用于确定在无细胞核酸中的肿瘤分数的系统及方法与流程
相关申请的交互引用
本申请主张2018年4月16日提交的标题为“使用在无细胞核酸中的变体的频率来分类受试者的系统及方法”的美国临时专利申请第62/658,479号的优先权权益,其内容通过引用并入本文中。
本说明书描述的是,确定在一受试者的无细胞核酸中的肿瘤分数,从而通知用于癌症分类的多种改良的分类器,其包括在较低的肿瘤分数下侦测癌症。
背景技术:
人类的基因组含有约三十亿个碱基对。多种大规模的定序技术,例如次世代定序(ngs),提供了以低于每百万碱基一美元的成本来实现定序的机会,并且实际上已经实现低于每百万碱基十美分的成本。这类的定序技术已经能够在多种异常的体细胞组织中,例如肿瘤样本,辨识出单核苷酸变体(snvs)、小的插入及缺失(indels)及大规模的拷贝数变体(cnvs)。
这类对多种异常的体细胞组织中的多个体细胞变体进行的分析为理解分子的扰乱提供一基础,所述分子的扰乱构成个体疾病表型或治疗反应的多个巨大差异的基础。然而,这些变体的辨识及这些变体的频率可能因受试者而异,此外,在任何特定的受试者中可能随着病况的进展而改变。并且,与例如癌症的多种疾病相关的多个变体由于一些变体的稀有性,需要对一生物样本中,例如组织活检或从一受试者抽取的血液,的核苷酸进行深度定序。例如,侦测源自于从一血液样本而来的多个肿瘤细胞的dna很困难,这是因为循环肿瘤dna(ctdna)相对于从所述血液萃取出的cfdna中的其他分子存在的水平很低。然而,对一受试者中的ctdna水平的了解,无论其水平多么低,皆有可能提供治疗决策,并改善预后及诊断。
在上述背景下,本领域需要多个用于确定一受试者中的ctdna的有效率且有成本效益的健全技术,其甚至可侦测非常低水平的ctdna。
技术实现要素:
本发明提供了利用侦测ctdna来解决上述多个经确认的问题的多个技术方案(例如,计算机系统、方法及非暂时性计算机可读存储介质)。
下面提供本发明的概论,以便提供对本发明的一些方面的一基本理解。此概论不是本发明的广泛概述。其并非旨在辨识本发明的多个主要/关键元素或描写本发明的范围。其唯一的目的是以一简化的形式呈现本发明的一些概念,以作为稍后呈现出的更详细描述的序言。
在所附权利要求的范围内的系统、方法及装置的各种实施例分别具有多个方面,其中没有单一的方面完全负责本文所描述的多个期望的特性。在不限制所附权利要求的范围的情况下,本文描述了一些突出的特征。在考虑到此讨论后,特别是在阅读完标题为“详细描述”的部分后,将理解如何使用各种实施例的多个特征。
本公开的一方面提供一种用于确定在一受试者的一液体生物样本的无细胞核酸中的肿瘤分数的方法。所述方法包括:在一计算机系统中具有一个或多个处理器及一记忆体,所述记忆体存储通过所述一个或多个处理器执行的一个或多个程序,在所述计算机系统中进行以下步骤。获得从所述受试者的所述液体生物样本而来的电子形式的多个第一序列读值,其中所述液体生物样本包括多个无细胞核酸分子;使用所述多个第一序列读值来辨识对一第一变体集合中的每个变体的支持,从而确定在所述第一变体集合中的每个变体的一观察到的频率。对于在所述第一变体集合中的每个个别的变体,获得在一第一参考集合中对于所述个别的变体的一对应的参考频率。在所述第一参考集合中的每个对应的参考频率是针对从所述受试者获得的一第一异常实体(solid)组织样本中的一个别的变体。对照在所述第一异常实体组织中的所述第一参考集合中的所述个别的变体的所述观察到的频率,来评估在所述第一变体集合中的每个个别的变体的所述观察到的频率,从而确定在所述受试者的所述液体生物样本的无细胞核酸中的一第一肿瘤分数。
在一些实施例中,所述第一变体集合中的一变体为与一预定的基因组位置相关的一单核苷酸变体、与一预定的基因组位置相关的一插入突变、与一预定的基因组位置相关的一缺失突变、一体细胞拷贝数的改变、与一预定的基因组位点相关的一核酸重组,或与一预定的基因组位置相关的任何异常的表观遗传修饰(例如,异常的甲基化模式)。
在一些实施例中,当所述多个第一序列读值中的一个别的序列读值包括所述第一变体集合中的一第一变体的全部或一部分时,将所述个别的序列读值视为支持所述第一变体;当所述多个第一序列读值中的一个别的序列读值不包括所述第一变体集合中的所述第一变体时,将所述个别的序列读值视为不支持所述第一变体;及支持所述第一变体的所述多个第一序列读值中的多个序列读值的一数量对上不支持所述第一变体的所述多个第一序列读值中的多个序列读值的一数量用以确定所述第一变体的所述观察到的频率,由所述第一变体的所述观察到的频率估计在所述液体生物样本中的所述第一变体的一变体频率。
在一些实施例中,所述受试者为人类。在一些实施例中,所述受试者患有来自一单一原发部位的癌症。在一些实施例中,所述受试者患有源自于两个或更多个不同器官的癌症。
在一些实施例中,所述受试者患有乳腺癌、肺癌、前列腺癌、结肠直肠癌、肾癌、子宫癌、胰腺癌、食道癌、淋巴瘤、头颈癌、卵巢癌、肝胆癌、黑色素瘤、子宫颈癌、多发性骨髓瘤、白血病、甲状腺癌、膀胱癌、胃癌或其组合。
在一些实施例中,所述受试者患有乳腺癌、肺癌、前列腺癌、结肠直肠癌、肾癌、子宫癌、胰腺癌、食道癌、头颈癌、卵巢癌、肝胆癌、子宫颈癌、甲状腺癌、膀胱癌或胃癌的一预定阶段。
在一些实施例中,所述第一异常的实体组织样本为一肿瘤样本。
在一些实施例中,所述第一变体集合是由对于位在所述受试者的基因组中的一单一位点处的一单一遗传变异的一单一变体组成。在多个可替代的实施例中,所述第一变体集合是由对于位在所述受试者的基因组中的一第一位点处的一第一遗传变异的一第一变体及对于位在所述受试者的基因组中的一第二位点处的一第二遗传变异的一第二变体组成。在另外的多个可替代的实施例中,所述第一变体集合是由对于位在所述受试者的基因组中的一第一位点处的一第一遗传变异的一第一变体、对于位在所述受试者的所述基因组中的一第二位点处的一第二遗传变异的一第二变体,及对于位在所述受试者的所述基因组中的一第三位点处的一第三遗传变异的一第三变体组成。
在一些实施例中,所述第一变体集合是由介于2至20个之间的变体组成,或由介于2至200个之间的变体组成,或包括1000个或更多个变体,或包括5000个或更多个变体,其中所述第一变体集合中的每个变体为所述受试者的基因组中的一不同的遗传变异。
在一些实施例中,所述使用所述多个序列读值来辨识对一变体集合中的每个变体的支持的步骤包括:将所述多个第一序列读值中的一序列读值与一参考基因组中的一区域比对,或者与多个变体的一查找表比对,以确定所述序列读值是否包括一第一变体的全部或一部分。
在一些实施例中,所述使用所述多个序列读值来辨识对一变体集合中的每个变体的支持的步骤包括:将所述多个第一序列读值中的一序列读值与一查找表中的每个条目(entry)比对,其中所述查找表中的每个条目代表一基因组的一不同部分。
在一些实施例中,所述受试者患有第二期、第三期或第四期乳腺癌,并且所述对照在所述第一异常实体组织中的所述第一参考集合中的所述个别的变体的所述观察到的频率,来评估在所述第一变体集合中的每个个别的变体的所述观察到的频率的步骤,确定了所述无细胞核酸的所述第一肿瘤分数小于1x10-3。
在一些实施例中,所述方法进一步包括步骤:使用所述多个第一序列读值,以辨识对于在一第二变体集合中的每个变体的支持,从而确定在所述第二变体集合中的每个变体的一观察到的频率;对于在所述第二变体集合中的每个个别的变体,获得在一第二参考集合中对于所述个别的变体的一对应的参考频率,其中在所述第二参考集合中的每个对应的参考频率是针对从所述受试者获得的一第二异常的实体组织样本中的一个别的变体;及对照在所述第二参考集合中的所述个别的变体的所述观察到的频率,来评估在所述第二变体集合中的每个个别的变体的所述观察到的频率,从而确定在所述受试者的所述液体生物样本的无细胞核酸中的一第二肿瘤分数。在一些这样的实施例中,当所述多个第一序列读值中的一个别的序列读值包括所述第二变体集合中的一变体的全部或一部分时,将所述个别的序列读值视为支持所述变体;及当所述多个第一序列读值中的一个别的序列读值不包括所述第二变体集合中的一变体时,将所述个别的序列读值视为不支持所述变体。在一些这样的实施例中,所述第一异常的组织样本是由一第一肿瘤分数组成,而所述第二异常的组织样本是由来自所述受试者的相同肿瘤的一第二肿瘤分数组成。在一些这样的实施例中,所述第一异常的组织样本为一第一癌症类型,且所述第二异常的组织样本为一第二癌症类型。在一些这样的实施例中,所述第一癌症类型与所述第二癌症类型相同。在多个可替代的实施例中,所述第一癌症类型不同于所述第二癌症类型。在一些这样的实施例中,所述第一癌症类型及所述第二癌症类型分别选自于由乳腺癌、肺癌、前列腺癌、结肠直肠癌、肾癌、子宫癌、胰腺癌、食道癌、淋巴瘤、头颈癌、卵巢癌、肝胆癌、黑色素瘤、宫颈癌、多发性骨髓瘤、白血病、甲状腺癌、膀胱癌及胃癌所组成的群组。
在一些实施例中,所述第一参考集合中的每个变体的所述频率是通过从所述第一异常实体组织样本共同取得的多个第二序列读值来获得。在一些这样的实施例中,从所述第一异常实体组织样本共同取得1000个以上的序列读值,或3000个以上的序列读值,或5000个以上的序列读值。在一些这样的实施例中,所述方法进一步包括:对照一变体候选者小组来分析从所述第一异常实体组织样本取得的所述多个第二序列读值。在一些这样的实施例中,所述变体候选者小组包括介于100个至1000个之间的变体。
在一些实施例中,从所述第一异常实体组织样本取得的所述多个第二序列读值代表对于各个细胞的全基因组数据。在一些实施例中,从所述第一异常实体组织样本取得的所述多个第二序列读值的一平均覆盖率为至少10倍、至少100倍,或至少2000倍。
在一些实施例中,所述液体生物样本包括所述受试者的血液、全血、血浆、血清、尿液、脑脊髓液、粪便、唾液、汗水、眼泪、胸水、心包液或腹膜液。
在一些实施例中,所述液体生物样本是由所述受试者的血液、全血、血浆、血清、尿液、脑脊髓液、粪便、唾液、汗水、眼泪、胸水、心包液或腹膜液组成。
在一些实施例中,所述针对所述第一参考集合中的所述个别的变体的一对应的参考频率来评估所述第一变体集合中的每个个别的变体的所述观察到的频率的步骤包括:使用所述观察到的频率及对于在一可能的肿瘤分数范围内的所述个别的变体的所述参考频率,来评估对于所述个别的变体的一累积密度函数或一累积分布函数。在一些实施例中,使用一累积密度函数,并且所述范围是从0%至110%。在一些这样的实施例中,将所述第一肿瘤分数视为所述累积密度函数的一中位数。
在一些实施例中,使用一累积分布函数。
在一些实施例中,所述累积分布函数具有以下形式:
其中x=a2i,支持所述液体生物样本中的所述个别的变体的所述多个序列读值的观察到的数量;p=t*f1i,其中t为所述估计的第一肿瘤分数,且f1i为在所述第一变体集合中的所述个别的变体的所述观察到的频率;及n=d2i,映射至对应于所述个别的变体的所述基因组位置的来自所述生物样本的多个序列读值的总数量。
在一些实施例中,所述累积分布函数具有以下形式:
其中x=a2i,支持所述液体生物样本中的所述个别的变体k的所述多个序列读值的观察到的数量;pk=t*f1i,其中t为所述估计的第一肿瘤分数,且f1i为在所述第一变体集合中的所述个别的变体k的所述观察到的频率;及nk=d2i,映射至对应于所述个别的变体k的所述基因组位置的来自所述生物样本的多个序列读值的总数量。
在一些实施例中,所述累积密度函数或所述累积分布函数是在负二项分布的假设下得出。
在一些实施例中,所述方法进一步包括:从在每个个别的时间点取得所述受试者的一个别的生物样本中,于一时期内的多个时间点中的每个个别的时间点重复所述获得所述多个第一序列读值的步骤,其中所述个别的生物样本包括多个无细胞核酸分子,从而获得在每个个别的时间点对于所述受试者的对应的多个第一序列读值。进一步地,在多个这样的实施例中,对于所述多个时间点中的每个个别的时间点,确定在所述每个个别的时间点对于所述受试者的所述对应的多个第一序列读值中的所述第一变体集合中的每个变体的支持,从而确定在所述多个时间点中的每个时间点来自于支持或不支持所述个别的变体的所述对应的多个第一序列读值中的所述多个序列读值之间的所述第一变体集合中的每个个别的变体的一观察到的频率。对照在所述第一异常实体组织中的所述个别的变体的所述观察到的频率,来评估在所述多个时间点中的每个时间点的所述第一变体集合中的每个个别的变体的所述观察到的频率,从而以在所述时期内的所述第一肿瘤分数的增加或减少的形式来确定所述时期期间的所述受试者的一病况的状态或进展。在一些这样的实施例中,所述时期为数个月的一时间段(例如,少于4个月、介于1个月至4个月之间等),并且在所述多个时间点中的每个时间点为所述数个月的时间段中的一不同的时间点。在一些这样的实施例中,所述时期为数个年的一时间段(介于2至10年之间),并且在所述多个时间点中的每个时间点为所述数个年的时间段中的一不同的时间点。在一些这样的实施例中,所述时期为数小时的一时间段(例如,介于1小时至6小时之间),并且在所述多个时间点中的每个时间点为所述数小时的时间段中的一不同的时间点。
在一些实施例中,所述方法进一步包括:当观察到所述受试者的所述第一肿瘤分数在所述时期内以一阈值量变化时(例如,在第一量测时间点,相对于一参考量改变百分之十、百分之二十、百分之三十),改变所述受试者的一诊断。
在一些实施例中,所述方法进一步包括:当观察到所述受试者的所述第一肿瘤分数在所述时期内以一阈值量变化时(例如,在第一量测时间点,相对于一参考量改变百分之十、百分之二十、百分之三十),改变所述受试者的一预后情形。
