反应2小时,得到式 12化合物;其中,每毫摩尔mmol式7化合物对应适用溶剂的用量范围为1ml到10ml;其
物质的量的比为式7化合物:戴斯马丁氧化剂=1:2。
[0108]
本发明还提出了所述式12多环不饱和缩酮化合物在用于制备式15化合物、abe、abx-a
ꢀꢀ
abx-e、abx-c、babx中的应用。
[0109]
本发明的有益效果包括:本发明首次报道了abx-a、abx-c、abx-e、babx等天然产物的首次全合成,利用此方法可以制备大量的天然产物用于药物化学研究。本发明的合成路线高效,合成的天然产物结构较为复杂,可改造的位点较多,本发明合成路线设计合理,原料便宜易得,且易于大量制备,关键反应中间体易于修饰,利用此方法可以实现多种芳香聚酮类天然产物及其类似物的合成,利于制备天然产物衍生物,为后续药物化学研究奠定基础。
具体实施方式
[0110]
结合以下具体实施例,对本发明作进一步的详细、完整的说明。实施本发明的过程、条件、 实验方法等,除以下专门提及的内容之外,均为本领域的普遍知识和公知常识,本发明没有特 别限制内容。
[0111]
化合物3的合成:
[0112][0113]
在干燥的100ml的圆底瓶中加入式1化合物(20g,108.7mmol),加入式2(17g,108.7mmol) 100℃反应5小时,冷却至室温,使用柱色谱(5%乙酸乙酯/石油醚)分离得式3化合物(22g, 60%)。r
f
=0.2(5%乙酸乙酯/石油醚)。
[0114]
经测试:1h nmr(500mhz,chloroform-d)δ9.83(d,j=3.2hz,1h,major),9.64 (d,j=4.2hz,1h,minor),5.74
–
5.71(m,2h,major),5.70
–
5.66(m,2h,minor), 4.63
–
4.59(m,1h,minor),4.53
–
4.51(m,1h,major),3.96
–
3.85(m,4h), 2.57
–
2.18(m,3h),1.93
–
1.57(m,3h),1.32(s,3h,major),1.28(s,3h,minor), 0.89
–
0.84(m,9h),0.08
–
0.02(m,6h).
[0115]
13
c nmr(125mhz,chloroform-d)δ207.1,204.6,129.4,128.9,128.8,127.5,109.9, 109.7,67.7,65.8,64.6,64.5,64.4,64.1,60.6,55.8,42.5,40.0,31.8,30.1,29.1, 27.2,25.72(3c),25.68(3c),24.2,24.0,18.0,17.9,-4.2,-4.3,-4.8,-5.0ppm.
[0116]
hrms
–
esi(m/z):[m+na]
+
calcd for c
18
h
32
o4nasi,363.1968;found,363.1942.
[0117]
化合物5的合成
[0118][0119]
在干燥的1000ml的圆底瓶中加入式4化合物(30g,148.5mmol)和丙酮(300ml),加入无水碳酸钾(61.6g,445.5mmol)室温反应30分钟,再加入4-甲基苯磺酰氯(29.7g,156mmol), 反应体系加热回流24小时后,冷却至室温,加入氯甲基甲醚,室温反应6小时,滤
除碳酸钾固体,用乙酸乙酯洗涤滤饼,收集滤液,加入水,旋除丙酮,乙酸乙酯萃取三次,合并有机相后,饱和氯化钠洗涤一次,无水硫酸钠干燥,过滤后浓缩,使用柱色谱(5%乙酸乙酯/石油醚) 分离得式5化合物(41.6g,70%)。rf=0.20(5%乙酸乙酯/石油醚)。
[0120]
经测试:1h nmr(400mhz,chloroform-d)δ7.73(d,j=8.3hz,2h),7.32(d,j=8.1 hz,2h),6.67(d,j=2.4hz,1h),6.55(d,j=2.5hz,1h),5.06(s,2h),3.41 (s,3h),2.45(s,3h),2.35(s,3h)ppm.
[0121]
13
c nmr(100mhz,chloroform-d)δ154.2,148.6,145.4,140.5,132.3,129.7(2c), 128.6(2c),117.7,113.6,107.7,95.2,56.3,23.4,21.7ppm.
[0122]
hrms
–
esi(m/z):[m+na]
+
calcd for c
16
h
17
o5brnas,422.9878;found,422.9851.
[0123]
化合物6的合成
[0124][0125]
在干燥的1000ml的圆底瓶中加入式5化合物(40g,100mmol)和四氢呋喃(300ml), 反应体系冷却至0℃,加入钠氢(12g,300mmol),0℃反应30分钟,加入溴化苄(23.7ml, 200mmol),50℃反应4小时,反应体系冷却至0℃,缓慢加入水淬灭反应,旋除四氢呋喃,水相用乙酸乙酯萃取三次,合并有机相后,饱和氯化钠洗涤一次,无水硫酸钠干燥,过滤后浓缩,使用柱色谱(1%乙酸乙酯/石油醚)分离得式6化合物(17.1g,51%)。r
f
=0.40(2%乙酸乙酯/石油醚).
[0126]
经测试:1h nmr(400mhz,chloroform-d)δ7.44
–
7.31(m,5h),6.70(d,j=2.7 hz,1h),6.59(d,j=2.6hz,1h),5.21(s,2h),5.02(s,2h),3.51(s,3h),2.39 (s,3h).
[0127]
13
c nmr(100mhz,chloroform-d)δ158.3,154.5,139.9,136.7,128.6(2c),128.0, 127.5(2c),110.3,106.6,101.5,95.3,70.2,56.3,23.6ppm.
[0128]
hrms
–
esi(m/z):[m+h]
+
calcd for c
16
h
18
o3br,337.0439;found,337.0419.
[0129]
化合物7的合成
[0130][0131]
在干燥的1000ml的圆底瓶中加入式6化合物(15g,44.6mmol)和四氢呋喃(300ml), 反应体系冷却至0℃,加入正丁基锂(36ml,89.2mmol,2.5m/n-hexane),0℃反应30分钟,加入式3化合物(30.3g,89.2mmol),0℃反应2小时,缓慢加入2n hcl,调ph为酸性,50℃反应10小时,反应体系冷却至室温,旋除四氢呋喃,水相用乙酸乙酯萃取三次,合并有机相后,饱和氯化钠洗涤一次,无水硫酸钠干燥,过滤后浓缩,使用柱色谱(50%乙酸乙酯/石油醚) 分离得式7化合物(8.9g,53%)。r
f
=0.38(30%乙酸乙酯/石油醚).
[0132]
经测试:1h nmr(400mhz,chloroform-d)δ7.42
–
7.30(m,5h),6.46(d,j=2.0 hz,
1h),6.33(d,j=2.2hz,1h),5.92(s,2h),5.19(s,1h),5.00(s,2h),4.33 (s,1h),2.19(s,3h),2.17
–
2.09(m,2h),1.73
–
1.65(m,2h),1.63(s,4h), 1.60
–
1.51(m,2h).
[0133]
13
c nmr(125mhz,chloroform-d)δ158.3,150.2,136.9,134.9,131.5,128.9,128.5 (2c),127.9,127.4(2c),117.8,110.3,100.4,98.7,69.9,68.5,66.8,49.8,38.9, 32.0,27.3,23.0,19.1ppm.hrms
–
esi(m/z):[m+h]
+
calcd for c
24
h
27
o4,379.1909;found, 379.1902.
[0134]
化合物9的合成
[0135][0136]
在干燥的1000ml的圆底瓶中加入式8化合物(40g,192mmol)和二氯甲烷(500ml), 反应体系冷却至0℃,加入三溴化硼(93ml,961mmol),室温反应12小时,反应体系冷却至0℃,将反应液缓慢倒入冰水中,水相用二氯甲烷萃取三次,合并有机相后,饱和氯化钠洗涤一次,无水硫酸钠干燥,过滤后浓缩,使用柱色谱(8%乙酸乙酯/石油醚)分离得式9化合物(30g,87%)。 r
f
=0.40(10%乙酸乙酯/石油醚).