在一些实施例中,所述方法进一步包括:当观察到所述受试者的所述第一肿瘤分数在所述时期内以一阈值量变化时(例如,在第一量测时间点,相对于一参考量改变百分之十、百分之二十、百分之三十),改变所述受试者的一治疗。
在一些实施例中,所述病况为一癌症(例如,乳腺癌、肺癌、前列腺癌、结肠直肠癌、肾癌、子宫癌、胰腺癌、食道癌、淋巴瘤、头颈癌、卵巢癌、肝胆癌、黑色素瘤、子宫颈癌、多发性骨髓瘤、白血病、甲状腺癌、膀胱癌、胃癌或其组合)。在一些实施例中,所述病况为一癌症的一阶段(例如,一乳腺癌的一阶段、一肺癌的一阶段、一前列腺癌的一阶段、一结肠直肠癌的一阶段、一肾癌的一阶段、一子宫癌的一阶段、一胰腺癌的一阶段、一食道癌的一阶段、一淋巴瘤的一阶段、一头颈癌的一阶段、一卵巢癌的一阶段、一肝胆癌的一阶段、一黑色素瘤的一阶段、一子宫颈癌的一阶段、一多发性骨髓瘤的一阶段、一白血病的一阶段、一甲状腺癌的一阶段、一膀胱癌的一阶段或一胃癌的一阶段)。
在一些实施例中,所述病况为一癌症的一预定亚型。
在一些实施例中,所述方法进一步包括步骤:将所述多个第一序列读值应用于一训练过的分类器,从而获得一分类器的结果,其中所述训练过的分类器的结果指出所述受试者是否具有一第一癌症病况;及当所述第一肿瘤分数介于0.003至1.0之间,且所述训练过的分类器的结果指出所述受试者具有所述第一癌症病况时,使用所述训练过的分类器的结果作为所述受试者对于所述第一癌症病况的诊断或预后情形的一基础。在一些实施例中,所述第一癌症病况为一癌症(例如,乳腺癌、肺癌、前列腺癌、结肠直肠癌、肾癌、子宫癌、胰腺癌、食道癌、淋巴瘤、头颈癌、卵巢癌、肝胆癌、黑色素瘤、子宫颈癌、多发性骨髓瘤、白血病、甲状腺癌、膀胱癌、胃癌或其组合)。在一些实施例中,所述第一癌症病况为一癌症的一亚型(例如,所述癌症为乳腺癌、肺癌、前列腺癌、结肠直肠癌、肾癌、子宫癌、胰腺癌、食道癌、淋巴瘤、头颈癌、卵巢癌、肝胆癌、黑色素瘤、子宫颈癌、多发性骨髓瘤、白血病、甲状腺癌、膀胱癌或胃癌的一亚型)。
在一些这样的实施例中,所述第一肿瘤分数介于0.003至1.0之间,且所述第一癌症病况为一癌症的一起源组织。
在一些实施例中,所述训练过的分类器为一神经网络、一支持向量机、一决策树、一非监督式聚类模型、一监督式聚类模型或一回归模型。
本公开的另一方面提供一种计算机系统,所述计算机系统包括:一个或多个处理器;一记忆体,存储通过所述一个或多个处理器执行的一个或多个程序;所述一个或多个程序,包括用于通过一方法来确定在一受试者的一液体生物样本的无细胞核酸中的肿瘤分数的多个指令,所述方法包括:获得从所述受试者的所述液体生物样本而来的电子形式的多个第一序列读值,其中所述液体生物样本包括多个无细胞核酸分子。所述方法进一步包括:使用所述多个第一序列读值来辨识对一第一变体集合中的每个变体的支持,从而确定在所述第一变体集合中的每个变体的一观察到的频率。所述方法进一步包括:对于在所述第一变体集合中的每个个别的变体,获得在一第一参考集合中对于所述个别的变体的一对应的参考频率,其中在所述第一参考集合中的每个对应的参考频率是针对从所述受试者获得的一第一异常实体组织样本中的一个别的变体。所述方法进一步包括:对照在所述第一异常实体组织中的所述第一参考集合中的所述个别的变体的所述观察到的频率,来评估在所述第一变体集合中的每个个别的变体的所述观察到的频率,从而确定在所述受试者的所述液体生物样本的无细胞核酸中的一第一肿瘤分数。
本公开的另一方面提供一种存储一个或多个程序的非暂时性计算机可读存储介质,所述一个或多个程序用以确定在一受试者的一液体生物样本的无细胞核酸中的肿瘤分数。所述一个或多个程序配置成通过一计算机来执行。所述一个或多个程序包括多个指令用于:获得从所述受试者的所述液体生物样本而来的电子形式的多个第一序列读值,其中所述液体生物样本包括多个无细胞核酸分子。所述一个或多个程序进一步包括多个指令用于:使用所述多个第一序列读值来辨识对一第一变体集合中的每个变体的支持,从而确定在所述第一变体集合中的每个变体的一观察到的频率。所述一个或多个程序进一步包括多个指令:对于在所述第一变体集合中的每个个别的变体,获得在一第一参考集合中对于所述个别的变体的一对应的参考频率,其中在所述第一参考集合中的每个对应的参考频率是针对从所述受试者获得的一第一异常实体组织样本中的一个别的变体。所述一个或多个程序进一步包括多个指令用于:对照在所述第一异常实体组织中的所述第一参考集合中的所述个别的变体的所述观察到的频率,来评估在所述第一变体集合中的每个个别的变体的所述观察到的频率,从而确定在所述受试者的所述液体生物样本的无细胞核酸中的一第一肿瘤分数。
本公开的另一方面提供一种用于确定在一受试者的一液体生物样本的无细胞核酸中的肿瘤分数的方法。所述方法包括:在一计算机系统中具有一个或多个处理器及一记忆体,所述记忆体存储通过所述一个或多个处理器执行的一个或多个程序,在所述计算机系统中获得从所述受试者的所述液体生物样本而来的电子形式的多个序列读值,其中所述液体生物样本包括多个无细胞核酸分子。所述方法进一步包括:使用所述多个序列读值来辨识对一变体集合中的每个变体的支持,从而确定在所述第一变体集合中的每个变体的一观察到的频率。所述方法进一步包括:将在所述变体集合中具有第n个最高等位基因频率的所述变体的所述观察到的频率视为所述受试者的所述液体生物样本的无细胞核酸中的所述肿瘤分数,其中n为1以外的正整数(例如,1、2、3、4、5等)。
在一些实施例中,所述变体集合中的一变体为与一预定的基因组位置相关的一单核苷酸变体、与一预定的基因组位置相关的一插入突变、与一预定的基因组位置相关的一缺失突变、一体细胞拷贝数的改变、与一预定的基因组位点相关的一核酸重组,或与一预定的基因组位置相关的一异常的表观遗传修饰模式(例如,甲基化模式)。
在一些实施例中,当所述多个序列读值中的一个别的序列读值包括所述变体集合中的一第一变体的全部或一部分时,将所述个别的序列读值视为支持所述第一变体;当所述多个序列读值中的一个别的序列读值不包括所述变体集合中的所述第一变体时,将所述个别的序列读值视为不支持所述第一变体;及支持所述第一变体的所述多个序列读值中的多个序列读值的一数量对上不支持所述第一变体的所述多个序列读值中的多个序列读值的一数量用以确定所述第一变体的所述观察到的频率,由所述第一变体的所述观察到的频率估计在所述液体生物样本中的所述第一变体的一变体频率。
在一些实施例中,所述受试者患有来自一单一原发部位的癌症。在一些实施例中,所述受试者患有源自于两个或更多个不同器官的癌症。在一些实施例中,所述受试者患有乳腺癌、肺癌、前列腺癌、结肠直肠癌、肾癌、子宫癌、胰腺癌、食道癌、淋巴瘤、头颈癌、卵巢癌、肝胆癌、黑色素瘤、子宫颈癌、多发性骨髓瘤、白血病、甲状腺癌、膀胱癌、胃癌或其组合。
在一些实施例中,所述变体集合包括五个或更多个变体,其中在所述变体集合中的每个个别的变体位在所述受试者的基因组的一不同的位点上。在一些实施例中,所述变体集合是由介于3至20个之间的变体组成,其中在所述变体集合中的每个变体为所述受试者的基因组中的一不同的遗传变异。
在一些实施例中,所述变体集合是由介于2至200个之间的变体组成,其中在所述变体集合中的每个变体为所述受试者的基因组中的一不同的遗传变异。在一些实施例中,所述变体集合包括1000个变体,其中在所述变体集合中的每个变体为所述受试者的基因组中的一不同的遗传变异。
在一些实施例中,所述使用所述多个序列读值来辨识对一变体集合中的每个变体的支持的步骤包括:将所述多个序列读值中的一序列读值与一参考基因组中的一区域比对,以确定所述序列读值是否包括一第一变体的全部或一部分。
在一些实施例中,所述使用所述多个序列读值来辨识对一变体集合中的每个变体的支持的步骤包括:将所述多个序列读值中的一序列读值与多个变体的一查找表比对,以确定所述序列读值是否包括一第一变体的全部或一部分。
在一些实施例中,所述使用所述多个序列读值来辨识对一变体集合中的每个变体的支持的步骤包括:将所述多个第一序列读值中的一序列读值与一查找表中的每个条目比对,其中所述查找表中的每个条目代表一基因组的一不同部分。
在一些实施例中,所述液体生物样本包括或由以下组成:所述受试者的血液、全血、血浆、血清、尿液、脑脊髓液、粪便、唾液、汗水、眼泪、胸水、心包液或腹膜液。
在一些实施例中,所述方法进一步包括步骤:从在每个个别的时间点取得所述受试者的一个别的生物样本中,于一时期内的多个时间点中的每个个别的时间点重复所述获得多个序列读值的步骤,其中所述个别的生物样本包括多个无细胞核酸分子,从而获得在每个个别的时间点对于所述受试者的对应的多个序列读值;及对于在所述多个时间点中的每个个别的时间点,确定对所述原先的认定步骤中的所述变体集合中具有所述第n个最高等位基因频率的所述变体的支持,从而以在所述时期内的所述变体的所述等位基因频率的增加或减少的形式来确定所述时期期间的所述受试者的一病况的状态或进展。
在一些实施例中,所述时期为数个月的一时间段(例如,介于1个月至4个月之间),并且在所述多个时间点中的每个时间点为所述数个月的时间段中的一不同的时间点。在一些实施例中,所述时期为数个年的一时间段(例如,介于2至10年之间),并且在所述多个时间点中的每个时间点为所述数个年的时间段中的一不同的时间点。在一些实施例中,所述时期为数小时的一时间段(例如,介于1小时至6小时之间),并且在所述多个时间点中的每个时间点为所述数小时的时间段中的一不同的时间点。
在一些实施例中,所述方法进一步包括:当观察到所述变体的所述等位基因频率在所述时期内以一阈值量变化时(例如,在第一量测时间点,相对于一参考量改变百分之十、百分之二十、百分之三十),改变所述受试者的一诊断。
在一些实施例中,所述方法进一步包括:当观察到所述变体的所述等位基因频率在所述时期内以一阈值量变化时(例如,在第一量测时间点,相对于一参考量改变百分之十、百分之二十、百分之三十),改变所述受试者的一预后情形。
在一些实施例中,所述方法进一步包括:当观察到所述变体的所述等位基因频率在所述时期内以一阈值量变化时(例如,在第一量测时间点,相对于一参考量改变百分之十、百分之二十、百分之三十),改变所述受试者的一治疗。
在一些实施例中,所述病况为一癌症(例如,乳腺癌、肺癌、前列腺癌、结肠直肠癌、肾癌、子宫癌、胰腺癌、食道癌、淋巴瘤、头颈癌、卵巢癌、肝胆癌、黑色素瘤、子宫颈癌、多发性骨髓瘤、白血病、甲状腺癌、膀胱癌、胃癌或其组合)。在一些实施例中,所述病况为一癌症的一阶段(例如,一乳腺癌的一阶段、一肺癌的一阶段、一前列腺癌的一阶段、一结肠直肠癌的一阶段、一肾癌的一阶段、一子宫癌的一阶段、一胰腺癌的一阶段、一食道癌的一阶段、一淋巴瘤的一阶段、一头颈癌的一阶段、一卵巢癌的一阶段、一肝胆癌的一阶段、一黑色素瘤的一阶段、一子宫颈癌的一阶段、一多发性骨髓瘤的一阶段、一白血病的一阶段、一甲状腺癌的一阶段、一膀胱癌的一阶段或一胃癌的一阶段)。在一些实施例中,所述病况为一癌症的一预定亚型。
在一些实施例中,所述方法进一步包括步骤:将所述多个序列读值应用于一训练过的分类器,从而获得一分类器的结果,其中所述训练过的分类器的结果指出所述受试者是否具有一第一癌症病况;及当所述肿瘤分数介于0.003至1.0之间,且所述训练过的分类器的结果指出所述受试者具有所述第一癌症病况时,使用所述训练过的分类器的结果作为所述受试者对于所述第一癌症病况的诊断的一基础。在一些这样的实施例中,所述第一癌症病况为一癌症(例如,乳腺癌、肺癌、前列腺癌、结肠直肠癌、肾癌、子宫癌、胰腺癌、食道癌、淋巴瘤、头颈癌、卵巢癌、肝胆癌、黑色素瘤、子宫颈癌、多发性骨髓瘤、白血病、甲状腺癌、膀胱癌、胃癌或其组合)。在一些这样的实施例中,所述第一癌症病况为一癌症的一亚型(例如,乳腺癌、肺癌、前列腺癌、结肠直肠癌、肾癌、子宫癌、胰腺癌、食道癌、淋巴瘤、头颈癌、卵巢癌、肝胆癌、黑色素瘤、子宫颈癌、多发性骨髓瘤、白血病、甲状腺癌、膀胱癌或胃癌的一亚型)。在一些这样的实施例中,所述第一肿瘤分数介于0.003至1.0之间,且所述第一癌症病况为一癌症的一起源组织。在一些实施例中,所述训练过的分类器为一神经网络、一支持向量机、一决策树、一非监督式聚类模型、一监督式聚类模型或一回归模型。
本公开的另一方面提供一种计算机系统,所述计算机系统包括:一个或多个处理器;及一记忆体,存储通过所述一个或多个处理器执行的一个或多个程序。所述一个或多个程序包括用于通过一方法来确定在一受试者的一液体生物样本的无细胞核酸中的肿瘤分数的多个指令,所述方法包括:获得从所述受试者的所述液体生物样本而来的电子形式的多个序列读值,其中所述液体生物样本包括多个无细胞核酸分子。所述方法进一步包括:使用所述多个序列读值来辨识对一变体集合中的每个变体的支持,从而确定在所述第一变体集合中的每个变体的一观察到的频率。所述方法进一步包括:将在所述变体集合中具有第n个最高等位基因频率的所述变体的所述观察到的频率视为所述受试者的所述液体生物样本的无细胞核酸中的所述肿瘤分数,其中n为1以外的正整数。
本公开的另一方面提供一种存储一个或多个程序的非暂时性计算机可读存储介质,所述一个或多个程序用于确定在一受试者的一液体生物样本的无细胞核酸中的肿瘤分数。所述一个或多个程序配置成通过一计算机来执行。所述一个或多个程序包括多个指令用于:获得从所述受试者的所述液体生物样本而来的电子形式的多个序列读值,其中所述液体生物样本包括多个无细胞核酸分子。所述一个或多个程序进一步包括多个指令用于:使用所述多个序列读值来辨识对一变体集合中的每个变体的支持,从而确定在所述第一变体集合中的每个变体的一观察到的频率。所述一个或多个程序进一步包括多个指令用于:将在所述变体集合中具有第n个最高等位基因频率的所述变体的所述观察到的频率视为所述受试者的所述液体生物样本的无细胞核酸中的所述肿瘤分数,其中n为1以外的正整数。
通过引用的合并
在此说明书中提及的所有出版物、专利及专利申请皆通过引用以其整体并入本文中,其程度与每个单独的出版物、专利或专利申请被具体地且单独地指出通过引用并入的程度相同。
附图说明
本文所公开的多个实施方式通过示例且非限制的方式在多个附图的图示中说明。类似的附图标记指的是整个附图中的多个视图的多个对应的部分。
图1a及1b根据本公开的一些实施例说明了描述一计算装置的一示例方块图;
图2a、2b、2c、2d、2e及2f根据本公开的一些实施例说明一种分类依受试者的方法的一示例流程图;
图3根据本公开的一些实施例说明一箱型图,在所述箱型图中,对于每种个别的癌症类型,提供患有所述个别的癌症类型的多个受试者的ctdna分数,其中对于每个个别的受试者,y轴提供一估计的ctdna分数,所述估计的ctdna分数是基于来自所述个别的受试者的一生物样本(例如血液)的一变体集合中的每个变体的一观察到的频率与从所述个别的受试者的一异常的组织样本(例如肿瘤分数)获得的每个这样的变体的一对应的参考频率的一配对组合(matchedpair)的比对;
图4根据本公开的一些实施例说明根据癌症阶段的一函数的罹患图3所说明的多种癌症的任一种的多个受试者的ctdna分数的一图示;
图5根据本公开的一些实施例说明根据乳腺癌阶段的一函数的多个受试者的ctdna分数的一图示,其分成三个类别:这些受试者的无细胞dna足以识别在此类受试者的一配对肿瘤中发现的一变体,而无需事先知道此变体在所述配对肿瘤中;这些受试者的无细胞dna支持在一配对肿瘤中发现的一变体;及这些受试者的无细胞dna不支持在一配对肿瘤癌症中发现的一变体;
图6根据本公开的一些实施例说明根据多个受试者的cfdna分数的函数的侦测所述多个受试者的癌症的能力;
图7a及7b根据本公开的一些实施例说明根据cfdna分数、分类器及乳腺癌亚型的函数的识别乳腺癌的能力;
图8根据本公开的一些实施例详细描述根据cfdna分数的一函数的针对跨越在图3中辨识出的不同癌症的范围(spectrum)的一受试者群体的wgbs多类别分类器的精确度;
图9根据本公开的一些实施例详细描述根据临床阶段的一函数的表现出一最小ctdna分数的多个受试者的百分比;
图10根据本公开的一些实施例说明横跨癌症的所有阶段的肿瘤尺寸与ctdna分数的正相关性;
图11根据本公开的一些实施例说明ctdna分数与针对增殖的ki67标记的相关性;
图12根据本公开的一些实施例说明一种用于制备定序用的一核酸样本的方法的一流程图;
图13为根据本公开的一些实施例的一个用于获得多个序列读值的流程的一图解表示法;
图14为根据本公开的一些实施例的一种用于确定多个序列读值的变体的方法的一流程图;
图15为根据本公开的一些实施例的目的在于辨识多个变体的一种用于获得一甲基化状态向量的方法的一流程图;
图16根据本公开的一些实施例提供在一试验的估计脱离率(sheddingrate)的范围内的一累计密度函数;
图17说明在本公开的一肿瘤配对实施例与一第二最高等位基因实施例之间进行的多个肿瘤分数量测的一致性;
图18根据本公开的一些实施例说明作为确定无细胞肿瘤分数的一基础的一ccga研究的多个细节;
图19a及19b根据本公开的一实施例针对按肿瘤来源划分的图18所总结的一训练集合(图19a,n=1,416)及一测试集合(图19b,n=847),提供了使用图18所总结的所述训练集合来训练的多个模式的敏感度的信息;
图19c及19d根据本公开的一实施例提供了在按肿瘤来源划分的图18所总结的所述训练集合(图19c)及所述测试集合(图19d)中的肿瘤分数的信息;及
图20a及20b根据本公开的一实施例说明通过比较cfdnawgs与肿瘤wgs的结果所计算出的cfdna肿瘤分数,所述cfdnawgs与肿瘤wgs的结果是根据乳腺癌、结肠直肠癌、肺癌及其他癌症的总集合的阶段(图20a),及根据各个癌症类型(图20b)。
具体实施方式
现在将详细说明多个实施例,所述多个实施例的示例在附图中阐明。在下面的详细描述中,阐述了许多具体细节,以便提供对本公开的一彻底理解。然而,对于本领域的普通技术人员将显而易见的是,本公开可在没有这些具体细节的情况下实践。在其他例子中,没有详细描述公知的方法、流程、组件、电路及网络,以免不必要地模糊所述多个实施例的各方面。
本文描述的多个实施方式提供各种用于确定在一受试者中的一肿瘤分数的技术方案。这样的信息可用于确定所述受试者的癌症状态,包括例如对所述受试者的起源组织进行分类。从一受试者的一生物样本获得多个序列读值。所述生物样本包括无细胞核酸。因此,所述多个序列读值为无细胞核酸的。所述多个序列读值用于辨识对一变体集合中的每个变体的支持,从而确定每个变体的一观察到的频率。将多个所述观察到的变体频率与在一参考集合中对于各个变体的对应的参考频率进行比较。每个这样的参考频率为来自所述受试者的一异常的组织样本(例如,一肿瘤)中的一个别的变体的一频率。以此种方式,确定了所述受试者的所述肿瘤分数。在一些实施例中,所述肿瘤分数与一分类器结合使用,以对所述受试者的一癌症病况进行分类。
图3提供对于本公开的多个实施方式的一基础。通常,从所述生物样本的所述无细胞核酸获得的所述变体集合中的所述多个变体的所述观察到的频率小于所述参考集合中对于这样的多个变体的所述观察到的参考频率。在不旨在受限于任何特定理论的情况下,假设含有这样的多个变体的所述无细胞核酸的来源是来自所述异常组织中的多个癌症细胞的降解或分解。因此,在一些实施例中,假设将含有本公开的所述多个公开的变体集合中的这样的多个变体的多个所述生物样本中的所述无细胞的核酸表示为ctdna,或“循环肿瘤dna”(ctdna)、所述无细胞核酸的一小部分(cfdna),它们被使用作为用于确定每个变体的所述观察到的频率的基础。因此,可预期的是,从所述生物样本的所述无细胞核酸获得的所述变体集合中的所述多个变体的所述观察到的频率小于所述参考集合中对于这样的多个变体的所述观察到的参考频率。图3中所总结的数据支持此论点,并且指出不同的癌症类型具有不同的对于多个特定受试者的所述变体集合中的所述多个变体的所述观察到的频率与相同的多个特定的受试者的一参考异常组织中对于这样的多个变体的所述观察到的参考频率的比率。
图3提供一箱型图,在所述箱型图中,对于在一ccga群体(cohort)中所研究的每个癌症类型,存在多个个体,并且对于每个个体的ctdna分数的一估计值在y轴上。图3显示出针对每个癌症类型的两类别受试者的对于每个个别的癌症类型所观察到的ctdna分数的分布的概述:(i)这些受试者具有所述个别的癌症类型,其中在他们的cfdna中没有(来自所述多个无细胞生物样本的所述多个序列读值中的)一变体的一量测到的证据(图3中称为“错误”);及(ii)这些受试者具有所述个别的癌症类型,其中在他们的cfdna中有(来自所述多个无细胞生物样本的所述多个序列读值中的)一变体的一量测到的证据(图3中称为“真实”)。对于每个特别的癌症类型,在所述真实类别中的所述多个受试者的所述量测到的ctdna的一第一分布形成一第一方箱(图3中的白色方箱),在所述错误类别中的所述多个受试者的所述预期的ctdna的一第二分布形成一第二方箱(图3中的填充灰色的方箱),其中第25个四分位数及第75个四分位数定义了每个这样的方箱,每个方箱的多个箱须(whisker)显示出多个极值。每个方箱中的黑线为对于一特定类别的一特定癌症类型的所有所述个体的一肿瘤分数估计值的中位数。例如,参考肾癌,对于在所述错误类别中的这些受试者有一ctdna分数的中位数,而对于在所述真实类别中的这些受试者有一不同的ctdna分数的中位数。
图3说明在所研究的ccga群体中,不同癌症的脱离率(sheddingrate)(ctdna分数)有一个大动态范围。下文的示例12提供所述ccga群体的多个细节。所述观察到的大动态范围可用于从所述参考集合中的所述多个变体的所述观察到的频率来为建立多个有意义且有益的阈值提供一基础。意即,例如,考虑到在一特定受试者的所述异常组织中的多个变体的观察到的频率,及可选择的关于具有一特别病况的多个受试者的预期的ctdna分数的信息,针对所述特定受试者的一变体集合中的所述多个变体的所述观察到的频率来确定并评估所述特定的癌症受试者的一阈值,以便根据具有或不具有所述病况将所述受试者分类。例如,参考图3,一阈值0.01可用于分析一受试者是否患有肾癌。在此示例中,一异常的组织,例如一肿瘤,从一患者来获得,并且用于确定在一第一参考集合中对于每个个别的变体的一参考频率。事实上,在一些实施例中,各种可能的变体的所述频率用于定义所述参考集合的所述多个变体。接下来,从所述异常组织以外的一生物样本获得无细胞核酸,并且从所述生物样本中的所述无细胞核酸的多个序列读值确定在所述参考集合中的多个相同变体的所述变体频率,从而形成在所述第一变体集合中的每个个别的变体的所述观察到的ctdna频率。接着,用于确定是否符合所述0.01的阈值状况的所述ctdna频率与所述参考频率的一比对提供了用于确定所述受试者是否患有肾癌的一基础。例如,假如所述比对指出所述ctdna分数高于0.01,则表明所述受试者未患有肾癌。另一方面,从约为1e-03的所述第一变体集合中的每个个别的变体的所述观察到的频率形成的一ctdna分数的观察与肾癌的发现为一致的。并且,在一些实施例中,不是在一绝对二进制的基础上使用本公开的多个系统及方法来指示一受试者是否具有一特定病况,而是提供一受试者具有一特定病况的一可能性或概率。在多个这样的实施例中,在所述第一变体集合中的每个个别的变体的所述观察到的频率与在一第一参考集合中对于所述个别的变体的一对应的参考频率的所述比对用于确定所述第一变体集合中的每个个别的变体的所述观察到的频率与对于所述个别的变体的所述对应的参考频率相距多远,并且基于此距离或此距离的函数确定一受试者具有一特定病况的所述概率或可能性。
在图3中,用于计算所述ctdna分数的方法为一种贝叶斯方法。例如,考虑到其中有对于一个别的癌症类型的多个变体的一肿瘤定序集合(参考集合)及针对患有所述癌症类型的多个受试者的一集合体的所述配对的无细胞dna的例子。假如对于所述个别的癌症类型的所述多个肿瘤变体皆无法与所述受试者的集合体中的所述受试者的任何一个的所述cfdna进行配对,则在没有任何支持性定序数据的情况下,所述受试者的集合体仍然可用于估计一个别的癌症类型的所述ctdna分数为何,从而提供一个关于有多少可用信号的上限值,即使它在所述无细胞核酸测定中被遗漏。对于图3的计算,平均而言,从来自每个受试者的一生物样本的所述无细胞dna获得的多个序列读值有3000个。假如所述多个序列读值中没有一个支持(包括)所述3000个序列读值中的所述变体,同时所述受试者属于图3的所述错误类别中,则此信息仍然可用于估计所述生物样本中的此变体的可能的潜在频率(后验概率)为多少,即使所述多个序列读值中没有一个支持(包括)所述变体。这样的分析形成了为图3所示的每种癌症的所述错误类别中的所述多个受试者提供所述估计的ctdna分数的基础。意即,图3中的所述多个灰色方盒代表所述多个生物样本,其中在所述多个个别的生物样本中没有已经量测到的与所述癌症相关的单一变体。这些生物样本用于独立地估计对于所述癌症的所述可能的潜在ctdna分数。如图3所证明,关于中位数,相对于使用所述相同癌症的样本群体所计算出的所述ctdna分数的中位数,这样的多个生物样本产生一减小的ctdna分数的中位数,其中在所述多个生物样本中已经观察到与所述癌症相关的多个变体。
定义
如本文所公开,术语“生物样本”指的是任何从一受试者取得的样本,其可反映出与所述受试者相关的一生物状态,并且包括无细胞dna。多个生物样本的示例包括,但不限于,所述受试者的血液、全血、血浆、血清、尿液、脑脊髓液、粪便、唾液、汗水、眼泪、胸水、心包液或腹膜液。
如本文所公开,多个术语“无细胞核酸”、“无细胞dna”及“cfdna”可互换地指的是多个核酸片段,所述多个核酸片段在一受试者的体内(例如,血流)循环,并且源自于一个或多个健康细胞及/或源自于一个或多个癌细胞。
如本文所公开,术语“循环肿瘤dna”或“ctdna”指的是源自于异常组织,例如一肿瘤或其他癌症类型的多个细胞,的多个核酸片段,所述多个核酸片段可被释放至一受试者的血流中,这是由于多种生物过程,例如,多个死亡细胞的雕亡或坏死,或通过多个活的肿瘤细胞进行的主动释放。
如本文所公开,术语“无细胞核酸”指的是可在细胞外发现的体液中的多个核酸分子,例如,一受试者的血液、全血、血浆、血清、尿液、脑脊髓液、粪便、唾液、汗水、眼泪、胸水、心包液或腹膜液。无细胞核酸可互换地用作为循环核酸。所述无细胞核酸的多个示例包括,但不限于,rna、粒线体dna或基因组dna。
如本文所使用,术语“甲基化”指的是脱氧核糖核酸(dna)的一种修饰,其中一胞嘧啶碱基的嘧啶环上的一氢原子被转化成一甲基,以形成5-甲基胞嘧啶。特别地,甲基化倾向于发生在胞嘧啶及鸟嘌呤的二核苷酸处,在本文中称为“cpg位点”。在其他例子中,甲基化可能发生在一胞嘧啶的非cpg位点的部分上,或是非胞嘧啶的其他核苷酸上;然而,这些情况很少发生。在本公开中,为了清楚起见,参考cpg位点讨论甲基化。异常的cfdna甲基化可被辨识为过甲基化或低甲基化,这两者皆可指示出癌症的状态。如本领域所熟知,dna甲基化异常(与健康的对照组相比)可引起不同的作用,其可能导致癌症。