[0137]
经测试:1h nmr(400mhz,chloroform-d)δ12.63(s,1h),10.19(s,1h),6.35(d, j=2.0hz,1h),6.21(d,j=2.0hz,1h),3.55(dt,j=13.7,6.8hz,1h),1.32(s, 3h),1.30(s,3h).
[0138]
13
c nmr(125mhz,chloroform-d)δ192.5,166.6,163.5,155.9,112.0,105.4,100.9, 27.6,24.0(2c)ppm.
[0139]
hrms
–
esi(m/z):[m+h]
+
calcd for c
10
h
13
o3,181.0864;found,181.0857.
[0140]
化合物10的合成
[0141][0142]
在干燥的1000ml的圆底瓶中加入式9化合物(30g,166.7mmol)和乙腈(500ml), 反应体系冷却至0℃,加入n,n-二异丙基乙胺(27.5ml,166.7mmol),再加入氯甲基加醚(12.6ml, 166.7mmol),室温反应12小时,用水淬灭反应,旋除乙腈,水相用乙酸乙酯萃取三次,合并有机相后,饱和氯化钠洗涤一次,无水硫酸钠干燥,过滤后浓缩,使用柱色谱(3%乙酸乙酯/ 石油醚)分离得式10化合物(26.1g,70%)。r
f
=0.50(5%乙酸乙酯/石油醚).
[0143]
经测试:1h nmr(300mhz,chloroform-d)δ12.58(s,1h),10.18(s,1h),6.46(d, j=2.4hz,1h),6.41(d,j=2.4hz,1h),5.18(s,2h),3.60
–
3.47(m,1h),3.45 (s,3h),1.30(s,3h),1.27(s,3h).
[0144]
13
c nmr(125mhz,chloroform-d)δ192.6,166.3,164.4,154.8,112.2,106.2,100.6, 93.9,56.3,27.5,23.9(2c)ppm.
[0145]
hrms
–
esi(m/z):[m+h]
+
calcd for c
12
h
17
o4,225.1127;found,225.1119.
[0146]
化合物11的合成
[0147][0148]
在干燥的1000ml的圆底瓶中加入式10化合物(20g,89.2mmol)和四氢呋喃(300ml), 反应体系冷却至0℃,加入碳酸钾(18.5g,133.8mmol)、碘甲烷(8.3ml,133.8mmol),室温反应4小时,滤除碳酸钾固体,滤饼用四氢呋喃洗涤一次,收集滤液,向滤液加入6n盐酸,调ph至酸性,50℃反应3小时,反应体系冷却至室温,旋除四氢呋喃,水相用乙酸乙酯萃取三次,合并有机相,用饱和氯化钠水洗,无水硫酸钠干燥,旋干浓缩后将粗产物用于n,n-二甲基甲酰胺(300ml),加入咪唑(7.9g,116mmol)、4-二甲氨基吡啶(1.09g,8.9mmol)、三异丙基氯硅烷(24.8ml,116mmol),室温反应12小时,加入饱和氯化铵淬灭反应,水相用乙酸乙酯萃取三次,合并有机相,用饱和氯化钠水洗,无水硫酸钠干燥,旋干浓缩,使用柱色谱 (2%乙酸乙酯/石油醚)分离得式11化合物(28.1g,90%)。r
f
=0.40(2%乙酸乙酯/石油醚).
[0149]
经测试:1h nmr(500mhz,chloroform-d)δ10.46(s,1h),6.48(d,j=2.1hz,1h), 6.28(d,j=2.2hz,1h),4.10
–
4.01(m,1h),3.80(s,3h),1.30
–
1.22(m,3h), 1.15(s,3h),1.14(s,3h),1.09(d,j=7.6hz,18h).