如本文所使用,术语“甲基化指数”对于每个基因组位点(例如,一cpg位点,为dna的一个区域,其中沿其5'→3'方向的碱基线性序列中的一胞嘧啶核苷酸后面为一鸟嘌呤核苷酸)可指的是在所述位点显示甲基化的多个序列读值占覆盖此位点的多个读值的总数量的比例。一区域的“甲基化密度”可为在显示甲基化的一区域内的多个位点处的多个读值的数量除以覆盖所述区域中的所述多个位点的多个读值的总数量。所述多个位点可具有多个特定的特性(例如,所述多个位点可为cpg位点)。一区域的“cpg甲基化密度”可为显示cpg甲基化的多个读值的数量除以覆盖所述区域中的多个cpg位点(例如,一特定的cpg位点、一cpg岛内的多个cpg位点,或一更大的区域)的多个读值的总数量。例如,人类基因组中每个100-kb数据箱(bin)的甲基化密度可从多个cpg位点处的多个未转化的胞嘧啶(其可对应于甲基化胞嘧啶)的总数量确定为通过映射至所述100-kb区域的多个序列读值所覆盖的所有的cpg位点的比例。在一些实施例中,对其他数据箱的尺寸进行此分析,例如50-kb或1-mb等。在一些实施例中,一区域可为一整个基因组或一染色体或一染色体的一部分(例如,一染色体臂)。当一区域仅包括一cpg位点时,所述cpg位点的一甲基化指数可与所述区域的甲基化密度相同。所述“甲基化胞嘧啶的比例”可指的是在所述区域中显示甲基化过(例如,在亚硫酸氢盐转化后未转化)的多个胞嘧啶位点“c's”的数量除以多个分析过的胞嘧啶残基的总数量,例如,包括cpg背景之外的多个胞嘧啶。所述甲基化指数、所述甲基化密度及甲基化胞嘧啶的比例为“甲基化水平”的多个示例。
如本文所公开,多个术语“核酸”及“核酸分子”可互换使用。所述多个术语指的是任何组成形式的核酸,例如,脱氧核糖核酸(dna,例如,互补dna(cdna)、基因组dna(gdna)等),及/或dna类似物(例如,含有碱基类似物、醣类似物及/或一非天然的骨架等),所有的这些可为单股或双股形式。除非另有限制,否则一核酸可包括天然核苷酸的多个已知的类似物,其中的一些可以一类似于天然存在的核苷酸的方式来起作用。一核酸可为能用于进行本文的多个过程的任何形式(例如,线性、环形、超螺旋、单股、双股等)。在一些实施例中的一核酸可来自一单一染色体或所述单一染色体的片段(例如,一核酸样本可来自于从一个二倍体生物体获得的一样本的一染色体)。在某些实施例中,核酸包括多个核小体、多个核小体的多个片段或部分,或多个类核小体的结构。核酸有时包括蛋白质(例如,组蛋白、dna结合蛋白等)。通过本文所描述的多个过程所分析的核酸有时基本上是分离的,并且基本上与蛋白质或其他分子不相关。核酸还包括从单股(“有义的”或“反义的”、“正股”或“负股”、“正向阅读框”或“反向阅读框”)及双股多核苷酸合成、复制或扩增的dna的衍生物、变异体及类似物。脱氧核糖核酸包括脱氧腺苷、脱氧胞苷、脱氧鸟苷及脱氧胸苷。通过使用从一受试者获得的作为一模板的核酸可制备核酸。
如本文所公开,术语“参考基因组”指的是可用于参考来自一受试者的多个辨识序列的任何生物体或病毒的任何特定的已知、定序过或表征的基因组,无论是部分的或完整的。在由国家生物技术信息中心(“ncbi”)或圣塔克鲁兹(santacruz)的加利福尼亚大学(ucsc)主持的在线基因组浏览器中提供了用于人类受试者以及许多其他生物体的多个示例性参考基因组。一“基因组”指的是以多个核酸序列表现的一生物体或病毒的完整遗传信息。如本文所使用,一参考序列或参考基因组通常为来自一个体或多个个体的一组装过或部分组装过的基因组序列。在一些实施例中,一参考基因组为来自一个或多个人类个体的一组装过或部分组装过的基因组序列。所述参考基因组可被视为一物种的基因集合的一代表性示例。在一些实施例中,一参考基因组包括分配给多个染色体的多个序列。多个示例性人类参考基因组包括但不限于ncbi构建34(ucsc等效物:hg16)、ncbi构建35(ucsc等效物:hg17)、ncbi构建36.1(ucsc等效物:hg18)、grch37(ucsc等效物:hg19)及grch38(ucsc等效物:hg38)。
如本文所公开,术语“一参考基因组的多个区域”、“基因组区域”或“染色体组区域”指的是一参考基因组的连续或不连续的任何部分。它也可被称为,例如,一数据箱(bin)、一分区、一基因组部分、一参考基因组的一部分、一染色体的一部分等。在一些实施例中,一基因组区段是基于基因组序列的一特定长度。在一些实施例中,一方法可包括对多个基因组区域的多个映射序列的分析。多个基因组区域可大致为相同长度,或者所述多个基因组区段可为不同长度。在一些实施例中,多个基因组区域的长度大致相同。在一些实施例中,对不同长度的多个基因组区域进行调整或加权。在一些实施例中,一基因组区域为约10千碱基(kb)至约500kb、约20kb至约400kb、约30kb至约300kb、约40kb至约200kb,且有时为约50kb至约100kb。在一些实施例中,一基因组区域为约100kb至约200kb。一基因组区域不限于连续延伸的序列。因此,多个基因组区域可由多个连续及/或不连续的序列组成。一基因组区域不限于一单一的染色体。在一些实施例中,一基因组区域包括一染色体的全部或部分,或两个或更多个染色体的全部或部分。在一些实施例中,多个基因组区域可跨越一个、两个或更多个完整的染色体。此外,所述多个基因组区域可跨越多个染色体的多个接合或未接合的部分。
如本文所公开,术语“序列读值”或“读值”指的是通过本文所描述的或本领域所知的任何定序过程来产生的多个核苷酸序列。多个读值可从多个核酸片段的一端产生(“单端读值”),且有时从核酸的两端产生(例如,双端读值、两端读值)。所述序列读值的长度时常与特定的定序技术相关。例如,多个高通量方法提供大小可从数十至数百个碱基对(bp)变化的多个序列读值。在一些实施例中,所述多个序列读值的一平均值、中位数或平均长度约为15bp至900bp长(例如,约20bp、约25bp、约30bp、约35bp、约40bp、约45bp、约50bp、约55bp、约60bp、约65bp、约70bp、约75bp、约80bp、约85bp、约90bp、约95bp、约100bp、约110bp、约120bp、约130、约140bp、约150bp、约200bp、约250bp、约300bp、约350bp、约400bp、约450bp、约500bp。在一些实施例中,所述多个序列读值的一平均值、中位数或平均长度约为1000bp或更长。例如,纳米孔定序法提供大小可从数十至数百至数千个碱基对变化的多个序列读值。伊卢米那(illumina)的平行定序法可提供变化不大的多个序列读值,例如,大部分的序列读值可小于200bp。
如本文所公开,本文所使用的多个术语“定序”或“序列的确定”等通常是指可用于确定如核酸或蛋白质的生物大分子等级的任何或所有的生化过程。例如,定序数据可包括如一dna片段的一核酸分子的全部或一部分的核苷酸碱基。
如本文所公开,术语“单核苷酸变体”或“snv”指的是在一核苷酸序列,例如来自一个体的一序列读值,的一位置(例如,位点)处将一核苷酸取代为一不同核苷酸。从一第一核碱基x至一第二核碱基y的取代可表示为“x>y”。例如,胞嘧啶至胸腺嘧啶的snv被表示为“c>t”。
如本文所公开,术语“受试者”指的是任何有生命或无生命的生物体,包括但不限于人类(例如,男性、女性、胎儿、怀孕的女性、儿童等)、非人类的动物、植物、细菌、真菌或原生生物。任何人类或非人类的动物可作为一受试者,包括但不限于哺乳动物、爬行动物、鸟类、两栖动物、鱼类、有蹄类动物、反刍动物、牛科动物(例如牛)、马科动物(例如马)、山羊及绵羊类(例如绵羊、山羊)、猪科动物(例如猪)、骆驼科动物(例如骆驼、美洲驼、羊驼)、猴子、猿类(例如大猩猩、黑猩猩)、熊科动物(例如熊)、家禽、狗、猫、小鼠、大鼠、鱼、海豚、鲸鱼及鲨鱼。在一些实施例中,一受试者为任何阶段的男性或女性(例如,男人、女人或儿童)。
示例性系统实施例
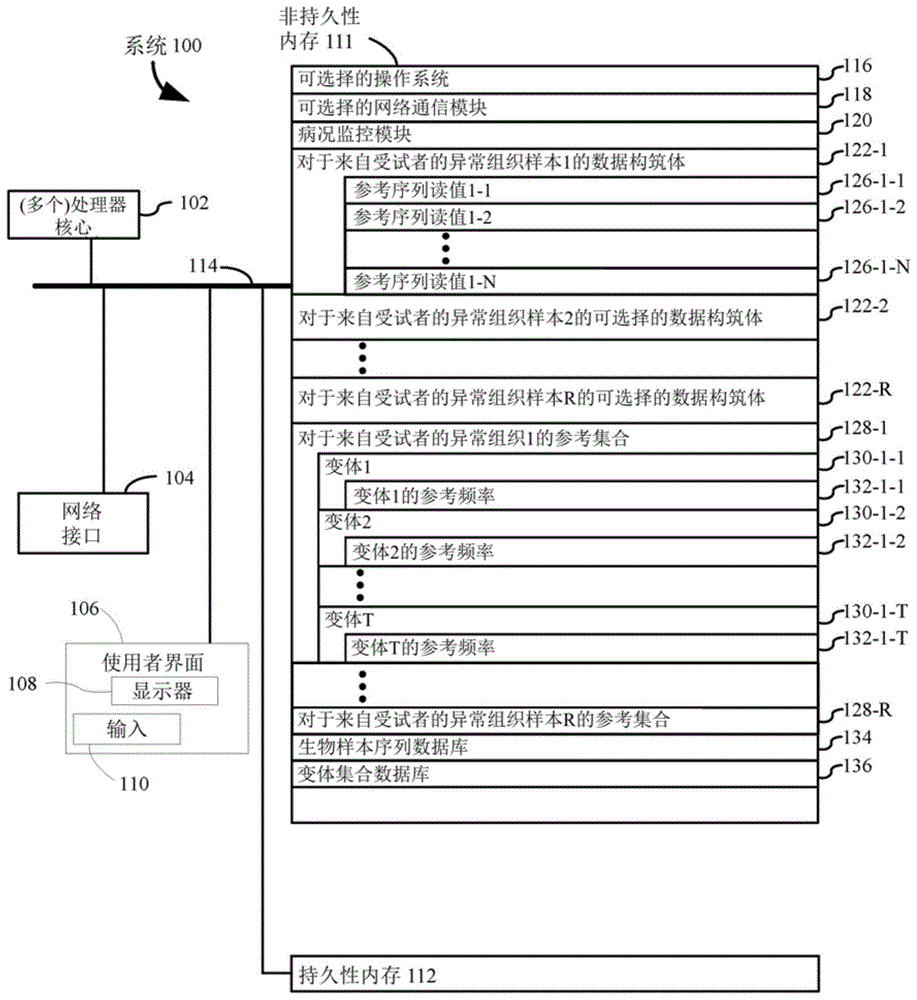
目前已经提供本公开的一些方面的一概论及本公开中使用的一些定义,现在将结合图1描述一示例性系统的多个细节。图1为根据一些实施方式的说明一系统100的一方块图。在一些实施方式中的一装置100包括:一个或多个处理单元cpu(s)102(也称为处理器);一个或多个网络接口104;一使用者界面106;一非持久性内存111;一持久性内存112;及一个或多个通信总线114,用于交互连接这些组件。所述一个或多个通信总线114可选择地包括电路(有时称为一芯片组),所述电路交互连接及控制多个系统组件之间的通信。所述非持久性内存111通常包括高速随机存取内存,例如dram、sram、ddrram、rom、eeprom、闪存,而所述持久性内存112通常包括cd-rom、数字多用光盘(dvd)或其他光学存储器、磁带盒、磁带、磁盘存储器或其他磁性存储装置、磁盘存储装置、光盘存储装置、闪存装置,或其他非易失性固态存储装置。所述持久性内存112可选择地包括一个或多个距离所述(多个)cpu102远程定位的存储装置。所述持久性内存112及在所述非持久性内存112中的所述(多个)非易失性内存装置包括非暂时性计算机可读存储介质。在一些实施方式中,所述非持久性内存111或可替代的所述非暂时性计算机可读存储介质存储下面的程序、模块,及数据结构或其子集合,有时与所述持久性内存112结合:
一可选择的操作系统116,包括多个用于处理各种基础系统服务及用于进行多个硬件相关任务的流程;
一可选择的网络通信模块(或多个指令)118,用于将连接所述系统100与其他装置或一通信网络连接;
一病况监控模块120,用于分类一受试者,及/或评估一受试者的一病况的一状态,及/或确定或监控一受试者的一ctdna肿瘤分数;
一个或多个数据构筑体122,针对来自一受试者的一个或多个异常的数据集组织样本,每个这样的数据构筑体122包括多个第二序列读值126;
一个或多个参考集合128,每个个别的参考集合128针对一异常组织样本的一对应的数据构筑体122,并包括在一组变体中的每个变体130的辨识及每个这样的变体的一参考频率132;
一生物样本序列库134,包括对于来自所述受试者的每个对应的生物样本的一个别的数据构筑体138,所述对应的生物样本包括多个无细胞核酸分子,所述个别的数据构筑体138包括这样的多个无细胞核酸分子的多个第一序列读值140;及
一变体集合数据库136,包括对于每个对应生物样本的一变体集合142,每个这样的变体集合142包括一组变体144,每个变体包括对所述对应的生物样本中的所述第一变体的支持的表示。
在一些实施方式中,一个或多个上文标识出的元件被存储在一个或多个先前所提及的存储装置中,并且对于应用于进行一上述功能的一组指令中。多个上文标识出的模块、数据或程序(例如,多组指令)不需要被实施作为多个个别的软件程序、流程、数据集或模块,因此,这些模块或数据的各种子集可在各种实施方式中组合或以其他方式重新布置。在一些实施方式中,所述非持久性内存111可选择地存储上文标识出的多个模块及数据结构的一子集。再者,在一些实施例中,所述记忆体存储上文未描述的多个额外的模块及数据结构。在一些实施例中,一个或多个上文标识出的元件被存储在除可视化系统100之外的一计算机系统中,所述计算机系统通过所述可视化系统100为可寻址的,使得所述可视化系统100可在需要时检索这类数据的全部或一部分。
虽然图1描绘出一“系统100”,但与本文所描述的多个实施方式的一结构示意图相比,此图示更多地旨在作为可能存在于多个计算机系统中的各种特征的一功能性描述。在实践上,及如本领域普通技术人员所认知,多个单独显示的项目可被结合,并且一些项目可被分开。并且,虽然图1描绘出在所述非持久性内存111中的某些数据及模块,但这些数据及模块中的一些或全部可位于所述持久性内存112中。
示例性方法实施例-基于异常组织的定序,发现一病况
当参考图1已揭露根据本公开的一系统时,现在参考图2详细说明根据本公开的一方法。
参考图2a的方块202至208,在一些实施例中,在一计算机系统中,例如图1的系统100,进行一种用于确定在一受试者的一液体生物样本的无细胞核酸中的一肿瘤分数的方法,所述系统100具有一个或多个处理器102及记忆体111/112,所述记忆体111/112存储一个或多个程序,例如病况监控模块120,所述病况监控模块120通过所述一个或多个处理器来执行。在一些这样的实施例中,从所述受试者的一生物样本获得电子形式的多个第一序列读值140,其中所述生物样本包括多个无细胞核酸分子。
参考方块204,在一些实施例中,所述受试者为人类或哺乳动物。在一些实施例中,所述受试者为任何有生命或无生命的生物体,包括但不限于人类(例如,男性、女性、胎儿、怀孕的女性、儿童等)、非人类的动物、植物、细菌、真菌或原生生物。在一些实施例中,所述受试者为哺乳动物、爬行动物、鸟类、两栖动物、鱼类、有蹄类动物、反刍动物、牛科动物(例如牛)、马科动物(例如马)、山羊及绵羊类(例如绵羊、山羊)、猪科动物(例如猪)、骆驼科动物(例如骆驼、美洲驼、羊驼)、猴子、猿类(例如大猩猩、黑猩猩)、熊科动物(例如熊)、家禽、狗、猫、小鼠、大鼠、鱼、海豚、鲸鱼及鲨鱼。在一些实施例中,一受试者为任何阶段的男性或女性(例如,男人、女人或儿童)。
在一些实施例中,所述生物样本包括所述受试者的血液、全血、血浆、血清、尿液、脑脊髓液、粪便、唾液、汗水、眼泪、胸水、心包液或腹膜液(方块206)。在多个这样的实施例中,所述生物样本可包括所述受试者的血液、全血、血浆、血清、尿液、脑脊髓液、粪便、唾液、汗水、眼泪、胸水、心包液或腹膜液,以及所述受试者的其他组成物(例如,实体(solid)组织等)。
在一些实施例中,所述生物样本由所述受试者的血液、全血、血浆、血清、尿液、脑脊髓液、粪便、唾液、汗水、眼泪、胸水、心包液或腹膜液(方块208)组成。在多个这样的实施例中,所述生物样本受限于所述受试者的血液、全血、血浆、血清、尿液、脑脊髓液、粪便、唾液、汗水、眼泪、胸水、心包液或腹膜液,并且不包括所述受试者的其他组成物(例如,实体组织等)。
在一些实施例中,将所述生物样本进行处理,以提取出准备用于定序分析的无细胞核酸。通过一非限制的示例,在一些实施例中,从在k2edta试管中的一受试者采集到的一血液样本中提取无细胞核酸。在收集后的两小时内,通过先以1000g对血液进行十分钟,接着以2000g对血浆进行十分钟的二次旋转来处理多个样本。接着,将所述血浆以1毫升等分的方式在-80℃下贮存。以此种方式,为了无细胞核酸提取的多个目的,从所述生物样本制备合适量的血浆(例如,1至5毫升)。在一些这样的实施例中,将无细胞核酸使用qiaamp循环核酸试剂组(凯杰(qiagen))提取出,并洗提到dna悬浮缓冲液(西格玛(sigma))中。在一些实施例中,将所述纯化过的无细胞核酸贮存在-20℃,直到使用为止。例如,参见swanton等人,2017,“系统发育ctdna分析描写早期肺癌的演变”nature,545(7655):446-451,其通过引用并入本文。为了定序的目的,其他等效的方法可用于制备从多个生物方法而来的无细胞核酸,并且所有的这类方法皆在本公开的范围内。
在一些实施例中,从一生物样本获得的所述无细胞核酸为任何形式的本公开所定义的核酸,或其组合物。例如,在一些实施例中,从一生物样本获得的所述无细胞核酸为rna及dna的一混合物。
任何形式的定序可用于从自所述生物样本获得的所述无细胞核酸中获得所述多个序列读值140,其包括但不限于多个高通量定序系统,例如,罗氏(roche)454平台、应用生物系统(appliedbiosystems)的solid平台、螺旋生物科学(helicos)的真实单一分子dna定序技术、来自艾菲矩阵(affymetrix)公司的杂交定序平台、太平洋生物科学(pacificbiosciences)的单一分子即时(smrt)技术、来自454莱富生命科技(lifesciences)、伊卢米那/索莱克斯(solexa)与螺旋生物科学的合成定序平台,及来自应用生物系统的连接(ligation)定序平台。来自莱富科技的离子倾注(iontorrent)技术及纳米孔定序法也可用于从自所述生物样本获得的所述无细胞核酸中获得多个序列读值140。
在一些实施例中,合成定序法及基于可逆终止子的定序法(例如,伊卢米那的基因组分析仪;基因组分析仪ii;hiseq2000;hiseq2500(加利福尼亚州圣地亚哥的伊卢米那))用于从自所述生物样本获得的所述无细胞核酸中获得多个序列读值140。在一些这样的实施例中,对数百万个无细胞核酸(例如dna)片段进行平行定序。在此类型的定序技术的一示例中,使用含有一光学透明载片的一流动槽,所述光学透明载片在结合多个寡核苷酸锚(例如,转接子(adaptor)引子)的表面上具有八个独立泳道。一流动槽时常为一固体支撑物,所述固体支撑物配置用以保持及/或允许试剂溶液依序通过多个经结合的分析物。在一些例子中,多个流动槽呈平面形状、光学透明、通常呈毫米或亚毫米级,且时常具有其中发生分析物/试剂交互作用的多个通道或泳道。在一些实施例中,一无细胞核酸样本可包括有助于侦测的一信号或标签。在一些这样的实施例中,从获自所述生物样本的所述无细胞核酸获取多个序列读值140包括:通过各种技术获得所述信号或标签的量化信息,所述各种技术为例如流式细胞术、定量聚合酶链反应(qpcr)、凝胶电泳、基因芯片分析、微阵列、质谱法、细胞萤光分析、萤光显微镜检查、共聚焦激光扫描显微镜检查、激光扫描细胞术、亲和层析法、手动分批模式分离、电场悬浮、定序及其组合。
在一些实施例中,以在示例10中公开的示例性测定实验流程中所描述的方式获得多个序列读值140。在一些实施例中,采取多个步骤,以确定每个这样的读值代表所述生物样本中的所述无细胞核酸中的一独特的核酸片段。取决于所使用的定序方法,每个这样的独特核酸片段可通过多个序列读值的一数量来表示。在多个典型的例子中,使用诸如条形码的多重定序技术解决了对于所述无细胞核酸中的多个独特的核酸片段的多个序列读值的冗余性,因此,对于一特定等位基因的所述多个序列读值的数量代表在所述生物样本中的所述无细胞核酸中的映射在由所述个别的等位基因代表的物种的基因组的不同部分上的所述多个独特的核酸片段的数量,而不是映射至所述个别的等位基因的所述多个序列读值中的多个序列读值的实际原始总数量。参见kircher等人,2012,nucleicacidsresearch40,no.1e3,其通过引用并入本文,例如,关于条形码的公开。在一些实施例中,这样的映射仅允许完美的配对。在一些实施例中,这样的映射允许一些错误的配对。在一些实施例中,诸如包提(bowtie)2的一程序用于进行这样的映射。例如,参见langmead及salzberg,2012,natmethods9,pp.357-359,例如,关于这类映射的公开。
在一些实施例中,在方块202从一生物样本的无细胞核酸获得的所述多个第一序列读值包括十个以上的所述无细胞核酸的序列读值、一百个以上的所述无细胞核酸的序列读值、五百个以上的所述无细胞核酸的序列读值、一千个以上的所述无细胞核酸的序列读值、两千个以上的所述无细胞核酸的序列读值、介于两千五百个以上至五千个之间的所述无细胞核酸的序列读值,或五千个以上的所述无细胞核酸的序列读值。在一些实施例中,这些序列读值的每一个是所述无细胞核酸的一不同部分。在一些实施例中,所述多个第一序列读值中的一序列读值140是所述无细胞核酸的全部,或与所述多个第一序列读值中的另一序列读值相同的一部分。
参考图2a的方块210至216,所述多个第一序列读值140用于辨识对一第一变体集合142中的每个变体144的支持146,从而确定所述第一变体集合中的每个变体的一观察到的频率。在一些实施例中,在进行噪声建模、利用白细胞(wbc)的接合建模,及/或边缘变体的伪影(artifact)建模之后,从所述多个第一序列读值获得在所述第一变体集合142中的每个变体144,如在2018年11月27日提交的标题为“用于靶向定序的模型”的美国专利申请第16/201,912号所公开,其通过引用并入本文中。
参考图2b的方块219,在一些实施例中,将所述多个第一序列读值中的一个别的序列读值140视为支持所述第一变体集合142中的一第一变体144,这是在所述个别的序列读值(i)涵括或位在与所述第一变体相关的一基因组位置中,并且(ii)含有所述第一变体的全部或一部分时。将所述多个第一序列读值中的一个别的序列读值视为不支持所述第一变体集合中的所述第一变体,这是在所述个别的序列读值(i)涵括或位在与所述第一变体相关的一基因组位置中,并且(ii)不含有所述第一变体的全部或一部分时。例如,考虑到一第一变体与一特定的基因组位置相关的例子。对这些涵括或位在此特定的基因组位置中的序列读值进行评估,以确定它们是否支持所述变体。换言之,对这些独特地映射至此特定的基因组位置的序列读值进行评估,以确定它们是否支持所述变体。假如一序列读值涵括或位在一基因组位置中,并编码所述变体,则将所述序列读值视为支持所述变体。例如,在所述变体为一单核苷酸变异的例子中,将这些(i)涵括与此单核苷酸变异对应的所述基因组位置,且(ii)具有所述单核苷酸变异的序列读值视为支持所述变异。在另一示例中,在所述变异为比所述多个序列读值的一平均长度长的一插入的例子中,这些位在与此变异对应的所述基因组位置(例如,映射至此插入欲被结合的所述基因组的位点)中,且(ii)具有所述插入的全部或一部分的序列读值将被视为支持所述变异。
在一些实施例中,通过将所述多个第一序列读值中的每个序列读值140与一参考基因组中的一区域比对,使用所述多个第一序列读值140来辨识对一第一变体集合中的每个变体144的支持,从而确定所述序列读值是否含有一第一变体144的全部或一部分(方块214)。一序列读值140与一参考基因组中的一区域的所述比对涉及:基于多个序列之间的完全或部分同一性,将来自一个或多个序列读值140的多个序列与所述参考基因组的多个序列进行配对。多次的比对可手动或通过一计算机演算法来完成,多个示例包括分配作为伊卢米那基因组学分析管道的一部分的核苷酸数据的高效局部比对(eland)的计算机程序。一序列读值与所述参考基因组的所述比对可为一种100%的序列配对。在一些实施例中,一比对为低于100%的序列配对(例如,不完美的配对、部分配对、部分比对)。在一些实施例中,一比对包括一错误配对。在一些实施例中,一比对包括1、2、3、4或5个错误配对。在一些实施例中,多个这样的错误配对指示出并支持一第一变体集合中的一变体144。例如,在一变体144为位于所述基因组中的一特定位置处的一单核苷酸变体的例子中,预期含有所述变体的一序列读值与所述基因组的一比对在所述序列读值与位在所述基因组中的与所述单核苷酸变体相关的位置处的所述基因组之间具有错误配对。使用任一股可比对两个或更多个序列。在一些实施例中,将一核酸序列与另一核酸的反向互补序列进行比对。
在多个可替代的实施例中,通过将所述多个第一序列读值中的一序列读值140与多个变体的一查找表比对,使用所述多个第一序列读值140来辨识对一第一变体集合中的每个变体144的支持,从而确定所述序列读值是否含有一第一变体144的全部或一部分(方块214)。因此,在这类的例子中,不是使用每个序列读值140来找寻一受试者的整个基因组中的任何地方的比对,而是将每个序列读值140与一查找表中的所述多个序列的每一个进行比对,其中所述查找表中的每个这样的序列代表所述第一变体集合142中的一变体144。作为一示例,再次考虑到一变体为与所述基因组中的一特定位置相关的一单核苷酸变体的例子。在此例子中,对于所述变体,所述查找表将包括所述基因组的所述相关位置附近的所述基因组的所述序列的一部分。在一些例子中,此部分的大小可取决于用于产生所述多个序列读值140的定序方法的类型。作为一非限制的示例,位在与所述单核苷胺酸变体相关的所述基因组中的所述位置的3’侧侧翼(flank)的50个碱基,及位在与所述单核苷胺酸变体相关的所述基因组中的所述位置的5’侧侧翼的50个碱基用于代表所述查找表中的所述变体。在一些实施例中,如下文关于方块218所讨论,在一些例子中,所述变体为一些其他种类的变体,例如,与所述基因组中的一特定位置相关的一插入突变。在多个这样的例子中,在所述查找表中,所述变体通过足以与含有此插入突变的全部或一重要部分的一序列读值比对的所述基因组的一部分来表示。
在一些实施例中,通过使用一变体识别(calling)过程,例如单倍型识别仪(haplotypecaller),使用所述多个第一序列读值140来辨识对所述第一变体集合142中的每个变体144的支持146。例如,参见mckenna等人,2010,“基因组分析工具包:用于分析次世代dna定序数据的映射归约(mapreduce)框架”,genomeresearch20:1297-303;及vanderauwera,2013,“从fastq数据到高可信度变异识别:基因组分析工具包的最佳实践管道”,currentprotocolsinbioinformatics43:11.10.1-11.10.33,其中的每一个皆通过引用并入本文中。
在一些实施例中,所述多个第一序列读值140用于通过使用varscan来辨识对所述第一变体集合142中的每个变体144的支持146。例如,参见koboldt等人,2012,“varscan2:通过外显子组定序发现癌症中的体细胞突变及拷贝数改变”,genomeresearch,pmid:22300766;及koboldt等人,2009,“varscan:单独及合并样本的大规模平行定序中的变体检测”,bioinformatics25(17):2283-5,其中的每一个皆通过引用并入本文中。
在一些实施例中,所述多个第一序列读值140用于通过使用史翠尔卡(strelka)来辨识对所述第一变体集合142中的每个变体的支持146。例如,参见kim等人,2017,“strelka2:用于临床定序应用的快速又准确的变体识别”,biorxivdoi:10.1101/192872,其通过引用并入本文中。
在一些实施例中,所述多个第一序列读值140用于通过使用体细胞思尼普(somaticsniper)来辨识对所述第一变体集合142中的每个变体的支持146。例如,参见larson等人,2012,“somaticsniper:全基因组定序数据中的体细胞点突变的辨识”,bioinformatics28(3),pp.311-317,其中的每一个皆通过引用并入本文中。
在一些实施例中,根据示例11,所述多个第一序列读值140用于辨识对所述第一变体集合142中的每个变体的支持146。在一些实施例中,使用一个或多个方法,例如归一化、gc偏差的校正及/或由于pcr过度扩增引起的偏差的校正,来对所述多个序列读值140进行预先处理,以校正多个偏差或多个错误。