[0150]
13
c nmr(125mhz,chloroform-d)δ190.6,165.1,162.0,154.9,116.3,109.6,100.4, 55.5,28.1,23.4(2c),17.8(6c),12.6(3c)ppm.
[0151]
hrms
–
esi(m/z):[m+h]
+
calcd for c
20
h
35
o3si,351.2355;found,351.2342.
[0152]
化合物12的合成
[0153][0154]
在干燥的500ml的圆底瓶中加入式7化合物(20g,52.9mmol)和二氯甲烷(300ml), 反应体系冷却至0℃,加入戴斯马丁氧化剂(44.9g,105.8mmol),室温反应2小时,向反应体系加入水淬灭反应,水相用二氯甲烷萃取三次,合并有机相后,饱和氯化钠洗涤一次,无水硫酸钠干燥,过滤后浓缩,使用柱色谱(15%乙酸乙酯/石油醚)分离得式12化合物(13.9g,70%)。 r
f
=0.50(20%乙酸乙酯/石油醚).
[0155]
经测试:1h nmr(400mhz,chloroform-d)δ7.44
–
7.29(m,5h),6.95(ddd,j=9.9, 5.8,1.9hz,1h),6.44(d,j=2.1hz,1h),6.33(d,j=2.4hz,1h),6.12(dd, j=10.0,2.2hz,1h),5.81(d,j=1.9hz,1h),5.00(s,2h),2.53
–
2.43(m, 1h),2.19
–
2.06(m,6h),2.01
–
1.89(m,1h),1.75(t,j=13.4hz,1h),1.60 (s,3h).
[0156]
13
c nmr(100mhz,chloroform-d)δ197.1,158.6,150.4,148.4,137.0,135.5,129.9, 128.5(2c),127.9,127.4(2c),117.0,110.4,100.5,97.9,69.9,63.4,56.4,39.5, 32.8,30.6,27.3,18.4ppm.
[0157]
hrms
–
ei(m/z):[m]
+
calcd for c
24
h
24
o4,376.1675;found,376.1672.
[0158]
化合物13的合成
[0159][0160]
在干燥的100ml的石英光反应光中加入式12化合物(500mg,1.33mmol)和无水二氧六环(66ml),加入式11化合物(1.4g,3.98mmol),钛酸四异丙酯(3.5ml,11.97mmol),用 300纳米波长紫外光照射4小时后,将反应液倒入饱和碳酸氢钠溶液中,室温搅拌2小时,水相用乙酸乙酯萃取三次,合并有机相后,饱和氯化钠洗涤一次,无水硫酸钠干燥,过滤后浓缩,使用柱色谱(10%乙酸乙酯/石油醚)分离得式13化合物(860mg,84%).r
f
=0.53(20%乙酸乙酯/石油醚).
[0161]
经测试:1h nmr(500mhz,chloroform-d)δ7.45
–
7.29(m,5h),6.48(d,j=2.2 hz,1h),6.45(d,j=2.5hz,1h),6.33(d,j=2.0hz,2h),5.83(s,1h),5.35(dd, j=6.5,2.5hz,1h),5.00(s,2h),3.82(s,3h),3.10
–
3.08(m,2h),2.46
–
2.39 (m,2h),2.14(s,3h),2.13
–
2.08(m,2h),1.77
–
1.66(m,3h),1.52(s,3h),1.31 (s,3h),1.29
–
1.23(m,3h),1.15(s,3h),1.12(d,j=7.3hz,18h).
[0162]
13
c nmr(125mhz,chloroform-d)δ210.0,158.4,158.4,156.9,150.6,144.7,137.0, 135.1,128.5(2c),127.9,127.4(2c),117.7,117.4,110.1,109.0,100.9,100.4,98.5, 69.8,63.7,61.8,55.3,54.0,47.5,41.6,38.9,36.9,34.0,27.2,27.1,26.3,23.9, 18.6,17.9(6c),12.7(3c)ppm.