在一些实施例中,根据本公开收集到的多个序列读值的umi及终点位置用于定义多个可能的pcr重复体的多个封包(bag),其被分裂(collapsed)(从而获得一平均分裂覆盖率)并缝合(stitched)成多个高精确度的片段序列。因此,在多个这样的实施例中,对于多个序列读值所回报的“覆盖率”为这类封包的所述平均分裂覆盖率。在一些实施例中,多个候选变体通过使用一德布鲁因(debruijn)组装器来产生,并且通过经多个35岁以下未诊断出癌症的未吸烟参与者的一群体训练过的一噪声模型进行评分,其用于量测来自所述定序测定的技术变异。所述噪声模型提供基于对每个变体的所述支持来估计的一校准过的质量得分数,从而允许将所述多个候选变体过滤成为纯技术变体不太可能发生的一高质量的变体子集合。例如,在诸如art定序的靶向定序的例子中,本公开的一些实施例使用了用于辨识多个变体的所述噪声模型及启发式算法,如在2018年11月27提交的标题为“用于靶向定序的模型”的美国专利申请第16/201912号所公开。在全基因组定序的例子中,本公开的一些实施例使用在2019年3月13提交的标题为“辨识拷贝数异常”的美国专利申请第16/352,214号中公开的用于辨识多个变体的所述噪声模型及启发式算法。针对聚集在多个读值的多个末端附近且发生在多个样本的一子集合中的多个dna损伤伪影,进一步过滤多个候选变体。将估计具有60或更高的phred得分且不可能为技术伪影的多个变体视为一些实施例中的多个变体。将估计具有40或更高、45或更高、50或更高、55或更高、60或更高、65或更高,或者70或更高的phred得分,且不可能为技术伪影的多个变体视为一些实施例中的多个变体。
使用诸如甲基化的多个表观遗传特征作为多个变体。在一些实施例中,根据示例13,及如同通过引用并入本文中的2018年3月13日提交的标题为“甲基化片段异常检测”的美国专利申请第62/642,480号,通过确定一个或多个甲基化状态向量,使用所述多个第一序列读值140来辨识对所述第一变体集合142中的每个变体的支持146。在多个这样的实施例中,5-胞嘧啶甲基化发生在cpg背景。一种用于确定甲基化状态的方法是通过亚硫酸氢盐转化定序(bs-seq)。在bs-seq的情况下,未甲基化的胞嘧啶被转化成尿嘧啶碱基,其在定序中以胸腺嘧啶读出。因此,在一些实施例中,将诸如在一个或多个核苷酸位置的甲基化状态的一表观遗传模式使用作为确定一变体等位基因的一基础,其中针对所述变体等位基因确定ctdna分数。在一些实施例中,所述甲基化可包括一cpg位点的一甲基化指数、一区域中的多个cpg位点(例如,包括2个或更多个、3个或更多个、4个或更多个、5个或更多个,或6个或更多个cpg位点)的一甲基化密度、跨越一连续区域的多个cpg位点的一分布、对于在含有一个以上的cpg位点的一区域内的每个单独的cpg位点的一甲基化的模式或程度,及/或非cpg的甲基化。哺乳动物基因组中的“dna甲基化”可以指在cpg二核苷酸中的胞嘧啶的杂环的位置5添加一甲基(例如,以产生5-甲基胞嘧啶)。胞嘧啶的甲基化可在其他序列背景中的多个胞嘧啶中发生,例如,5’-chg-3’及5’-chh-3’,其中h为腺嘌呤、胞嘧啶或胸腺嘧啶。胞嘧啶甲基化也可为5-羟甲基胞嘧啶的形式。dna的甲基化可包括非胞嘧啶核苷酸的甲基化,例如n6-甲基腺嘌呤。在一些实施例中,将所述多个无细胞核酸片段进行处理,以使多个未甲基化的胞嘧啶转化为多个尿嘧啶。在一实施例中,所述方法使用一种dna的亚硫酸氢盐处理法,这是将所述多个未甲基化的胞嘧啶转化为多个尿嘧啶,而不会转化多个甲基化过的胞嘧啶。例如,将诸如ezdna甲基化tm-金色、ezdna甲基化tm-直接或ezdna甲基化tm-快速(lightning)试剂盒(可从立莫(zymo)研究公司(尔湾,加利福尼亚州)取得)的一商业试剂盒用于进行所述亚硫酸氢盐转化法。在另一实施例中,使用一酶促反应来完成多个未甲基化的胞嘧啶变为尿嘧啶的转化。例如,所述转化可使用一种用于将多个未甲基化的胞嘧啶转化为多个尿嘧啶的市售试剂盒,例如apobec-seq(新英格兰生物实验室(nebiolabs),伊普斯维奇,马萨诸塞州),或者通过使用在schutsky等人,2018,“使用dna脱氨酶对5-羟甲基胞嘧啶进行无损坏碱基分辨率的定序”,naturebiotechnology36,1083-1090,或liu等人,2019,“在碱基分辨率下对5-甲基胞嘧啶及5-羟甲基胞嘧啶进行无亚硫酸氢盐的直接侦测”,naturebiotechnology37,pp.424-429中公开的多个技术来进行。从所述多个转化过的无细胞核酸片段制备一定序文库。可选择地,所述定序文库中富集了多个无细胞核酸片段或多个基因组区域,其可使用多个杂交探针为细胞起源提供信息。所述多个杂交探针为多个短的寡核苷酸,所述多个短的寡核苷酸与多个特别指定的无细胞核酸片段或目标区域杂交,并富集这些片段或区域,以进行随后的定序及分析。在一些实施例中,多个杂交探针用于对为细胞起源提供信息的一组指定的cpg位点进行有针对性且高深度的分析。一旦制备完,就对所述定序文库或所述定序文库的一部分进行定序,以获得多个序列读值。在多个可替代的实施例中,如在示例12(wgbs;34倍)中对于所述ccga研究的描述,进行全基因组亚硫酸氢盐定序。
使用甲基化定序来查明多个变体。在一些实施例中,全基因组亚硫酸氢盐定序(wgsb)或靶向亚硫酸氢盐定序用于取得所述多个序列读值140。例如,在一些实施例中,使用示例12中所描述的ccga研究的一覆盖率为34倍的所述wgbs。在一些实施例中,这类(wgbs)的所述覆盖率为100倍或更低、50倍或更低,或介于30倍与200倍之间。在多个典型的实施例中,序列读值的多个独特分子指标(umis)及多个终点位置用于定义多个可能的pcr重复体,所述多个可能的pcr重复体被分裂成多个封包,以达成这种覆盖率统计。在一些实施例中,将来自每个封包的一单一的序列读值用于所公开的分析中。在一些实施中,此单一的序列读值为一共有(consensus)序列读值。在一些实施例中,此单一的序列读值为一封包中的任何序列读值。因此,以此种方式,100倍指的是覆盖每个等位基因位置的多个独特片段的数量,而不是覆盖每个等位基因位置的多个序列读值的数量,因为这样的多个序列读值可包括多个pcr重复体。来自所述多个分裂封包的这样的多个序列读值可用于侦测多个定序变异(例如,单核苷酸变体、插入、缺失)或多个拷贝数变异。在将多个序列读值用于辨识多个单核苷酸变体的一些实施例中,无法使用介于非癌症与癌症之间的c->t或t->c的多个变体,这是因为多个未甲基化的包嘧啶转化为多个脲嘧啶,其在定序中以胸腺嘧碇读出;例如,通过包括用于变体识别的一噪声模型中的一变体噪声过滤器。在一些实施例中,将所述噪声模型修改为包括一个或多个参数,以说明一序列读值的股起源(例如,所述读值是来自于原始目标分子的正向股或反向股)。可考虑到多个另外的因子,包括但不限于三核苷酸背景、所述变体的所述片段中的位置,及不同类型的其他协变量。在将多个序列读值用于辨识多个单核苷酸变体的一些实施例中,事实上,只要所述dna的亚硫酸氢盐处理法将所述多个未甲基化的胞嘧啶转化为多个脲嘧啶,而不转化多个甲基化过的胞嘧啶,就可使用介于非癌症与癌症之间的c->t或t->c的多个变体。例如,在将诸如ezdna甲基化tm-金色、ezdna甲基化tm-直接或ezdna甲基化tm-快速(lightning)试剂盒(可从立莫研究公司(尔湾,加利福尼亚州)取得)的一商业试剂盒,或者将schutsky等人,2018,“使用dna脱氨酶对5-羟甲基胞嘧啶进行无损坏碱基分辨率的定序”,naturebiotechnology36,1083-1090,或liu等人,2019,“在碱基分辨率下对5-甲基胞嘧啶及5-羟甲基胞嘧啶进行无亚硫酸氢盐的直接侦测”,naturebiotechnology37,pp.424-429中所公开的多个技术,用于进行所述亚硫酸氢盐转化法时,这是可以做到的。从所述多个转化过的无细胞核酸片段制备一定序文库。可选择地,所述定序文库中富集了多个无细胞核酸片段或多个基因组区域,其可使用多个杂交探针为细胞起源提供信息。所述多个杂交探针为多个短的寡核苷酸,所述多个短的寡核苷酸与多个特别指定的无细胞核酸片段或目标区域杂交,并富集这些片段或区域,以进行随后的定序及分析。在一些实施例中,多个杂交探针用于对为细胞起源提供信息的一组指定的cpg位点进行有针对性且高深度的分析。一旦制备完,就对所述定序文库或所述定序文库的一部分进行定序,以获得多个序列读值。在多个可替代的实施例中,如在示例12(wgbs;34倍)中对于所述ccga研究的描述,进行全基因组亚硫酸氢盐定序。
全基因组血浆测定法。在一些实施例中,所述受试者为人类,并且从所述生物样本取得的所述多个第一序列读值140为一全基因组血浆测定法的一部分。
在一些这样的实施例中,使用从一改良过的qiaamp循环核酸试剂盒(凯杰;日耳曼敦,马里兰州)的两管血浆提取出的cfdna来实行所述全基因组血浆测定法。来自血沉棕黄层的基因组dna(gdna)是使用凯杰的dneasy血液与组织试剂盒提取出,并使用纳米微滴(nanodrop)(赛默飞世尔;沃尔瑟姆,麻萨诸塞州)进行定量。提取出的gdna使用科菲乐斯(covaris)的e220超声破碎仪(沃本,麻萨诸塞州)进行片段化,并使用阿让库尔(agencourt)ampurexp磁珠(贝克曼库尔特(beckmancoulter);贝弗利,麻萨诸塞州)进行尺寸筛选。血浆的cfdna(高达75纳克)及血沉棕黄层的gdna(75纳克)用于次世代定序(ngs)文库的建构。衔接子(adapter)包括一组218个独特分子指标(umi)序列,以减少测定及定序的错误。使用耶库克利儿(accuclear)超高敏感度dsdna定量试剂盒(拜欧廷(biotium);弗里蒙特,加利福尼亚州)来稀释及量化多个经扩增的文库的一小部分(25微升中的4微升)。剩余部分用在一靶向定序的实验流程中(参见下文)。将三个或四个经稀释的文库归一化、合并、聚集在一流动槽中,并且在伊卢米那的hiseqx(30倍)上进行定序。
将所述多个序列读值140与整个人类基因组进行比较,以辨识多个变体。在一些实施例中,从所述生物样本取得的所述多个第一序列读值140具有对一目标基因小组的至少30倍的覆盖率、对一目标基因小组的至少40倍的覆盖率、对一目标基因小组的至少50倍的覆盖率、对一目标基因小组的至少60倍的覆盖率,或对一目标基因小组的至少70倍的覆盖率。在一些这样的实施例中,所述目标基因小组在介于450至550个基因之间。在一些实施例中,所述目标基因小组在500±5个基因的范围内、在500±10个基因的范围内,或在500±25个基因的范围内。在一些实施例中,所述全基因组测定血浆寻找所述基因组中的多个体细胞拷贝数的改变(scna)或多个片段化的特征。
靶向血浆测定法。在一些实施例中,所述受试者为人类,并且从所述生物样本取得的所述多个第一序列读值140为一靶向血浆测定法的一部分。
在一些这样的实施例中,作为示例12中所公开的art测定法的一部分,所述多个经扩增的文库(参见上文的全基因组血浆测定法)用于利用一针对507个癌症相关基因的小组进行靶向富集。每个文库最多3.5微克经历基于杂交的捕捉。使用耶库克利儿超高敏感度dsdna定量试剂盒来量化所述多个经富集的文库。将三个或四个经富集的文库归一化、合并、聚集在一流动槽中,并且在伊卢米那的hiseqx(150-bp成对末端定序,60,000倍)上进行定序。
将以此方式获取的所述多个序列读值140与所述靶向血浆测定法的一目标基因小组进行比较,以辨识多个变体。在一些这样的实施例中,所述目标基因小组在介于450至550个基因之间。在一些实施例中,所述目标基因小组在500±5个基因的范围内、在500±10个基因的范围内,或在500±25个基因的范围内。在一些实施例中,从所述生物样本取得的所述多个第一序列读值140具有对此目标基因小组的至少50,000倍的覆盖率、对此目标基因小组的至少55,000倍的覆盖率、对此目标基因小组的至少60,000倍的覆盖率,或对此目标基因小组的至少70,000倍的覆盖率。在一些这样的实施例中,所述靶向血浆测定法寻找在所述目标基因小组中的多个单核苷酸变体、在所述目标基因小组中的多个插入、在所述目标基因小组中的多个缺失、在所述目标基因小组中的多个体细胞拷贝数的改变(scnas)、多个异常的甲基化模式,或影响所述目标基因小组的重组。
靶向白细胞测定法。在一些实施例中,所述受试者为人类,并且从所述生物样本取得的所述多个第一序列读值140为一靶向白细胞测定法的一部分。意即,所述生物样本为来自所述受试者的多个白细胞,并将所述多个序列读值140与所述靶向白细胞测定法的一目标基因小组进行比较,以辨识多个变体。在一些这样的实施例中,所述目标基因小组在介于450至550个基因之间。在一些实施例中,所述目标基因小组在500±5个基因的范围内、在500±10个基因的范围内,或在500±25个基因的范围内。在一些实施例中,从所述生物样本取得的所述多个第一序列读值140具有对此目标基因小组的至少50,000倍的覆盖率、对此目标基因小组的至少55,000倍的覆盖率、对此目标基因小组的至少60,000倍的覆盖率,或对此目标基因小组的至少70,000倍的覆盖率。在一些这样的实施例中,所述靶向白细胞测定法寻找在所述目标基因小组中的多个单核苷酸变体、在所述目标基因小组中的多个插入、在所述目标基因小组中的多个缺失,或在所述目标基因小组中的多个体细胞拷贝数的改变(scnas)。