[0163]
hrms
–
esi(m/z):[m+na]
+
calcd for c
44
h
58
o7nasi,749.3850;found,749.3833.
[0164]
化合物14的合成
[0165][0166]
在干燥的100ml的圆底瓶中加入式13化合物(4.1g,5.64mmol)和无水二氯甲烷(50ml), 加入戴斯马丁氧化剂(7.2g,16.92mmol)室温反应2小时,加入饱和碳酸氢钠溶液,水相用二氯甲烷萃取三次,合并有机相后,饱和氯化钠洗涤一次,无水硫酸钠干燥,过滤后浓缩,使用柱色谱(10%乙酸乙酯/石油醚)分离得式14化合物(2.86g,70%).r
f
=0.5(20%乙酸乙酯/石油醚).
[0167]
经测试:1h nmr(400mhz,chloroform-d)δ17.57(s,1h,major),17.27(s,1h,minor), 7.43
–
7.30(m,5h),6.55(d,j=2.0hz,1h,major),6.53(d,j=2.0hz,1h,minor), 6.46(dd,j=4.6,2.4hz,1h),6.38(dd,j=3.9,2.1hz,1h),6.33(d,j=2.3hz, 1h),5.91(d,j=1.7hz,1h,major),5.73(d,j=2.1hz,1h,minor),5.00(s,2h), 3.89(s,3h),2.75(t,j=9.6hz,1h),2.33(s,3h,minor),2.20(s,3h,major),2.17
ꢀ–
2.09(m,3h,major),
2.04
–
1.90(m,3h,minor),1.72
–
1.67(m,1h),1.60(s, 3h),1.49
–
1.41(m,1h),1.34(s,3h,major),1.28(s,3h,minor),1.32
–
1.21(m, 4h),1.12(d,j=7.3hz,18h),1.02(s,3h,major),0.91(s,3h,minor).
[0168]
13
c nmr(125mhz,chloroform-d)δ192.4,189.6,180.9,179.2,162.1,161.7,161.3, 161.1,158.5,155.7,154.1,150.5,150.4,137.0,137.0,136.1,135.7,128.6(2c),127.9, 127.5(2c),117.7,117.3,112.2,111.6,110.3,110.3,108.8,107.9,104.6,103.7,102.4, 102.4,100.4,98.1,98.1,69.9,69.9,65.2,63.7,56.2,56.2,54.0,53.4,41.8,39.9, 39.6,38.2,37.8,30.0,29.7,29.1,28.7,28.1,27.3,25.2,24.1,22.8,22.2,19.0, 18.5,17.9(6c),12.7(3c)ppm.
[0169]
hrms
–
esi(m/z):[m+h]
+
calcd for c
44
h
57
o7si,725.3873;found,725.3853.
[0170]
化合物15的合成
[0171][0172]
在干燥的100ml的圆底瓶中加入式14化合物(2.33g,3.2mmol)和无水二氧六环(50ml), 加入2,3-二氯-5,6-二氰基-1,4-苯醌(2.2g,9.6mmol),80℃反应2小时,反应体系冷却至室温,用饱和碳酸氢钠洗涤有机相至水相无色,有机相用饱和氯化钠洗涤一次,无水硫酸钠干燥,过滤后浓缩,使用柱色谱(10%乙酸乙酯/石油醚)分离得式15化合物(1.78g,79%).r
f
= 0.30(10%乙酸乙酯/石油醚).
[0173]
经测试:1h nmr(500mhz,chloroform-d)δ14.44(s,1h),7.40
–
7.28(m,5h),6.78 (s,1h),6.71(d,j=2.1hz,1h),6.45(d,j=2.1hz,1h),6.41(d,j=2.5hz,1h), 6.35(s,1h),6.33(d,j=2.6hz,1h),4.97(s,2h),3.97(s,3h),3.23(d,j=17.8 hz,1h),3.13(d,j=17.8hz,1h).2.48(s,3h),1.73(s,3h),1.62(s,3h),1.57 (s,3h),1.36
–
1.26(m,3h),1.14(d,j=7.5hz,18h).
[0174]
13
c nmr(125mhz,chloroform-d)δ188.2,163.3,162.0,158.5,158.4,155.7,152.2, 148.8,140.1,137.0(2c),128.5(2c),127.8,127.4(2c),123.3,115.7,115.3,113.5, 113.0,110.0,110.0,102.3,100.1,98.0,69.8,65.4,56.2,40.5,38.6,34.4,33.4, 27.8,19.5,17.9(6c),12.7(3c)ppm.
[0175]
hrms
–
esi(m/z):[m+h]
+
calcd for c
44
h
53
o7si,721.3560;found,721.3538.
[0176]
化合物16的合成
[0177][0178]
在干燥的50ml的圆底瓶中加入式15化合物(400mg,0.57mmol)和四氢呋喃(20ml),加入碘化镁(793mg,2.85mmol),70℃反应4小时后,反应体系冷却至室温,加入1n盐酸,旋除
四氢呋喃,水相用乙酸乙酯萃取三次,合并有机相,有机相用饱和氯化钠洗涤一次,无水硫酸钠干燥,过滤后浓缩,使用柱色谱(10%乙酸乙酯/石油醚)分离得式16化合物(378mg,94%). r
f
=0.61(20%乙酸乙酯/石油醚).
[0179]
经测试:1h nmr(400mhz,chloroform-d)δ13.40(s,1h),12.82(s,1h),7.42
–ꢀ
7.28(m,5h),6.85(s,1h),6.59(d,j=2.3hz,1h),6.44(d,j=2.5hz,1h),6.38 (d,j=2.2hz,1h),6.35(d,j=2.6hz,1h),6.34(s,1h),4.98(s,2h),3.24(d, j=18.0hz,1h),3.16(d,j=17.9hz,1h),2.51(s,3h),1.73(s,3h),1.64(s,3h), 1.56(s,3h),1.36
–
1.26(m,3h),1.13(d,j=7.4hz,18h).
[0180]
13
c nmr(125mhz,chloroform-d)δ191.0,165.4,163.7,158.6,158.3,154.0,152.3, 150.2,141.5,137.0,136.8,128.5(2c),127.9,127.4(2c),123.5,117.0,115.2,111.8, 110.4,110.0,108.6,105.9,100.2,98.1,69.8,65.4,40.5,38.5,34.0,33.8,27.7, 19.5,17.8(6c),12.7(3c)ppm.
[0181]
hrms
–
esi(m/z):[m+h]
+
calcd for c
43
h
51
o7si,707.3404;found,707.3387.
[0182]
化合物17的合成
[0183][0184]
在干燥的50ml的圆底瓶中加入式16化合物(181mg,0.256mmol)和无水四氢呋喃(20ml), 加入tbaf(0.51ml,0.512mmol,1m/thf),室温反应2小时后,加入水,旋除四氢呋喃,使用柱色谱(20%乙酸乙酯/石油醚)分离得式17化合物(141mg,100%).r
f
=0.30(20%乙酸乙酯/石油醚).
[0185]
经测试:1h nmr(400mhz,chloroform-d)δ13.37(s,1h),13.05(s,1h),7.42
–ꢀ
7.29(m,5h),6.85(s,1h),6.54(d,j=2.3hz,1h),6.48(d,j=2.5hz,1h),6.40 (d,j=2.5hz,1h),6.38(s,1h),6.29(d,j=2.2hz,1h),5.01(s,2h),3.26(d, j=17.9hz,1h),3.18(d,j=18.0hz,1h),2.54(s,3h),1.77(s,3h),1.60(s,3h), 1.49(s,3h).
[0186]
13
c nmr(100mhz,chloroform-d)δ190.9,165.4,163.2,158.6,158.2,154.8,152.3, 150.3,141.5,136.8,136.8,128.5(2c),127.9,127.4(2c),123.4,117.0,115.4,111.6, 110.2,108.2,106.0,101.6,100.5,98.2,70.0,65.5,40.5,38.6,33.8,33.6,27.6, 19.4ppm.