全基因组白细胞测定法。在一些实施例中,所述受试者为人类,并且从所述生物样本取得的所述多个第一序列读值140为一全基因组白细胞测定法的一部分。意即,所述生物样本为来自所述受试者的多个白细胞,并将所述多个序列读值140与整个人类基因组进行比较,以辨识多个变体。在一些实施例,从所述生物样本取得的所述多个第一序列读值140具有对一目标基因小组的至少30倍的覆盖率、对一目标基因小组的至少40倍的覆盖率、对一目标基因小组的至少50倍的覆盖率、对一目标基因小组的至少60倍的覆盖率,或对一目标基因小组的至少70倍的覆盖率。在一些这样的实施例中,所述目标基因小组在介于450至550个基因之间。在一些实施例中,所述目标基因小组在500±5个基因的范围内、在500±10个基因的范围内,或在500±25个基因的范围内。在一些实施例中,所述全基因组白细胞测定法寻找在所述基因组中的多个体细胞拷贝数的改变(scnas)或多个片段特征。
全基因组亚硫酸氢盐定序测定法。在一些实施例中,所述受试者为人类,且所述多个第一序列读值140通过进行亚硫酸氢盐定序来获得,并在一全基因组(genome-wide)基础上对多个变体进行评估。在一些实施例中,所述全基因组亚硫酸氢盐定序测定法寻找在所述基因组中的多个甲基化模式的变体。例如,参见示例13。还可参见2018年3月13日提交的标题为“甲基化片段异常检测”的美国专利申请第62/642,480号,其通过引用并入本文中。
在一些实施例中,参考图2a的方块216,通过将所述多个第一序列读值中的每个序列读值140与一查找表中的每个条目(entry)进行比对,使用所述多个第一序列读值140来辨识对一第一变体集合中的每个变体144的支持146,其中所述查找表中的每个条目代表一基因组(例如,一参考基因组)的一不同部分。在一些例子中,将这样的实施例用于利用所述基因组中的多个热点来填充(populate)所述查找表。因此,不是通过搜寻整个基因组中的比对来比对每个序列读值,而是仅将每个序列读值与所述基因组的那些与感兴趣的病况相关的部分,例如所述基因组内的多个基因,进行比对。例如,考虑到一特定基因的突变与一临床病况相关的例子。在此例子中,根据方块216的实施例,所述基因的所述基因组序列可被包括作为所述查找表中的一条目,并且可将多个序列读值140与此条目进行比对,以辨识对于一变体对上所述基因的支持。在此例子的一变体中,所述基因的每个已知的突变可被列表作为所述查找表中的一单独的条目,并且可将所述多个第一序列读值中的每个序列读值140与这些单独的条目中的每一个进行比对,以确定所述序列读值与所述多个基因的所述多个突变的其中一个之间是否有配对,从而辨识对所述变体集合中的一变体144的支持。
在又另一个示例中,仅有在一特定受试者的一异常组织(例如肿瘤)中发现的这些变体(及这类变体附近足够的基因组序列)被包括在所述查找表中。以此种方式,与其中将所述多个序列读值与一参考基因组的整体进行比对,以辨识对此类变体的支持的多个实施例相比,大大加快了与所述肿瘤中的所述多个变体的其中一个配对,从而辨识对所述变体的支持的所述辨识多个序列读值126的过程。例如,考虑到对一受试者的一特定肿瘤进行的定序辨识出三个变体的例子。在此例子中,所述三个变体被提供作为所述查找表中的多个单独的条目,并将所述多个第一序列读值中的所述多个序列读值140的每一个独立地与所述查找表中的所述多个条目的每一个进行比对,以确定它们是否与所述多个变体的其中一个进行比对,从而支持所述变体。
在一些实施例中,所述查找表由一单一的条目组成,其中所述单一的条目为已经在一受试者的一异常组织中辨识到的一变体。在一些实施例中,所述查找表由两个条目组成,其中每个条目代表已经在一受试者的一异常组织中辨识到的一变体。在一些实施例中,所述查找表由三个条目组成,其中每个条目代表已经在一受试者的一异常组织中辨识到的一变体。在一些实施例中,所述查找表由介于三至十个之间的条目组成,其中每个条目代表已经在一受试者的一异常组织中辨识到的一变体。
在一些实施例中,所述查找表包括介于两个至一千个之间的条目,其中每个条目代表所述人类基因组中的一不同的基因。
参考图2a的方块218,在一些实施例中,所述第一变体集合142中的一变体144为与一预定的基因组位置相关的一单核苷酸变体、与一预定的基因组位置相关的一插入突变、与一预定的基因组位置相关的一缺失突变、一体细胞拷贝数的改变、与一预定的基因组位点相关的一核酸重组,或与一预定的基因组位置相关的一异常的甲基化模式。通过示例的方式,在一些实施例中,一变体144为所述基因组中的一特定基因的一体细胞突变,因此与所述基因组中的所述特定基因的所述基因组位置相关。
在一些实施例中所述变体集合142包括一个以上的变体类型。例如,在一些实施例中,所述变体集合142包括与一基因组位置相关的一单核苷酸变体及与一基因组中的另一个基因组位置相关的一缺失突变。
在一些实施例中,一变体144为体细胞突变的任何形式。
在一些实施例中,所述第一变体集合中的所述多个变体144的每一个也被发现在所述参考集合128中。在一些实施例中,在所述变体集合142中的多个变体144与所述参考集合128中的多个变体130之间具有一对一的对应性。在多个这样的实施例中,所述变体集合142包括对所述受试者的所述生物样本(例如血液)中的所述多个样本的所述辨识过的支持146,而所述参考集合128包括在所述受试者的所述异常组织(例如肿瘤)中的这样的多个变体的所述参考频率132。
在一些实施例中,所述第一变体集合142是由对于位在所述受试者的所述基因组中的一单一位点处的一单一的遗传变异的一单一变体144组成(方块220)。例如,考虑到在一特定的基因中以来自一受试者的所述异常组织(例如肿瘤)的映射到此特定基因上的所述多个序列读值的一百分比发现一特定的单一核苷酸变体的例子。在此例子中,所述变体集合142也将包括所述特定的单一核苷酸变体,及在从所述受试者的所述生物样本(例如血液)获得的所述多个第一序列读值中发现的任何对于在所述特定基因中的此特定的单一核苷酸变体所辨识到的支持146。
在一些实施例中,所述第一变体集合142是由对于位在所述受试者的所述基因组中的一第一位点处的一第一遗传变异的一第一变体144-1及对于位在所述受试者的所述基因组中的一第二位点处的一第二遗传变异的一第二变体144-2组成(方块222)。例如,考虑到一例子,其中在一第一基因中以一些可估计的百分比(例如,大于百分之一、大于百分之二、大于百分之五)或一受试者的一异常组织(例如肿瘤)的映射到所述第一基因上的所述多个序列读值的数量,来发现一第一变体;以及在一第二基因中以一些可估计的百分比或所述异常组织的映射到所述第二基因上的所述多个序列读值的数量,来发现一第二变体。在此例子中,所述变体集合142将包括所述第一变体,及在从所述受试者的所述生物样本(例如血液)获得的所述多个第一序列读值中发现的任何对于所述第一变体所辨识到的支持146。所述变体集合142也将包括所述第二变体,及在从所述生物样本获得的所述多个第一序列读值中发现的任何对于所述第二基因中的所述第二变体所辨识到的支持146。
在一些实施例中,当来自所述异常组织的至少一序列读值支持一变体时,所述变体被包括在所述参考集合中。在多个这样的实施例中,当所述序列读值(i)映射到与一变体相关的一基因组位置上,并(ii)包括所述变体时,来自所述异常组织的一序列读值支持所述变体。在一些实施例中,当来自所述异常组织的至少两个序列读值支持一变体时,所述变体被包括在所述参考集合中。在一些实施例中,当来自所述异常组织的至少2个序列读值、至少5个序列读值、至少10个序列读值、至少100个序列读值、至少200个序列读值,或至少1000个序列读值支持一变体时,所述变体被包括在所述参考集合中。
在一些实施例中,所述第一变体集合142是由对于位在所述受试者的所述基因组中的一第一位点处的一第一遗传变异的一第一变体144-1、对于位在所述受试者的所述基因组中的一第二位点处的一第二遗传变异的一第二变体144-2,及对于位在所述受试者的所述基因组中的一第三位点处的一第三遗传变异的一第三变体144-1所组成(方块224)。例如,考虑到一例子,其中在一第一基因中以一些可估计的百分比或一受试者的一异常组织(例如肿瘤)的包括所述第一基因的所述多个序列读值的数量,来发现一第一变体;在一第二基因中以一些可估计的百分比或所述异常组织的包括所述第二基因的所述多个序列读值的数量,来发现一第二变体;以及在一第三基因中以一些可估计的百分比或所述异常组织的包括所述第三基因的所述多个序列读值的数量,来发现一第三变体。在此例子中,所述变体集合142将包括所述第一变体,及在从所述受试者的所述生物样本(例如血液)获得的所述多个第一序列读值中发现的任何对于所述第一变体所辨识到的支持146。所述变体集合142也将包括所述第二变体,及在从所述生物样本获得的所述多个第一序列读值中发现的任何对于所述第三基因中的所述第三变体所辨识到的支持146。
在一些实施例中,所述第一变体集合142是由介于2至20个之间的变体组成,其中所述第一变体集合中的每个变体为(代表)所述受试者的所述基因组中的一不同位点处的一不同的遗传变异(方块226)。在一些实施例中,所述第一变体集合142是由介于2至20个之间的变体组成,其中所述第一变体集合中的每个变体为(代表)所述受试者的所述基因组中的一不同的遗传变异(方块226)。在一些实施例中,所述第一变体中的每个个别的变体也以一可估计的百分比(例如,高于百分之一、高于百分之二、高于百分之五)或一受试者的一异常组织(例如肿瘤)的映射至所述个别的变体的所述基因组位置的所述多个序列读值的数量来被发现。在一些实施例中,所述第一变体集合142是由介于1至10个之间的变体组成,其中所述第一变体集合中的每个变体为(代表)所述受试者的所述基因组中的(可选择地位在一不同位点处的)一不同的遗传变异。在一些实施例中,所述第一变体集合142是由介于1至100个之间的变体组成,其中所述第一变体集合中的每个变体为(代表)所述受试者的所述基因组中的(可选择地位在一不同位点处的)一不同的遗传变异。在一些实施例中,所述第一变体集合142是由介于2至100个之间的变体组成,其中所述第一变体集合中的每个变体为(代表)所述受试者的所述基因组中的(可选择地位在一不同位点处的)一不同的遗传变异。在一些实施例中,所述第一变体集合142是由介于1至1000个之间的变体组成,其中所述第一变体集合中的每个变体为(代表)所述受试者的所述基因组中的(可选择地位在一不同位点处的)一不同的遗传变异。
在一些实施例中,所述变体集合中的一第一变体及一第二变体与一受试者的所述基因组中的相同位点相关。例如,所述第一及第二变体可代表相同基因的两个不同的异常等位基因。
对于所述第一变体集合中的每个个别的变体,获得在一第一参考集合中对于所述个别的变体的一对应的参考频率,其中所述第一参考集合中的每个对应的参考频率是针对从所述受试者获得的一第一异常实体(solid)组织样本中的一个别的变体。参考图2b的方块228,在多个公开的方法中,将所述第一变体集合142中的每个个别的变体144的所述观察到的频率(例如,支持146)与一第一参考集合128中对于所述个别的变体的一对应的参考频率132进行比较。所述第一参考集合128中的每个对应的参考频率132为从所述受试者获得的一第一异常组织样本中的一个别的变体130的一频率。
参考图2b的方块230,在一些实施例中,所述第一异常组织样本为一肿瘤样本,或所述肿瘤样本的一小部分。在一些实施例中,所述第一异常组织样本为肾上腺皮质癌、儿童肾上腺皮质癌、aids相关癌症的肿瘤、卡波西肉瘤、肛门癌相关的肿瘤、阑尾癌相关的肿瘤、星形细胞瘤、儿童(脑癌)肿瘤、非典型畸胎/类横纹肌细胞瘤、中枢神经系统(脑癌)肿瘤、皮肤的基底细胞癌、胆管癌(bileductcancer)相关的肿瘤、膀胱癌肿瘤、儿童膀胱癌肿瘤、骨癌(例如,尤文氏肉瘤及骨肉瘤及恶性纤维组织细胞瘤)组织、脑瘤、乳癌组织、儿童乳癌组织、儿童支气管肿瘤、伯基特淋巴瘤组织、类癌肿瘤(胃肠道)、儿童类癌肿瘤、原发性未知癌、儿童原发性未知癌、儿童心脏(心)肿瘤、中枢神经系统(例如,如儿童非典型畸胎/类横纹肌的脑癌)肿瘤、儿童胚胎肿瘤、儿童生殖细胞瘤、子宫颈癌组织、儿童子宫颈癌组织、胆管癌(cholangiocarcinoma)组织、儿童脊索瘤组织、慢性骨髓增生性肿瘤、结肠直肠癌肿瘤、儿童结肠直肠癌肿瘤、儿童颅咽管瘤组织、原位乳管癌(dcis)、儿童胚胎肿瘤、子宫内膜癌(子宫癌)组织、儿童室管膜瘤组织、食道癌组织、儿童食道癌组织、嗅神经母细胞瘤(头颈癌)组织、儿童颅外生殖细胞瘤、生殖腺外生殖细胞瘤、眼癌组织、眼内黑色素瘤、视网膜母细胞瘤、输卵管癌组织、胆囊癌组织、胃(胃(stomach))癌组织、儿童胃(胃(stomach))癌组织、胃肠道类癌肿瘤、胃肠道间质瘤(gist)、儿童胃肠道间质瘤、生殖细胞瘤(例如,儿童中枢神经生殖细胞瘤、儿童颅外生殖细胞瘤、生殖腺外生殖细胞瘤、卵巢生殖细胞瘤,或睪丸癌组织)、头颈癌组织、儿童心脏肿瘤、肝细胞癌(hcc)组织、胰岛细胞瘤(胰腺神经内分泌肿瘤)、肾脏或肾细胞癌(rcc)组织、喉癌组织、白血病、肝癌组织、肺癌(非小细胞及小细胞)组织、儿童肺癌组织、男性乳腺癌组织、骨头的恶性纤维组织细胞瘤与骨肉瘤、黑色素瘤、儿童黑色素瘤、眼内黑色素瘤、儿童眼内黑色素瘤、默克尔细胞癌、恶性间皮瘤、儿童间皮瘤、转移性癌组织、具有潜藏原发性的转移性鳞状颈部癌组织、具有nut基因改变的中线道癌、口腔(mouth)癌(头颈癌)组织、多发性内分泌腺瘤综合症组织、多发性骨髓瘤/浆细胞瘤、骨髓增生异常综合症组织、骨髓增生异常/骨髓增生性肿瘤、慢性骨髓增生性肿瘤、鼻腔与鼻旁窦癌组织、鼻咽癌(npc)组织、神经母细胞瘤组织、非小细胞肺癌组织、口腔(oral)癌组织、唇部与口腔癌及口咽癌组织、骨肉瘤与骨头的恶性纤维组织细胞瘤组织、卵巢癌组织、儿童卵巢癌组织、胰腺癌组织、儿童胰腺癌组织、乳头状瘤症(儿童喉头)组织、副神经节瘤组织、儿童副神经节瘤组织、鼻旁窦和鼻腔癌组织、副甲状腺癌组织、阴茎癌组织、咽癌组织、嗜铬细胞瘤组织、儿童嗜铬细胞瘤组织、垂体腺瘤、浆细胞瘤/多发性骨髓瘤、胸膜肺母细胞瘤、原发性中枢神经系统(cns)淋巴瘤、原发性腹膜癌组织、前列腺癌组织、直肠癌组织、视网膜母细胞瘤、儿童横纹肌肉瘤、唾液腺癌组织、肉瘤(例如,儿童血管瘤、骨肉瘤、子宫肉瘤等)、塞泽里(sézary)综合症(淋巴瘤)组织、皮肤癌组织、儿童皮肤癌组织、小细胞肺癌组织、小肠癌组织、皮肤的鳞状细胞癌、具有潜藏原发性的鳞状颈部癌、皮肤t细胞淋巴瘤、睪丸癌组织、儿童睪丸癌组织、喉癌(例如,鼻咽癌、口咽癌、下咽癌)组织、胸腺瘤或胸腺癌、甲状腺癌组织、肾盂与输尿管组织的移行细胞癌、原发性未知癌组织、输尿管或肾盂组织、移行细胞(肾脏(肾细胞))癌组织、尿道癌组织、子宫内膜子宫癌组织、子宫肉瘤组织、阴道癌组织、儿童阴道癌组织、血管瘤、外阴癌组织、威尔姆氏肿瘤或其他儿童肾脏肿瘤。