[0187]
hrms
–
esi(m/z):[m+h]
+
calcd for c
34
h
31
o7,551.2070;found,551.2058.
[0188]
天然产物abx-a的合成
[0189][0190]
在干燥的50ml的圆底瓶中加入式17化合物(50mg,0.091mmol)和甲醇(8ml),加入
氯化钯(8.1mg,0.045mmol),室温反应10小时后,旋除甲醇,使用柱色谱(30%乙酸乙酯/石油醚)分离得式天然产物abx-a(41mg,98%).r
f
=0.52(40%乙酸乙酯/石油醚).
[0191]
经测试:1h nmr(400mhz,methanol-d4)δ6.98(s,1h),6.60(d,j=2.2hz,1h),6.22 (d,j=2.2hz,1h),6.20(d,j=2.5hz,1h),6.18(s,1h),6.10(d,j=2.4hz,1h), 3.20(d,j=18.1hz,1h),3.04(d,j=18.1hz,1h),2.39(s,3h),1.61(s,3h),1.60 (s,3h),1.52(s,3h).
[0192]
13
c nmr(100mhz,methanol-d4)δ192.0,166.9,166.8,158.7,157.9,156.0,153.5, 151.7,142.9,137.3,124.7,118.3,115.0,112.5,111.0,108.2,107.3,102.0,101.8, 98.9,66.4,41.0,39.6,34.2,33.8,27.7,19.4ppm.
[0193]
hrms
–
esi(m/z):[m+h]
+
calcd for c
27
h
25
o7,461.1600;found,461.1588.
[0194]
化合物18的合成
[0195][0196]
在干燥的50ml的圆底瓶中加入式15化合物(200mg,0.28mmol)和甲醇(15ml),加入氯化钯(25mg,0.045mmol),室温反应6小时后,旋除甲醇,使用柱色谱(10%乙酸乙酯/ 石油醚)分离得式18化合物(172mg,97%).r
f
=0.42(20%乙酸乙酯/石油醚).
[0197]
经测试:1h nmr(500mhz,chloroform-d)δ14.35(s,1h),6.80(s,1h),6.72(d, j=2.3hz,1h),6.45(d,j=2.3hz,1h),6.32(s,1h),6.24(s,1h),6.23(s,1h), 3.95(s,3h),3.22(d,j=17.9hz,1h),3.13(d,j=17.9hz,1h).2.37(s,3h),1.71 (s,3h),1.63(s,3h),1.58(s,3h),1.34
–
1.29(m,3h),1.15(d,j=7.6hz,18h).
[0198]
13
c nmr(125mhz,chloroform-d)δ188.3,163.3,162.1,158.4,155.7,155.6,152.2, 148.8,140.2,137.0,123.4,115.8,114.6,113.4,112.9,110.2,110.0,102.2,101.0, 97.8,65.4,56.1,40.4,38.6,34.2,33.5,27.7,19.3,17.8(6c),12.7(3c)ppm.
[0199]
hrms
–
esi(m/z):[m+h]
+
calcd for c
37
h
47
o7si,631.3091;found,631.3070.
[0200]
化合物19的合成
[0201][0202]
在干燥的50ml的圆底瓶中加入式18化合物(100mg,0.16mmol)和氯仿(20ml),加入1,8-双二甲氨基萘(274mg,1.28mmol),再加入三甲基氧鎓地氟硼酸(237mg,1.6mmol), 室温反应4小时后,加入水,水相用二氯甲烷萃取三次,合并有机相,用饱和氯化钠溶液洗涤一次,无水硫酸钠干燥,过滤,旋除二氯甲烷,浓缩后,使用柱色谱(2%乙酸乙酯/石油醚) 分离得式19化合物(82.5mg,80%).r
f
=0.52(5%乙酸乙酯/石油醚).
[0203]
经测试:1h nmr(400mhz,chloroform-d)δ14.43(s,1h),6.78(s,1h),6.71(d, j=
1h),6.53(d,j=2.4hz,1h),6.43(s,1h),6.36(d,j=2.6hz,1h),6.35(s,1h), 6.30(d,j=2.6hz,1h),6.29(d,j=2.3hz,1h),3.74(s,3h),3.25(d,j=18.0 hz,1h),3.16(d,j=18.0hz,1h),2.51(s,3h),1.75(s,3h),1.59(s,3h),1.49 (s,3h).
[0216]
13
c nmr(100mhz,chloroform-d)δ190.9,165.5,163.1,159.3,158.3,154.9,152.4, 150.3,141.6,136.8,123.5,117.0,115.2,111.7,109.4,108.3,106.0,101.7,99.6, 98.2,65.5,55.2,40.5,38.6,33.9,33.7,27.6,19.4ppm.
[0217]
hrms
–
esi(m/z):[m+h]
+
calcd for c
28
h
27
o7,475.1757;found,475.1746.
[0218]
天然产物abx-c、babx的合成
[0219][0220]
在干燥的10ml的圆底瓶中加入abx-e(10mg,0.021mmol)和四氢呋喃(2.0ml),加入正丁基锂(25μl,0.063mmol),0℃反应1小时后,再加入次氯酸叔丁酯(4.7μl,0.042 mmol),0℃反应2小时,加入水淬灭反应,水相用乙酸乙酯萃取三次,合并有机相,有机相用饱和氯化钠洗涤一次,无水硫酸钠干燥,过滤后浓缩,使用柱色谱(30%乙酸乙酯/石油醚) 分离得abx-c化合物(3.3mg,31%).r
f
=0.36(30%乙酸乙酯/石油醚)。使用柱色谱(30%乙酸乙酯/石油醚)分离得babx(1.9mg,17%).r
f
=0.19(30%乙酸乙酯/石油醚)。
[0221]
经测试:
[0222]
abx-c:1h nmr(500mhz,chloroform-d)δ13.62(s,1h),13.14(s,1h),6.86(s,1h), 6.80(s,1h),6.35(d,j=2.6hz,1h),6.32(s,1h),6.27(d,j=2.5hz,1h),3.72 (s,3h),3.24(d,j=18.0hz,1h),3.16(d,j=17.9hz,1h),2.51(s,3h),1.73(s, 3h),1.64(s,3h),1.56(s,3h).
[0223]
13
c nmr(125mhz,chloroform-d)δ190.9,160.0,159.4,158.4,158.0,152.4,152.3, 150.0,142.3,136.6,123.7,117.3,114.9,111.4,109.2,108.7,105.6,105.5,99.4, 98.1,65.3,55.1,40.6,38.6,34.0,33.7,27.6,19.4ppm.
[0224]
hrms
–
esi(m/z):[m+h]
+
calcd for c
28
h
26
o7cl,509.1367;found,509.1356.
[0225]
babx:1h nmr(500mhz,chloroform-d)δ14.29(s,1h),13.08(s,1h),6.83(s,1h), 6.34(d,j=2.6hz,1h),6.32(s,1h),6.27(d,j=2.6hz,1h),3.72(s,3h),3.25 (d,j=17.9hz,1h),3.17(d,j=17.8hz,1h),2.50(s,3h),1.93(s,3h),1.85(s, 3h),1.73(s,3h).
[0226]
13
c nmr(125mhz,chloroform-d)δ190.7,159.9,159.4,157.6,152.5,152.3,146.4, 142.7,136.6,123.2,117.7,114.8,110.2,109.2(2c),107.9,99.4,98.0,65.3,55.1, 40.6,39.5,29.0,28.7,27.6,19.4ppm.
[0227]
hrms
–
esi(m/z):[m+h]
+
calcd for c
28
h
25
o7cl2,543.0977;found,543.0966。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1