在一些实施例中,来自所述第一异常组织样本的所述多个序列读值为多个福尔马林固定与石蜡包埋(ffpe)的肿瘤组织切片,所述多个ffpe的肿瘤组织切片被刮取并送至哈德森阿尔法(hudsonalpha)生物技术研究所(亨茨维尔,阿拉巴马州汉茨维尔)的基因组服务实验室,在所述实验室中,从所述多个刮取物中提取dna,并将所述dna转化为多个ngs文库,以便在伊卢米那的hiseqx(30倍)上进行全基因组定序。对于每个组织刮取物,将一管对应的血沉棕黄层运送至哈德森阿尔法,以便进行提取、文库制备及在伊卢米那的hiseqx(60倍)上的全基因组定序。接着,根据本公开分析定序数据。
参考方块234至240,在一些实施例中,从由所述第一异常组织样本取得的多个第二序列读值(多个参考序列读值)126中获得所述第一参考集合128中的每个变体130的所述频率(参考频率132)(方块234)。在一些实施例中,一个别的变体的频率为所述变体所属的受试者的所述第一异常组织中多个细胞的比例的一量测值。例如,参见lu等人,2015“个体中的体细胞突变的等位基因频率揭示了癌症相关基因的特征”,actabiochimbiophyssin.47(8),657-680,其通过引用并入本文中,以便根据一些实施例将关于确定一异常组织中的多个体细胞变体的频率的步骤公开。
在一些实施例中,通过首先辨识可能具有一个别的变体130的所述多个序列读值来确定所述个别的变体130的频率。例如,假如所述个别的变体为一单核苷酸变体,则辨识出来自所述第一异常组织的映射至对应于此个别的变体的所述基因组位置的所述多个变体。接着,包括有所述变体的这些被辨识出的序列读值的比例代表所述个别的变体的所述频率。因此,假如有200个来自所述异常组织的映射至与所述变体相关的所述基因组位置的多个序列读值,且这些序列读值中的50个包括对于所述变体的等位基因,而剩余的150个序列读值具有一野生型等位基因,而不是对于所述变体的所述等位基因,则对于所述个别的变体130的所述频率为百分之25。在一些这样的实施例中,采取多个步骤以确保每个这样的序列读值代表所述异常组织中的一独特的核酸片段。取决于所使用的定序方法,每个这类独特的核酸片段可由多个序列读值的数量来表示。在多个典型的例子中,使用诸如条形码的多重定序技术解决了在所述异常的实体组织样本中的多个独特的核酸片段的多个序列读值的冗余性,因此,对于一特定等位基因的所述多个序列读值的数量代表在所述异常的实体组织样本中的映射在由所述个别的等位基因代表的物种的基因组的不同部分上的多个独特的核酸片段的数量,而不是映射至所述个别的等位基因的所述多个序列读值中的多个序列读值的实际原始总数量。参见kircher等人,2012,nucleicacidsresearch40,no.1e3,其通过引用并入本文,例如,关于条形码的公开。
在一些实施例中,从所述异常组织取得超过1000、2000、3000、4000、5000、10,000、20,000、100,000或一百万个参考序列读值126。在一些实施例中,从所述异常组织取得的所述多个参考序列读值126为所述受试者的所述基因组的至少百分之二、至少百分之五、至少百分之十、至少百分之二十、至少百分之三十、至少百分之四十、至少百分之五十、至少百分之六十、至少百分之七十、至少百分之八十、至少百分之九十、至少百分之九十八,或至少百分之九十九提供1倍或更高的、2倍或更高的、5倍或更高的、10倍或更高的,或者50倍或更高的一覆盖率。在一些实施例中,从所述异常组织取得的所述多个参考序列读值126为所述受试者的所述基因组的至少三个基因、至少五个基因、至少十个基因、至少二十个基因、至少三十个基因、至少四十个基因、至少六十个基因、至少七十个基因、至少八十个基因、至少200个基因、至少300个基因、至少400个基因、至少500个基因,或至少1000个基因提供1倍或更高的、2倍或更高的、5倍或更高的、10倍或更高的,或者50倍或更高的一覆盖率。
在一些实施例中,对照一变体候选者小组来分析(比对)从所述第一异常组织取得的所述多个参考序列读值126。例如,在一些实施例中,所述变体候选者小组包括对于所述受试者的至少三个基因、至少五个基因、至少十个基因、至少二十个基因、至少三十个基因、至少四十个基因、至少五十个基因、至少六十个基因、至少七十个基因、至少八十个基因、至少九十个基因、至少200个基因、至少300个基因、至少400个基因、至少500个基因,或至少1000个基因的多个变体候选者的多个序列。为了进行这样的分析,一特定的参考序列读值126与所述变体候选者小组中的一变体候选者的所述序列的比对涉及:将所述参考序列读值126的所述序列与所述变体候选者的所述序列进行配对,以查看在所述多个序列之间是否存在完全或部分同一性。多次这样的比对(分析)可手动或通过一计算机演算法来完成,多个示例包括分配作为伊卢米那基因组学分析管道的一部分的核苷酸数据的高效局部比对(eland)的计算机程序。在一些实施例中,当100%的一参考序列读值126与所述变体候选者的所述序列的一对应部分配对时,将所述参考序列读值126与所述变体候选者小组中的所述变体候选者的所述序列视为匹配的。在一些实施例中,当100%的所述变体候选者126的所述序列与一参考序列读值126的所述序列的一对应部分配对时,将所述参考序列读值126与所述变体候选者小组中的所述变体候选者的所述序列视为匹配的。在一些实施例中,一比对为低于100%的序列配对(例如,不完美的配对、部分配对、部分比对)。在一些实施例中,一比对包括一错误配对。在一些实施例中,一比对包括1、2、3、4或5个错误配对。使用任一股可比对两个或更多个序列。在一些实施例中,将一核酸序列与另一核酸的反向互补序列进行比对。
参考图2c的方块244至246,在一些实施例中,从所述第一异常组织样本取得的所述多个参考序列读值126代表对于所述个别的细胞的所述全基因组数据。在一些这样的实施例中,从所述第一异常组织样本取得的所述多个参考序列读值126的一平均覆盖率为所述受试者的整个基因组的至少1倍、2倍、3倍、4倍、5倍、6倍、7倍、8倍、9倍、10倍、至少20倍、至少30倍,或至少40倍。在一些实施例中,整个所述第一参考集合128的所述多个第二序列读值的所述平均覆盖率为至少10倍、至少100倍或至少2000倍。
参考图2c的方块248,在一些实施例中,将在所述多个第二序列读值中的一个别的序列读值126视为支持所述参考集合128中的一第一变体130,这是在所述个别的序列读值(i)映射至与所述第一变体相关的所述基因组的一部分,且(ii)所述个别的序列读值126含有所述第一变体130的全部或一部分时。将所述多个第二序列读值中的一个别的序列读值126视为不支持所述参考集合128中的一第一变体130,这是在所述个别的序列读值126(i)映射至与所述第一变体130相关的所述基因组的一部分(对应于所述第一变体的基因组位置),且(ii)不含有所述第一变体130时。例如,考虑到所述变体为与一预定的基因组位置相关的一单核苷酸变体的例子。在此例子中,当所述第一变体映射到所述预定的基因组位置,并含有此单核苷酸变体时,一序列读值126支持所述变体。在实践上,为了确定所述第一变体是否含有此单核苷酸变体,所述序列读值还包括位在所述受试者的物种的基因组中的此单核苷酸变体两侧的5’及3’序列,以便将所述序列读值映射至所述基因组,从而确定其是否映射至对应于所述变体的所述基因组位置。接下来,考虑到所述变体为插入至一特定基因中的38个插入碱基的例子。当一序列读值含有所述38个插入碱基(以及位在所述特定基因中的此插入两侧的5’及3’区域)时,所述序列读值将支持此变体。在一些例子中,当所述序列读值包括的变体少于所述变体的全部时,所述序列读值仍可能支持此变体。例如,所述序列读值可终止在所述38个插入碱基中的25个碱基。然而,位在此变体两侧的所述序列读值的区域可与所述基因及所述插入的前25个碱基配对,因此,可将所述序列读值视为支持所述变体。进一步地,支持所述参考集合128中的一第一变体130的所述多个第二序列读值中的多个序列读值126的一数量对上不支持所述第一变体130的所述多个第二序列读值中的多个序列读值126的一数量,确定了所述第一变体130的所述观察到的频率(支持132)。例如,再次考虑到一变体与所述基因组中的一特定的基因组位置相关的例子,其中来自所述第一异常样本的所述多个第二参考序列读值126是由1000个序列读值组成,然而这1000个序列读值中仅有100个覆盖(映射至、有关)与所述变异相关的所述基因组位置。对覆盖与所述变体相关的所述基因组位置的所述100个序列读值进行分析,以查看它们是否支持或不支持所述变体。将所述100个序列读值中含有所述变体的全部或一部分的这些序列读值视为支持所述变体,并将所述100个序列读值中不含有所述变体的这些序列读值视为不支持所述变体。其他的900个序列读值不符合支持或不支持所述变体,因为它们没有覆盖所讨论的与所述变体相关的所述基因组位置。进一步地,考虑到一例子,其中所述100个序列读值中有3个含有所述变体的全部或一部分,并将其视为支持所述变体,而所述100个序列读值中的其余97个序列读值不含有所述变体,因此不支持所述变体。根据此实施例,在此示例中,对于所述第一变体的所述观察到的频率(支持146)为3/100或百分之三。
参考图2d的方块256,继续进行所述方法:通过对照所述第一异常实体组织的所述第一参考集合128中的所述个别的变体的所述观察到的频率,来评估所述第一变体集合142中的每个个别的变体的所述观察到的频率,从而确定在所述受试者的所述液体生物样本的无细胞核酸中的一第一肿瘤分数。
在一些实施例中,当所述第一变体集合142中的每个变体144的所述观察到的频率(支持14)满足一第一阈值时,通过将一受试者视为患有一第一病况来使用此第一肿瘤分数对所述受试者进行分类,其中所述第一阈值是通过所述第一异常组织样本的所述第一参考集合128中的每个变体130的一频率来确定。例如,参考图2d的方块258,在一些实施例中,所述方块256的评估步骤包括:从所述多个第一序列读值的所述第一变体集合142中的每个变体144的所述观察到的频率(支持146)计算出在所述受试者的所述cfdna中的一单一的估计出的ctdna分数。进一步地,在多个这样的实施例中,所述第一阈值为所述受试者的所述cfdna中的从对于所述第一异常组织样本的所述参考集合128中的每个变体130的所述频率(参考频率132)确定的一单一的预期ctdna分数。例如,考虑到在所述方块256的评估步骤中比较一单一变体的例子。因此,将在来自所述生物样本(例如血液)的所述变体集合142中的对此变体的所述支持146与在对于所述异常组织的所述参考集合128中的相同变体的所述参考频率132进行比较。假定所述无细胞核酸中的所述单一变体的唯一来源来自所述异常组织。因此,在此假设下,根据对所述变体集合中的所述变体的所述支持146与对于在所述参考集合中的相同变体的所述参考频率132的比率,计算所述单一的估计出的ctdna分数。例如,假如对所述变体的所述支持146为所述变体集合142中的100个序列读值中的3个,且相同变体的所述参考频率132在所述参考集合128中为0.10,则所述单一的估计出的ctdna分数为(3/100)/(0.10)或0.3。
接下来,考虑到一例子,其中在方块256的所述评估步骤中比较两个变体,即一第一变体及一第二变体。将对来自所述生物样本(例如血液)的所述变体集合142中的所述第一变体的所述支持146与在对于所述异常组织的所述参考集合128中的相同变体的所述参考频率132进行比较。同样地,将对来自所述生物样本的所述变体集合142中的所述第二变体的所述支持146与在所述参考集合128中的相同变体的所述参考频率132进行比较。假定所述无细胞核酸中的所述第一及第二变体的唯一来源来自所述异常组织。因此,在此假设下,对于所述第一变体的一比率是根据对所述变体集合142中的所述第一变体的所述支持146对上针对在所述参考集合中的所述第一变体的所述参考频率132来计算出。例如,假如对所述第一变体的所述支持146为所述变体集合142中的100个序列读值中的3个,且所述第一变体的所述参考频率132在所述参考集合128中为0.10,则对于所述第一变体的所述比率为(3/100)/(0.10)或0.3。进一步地,对于所述第二变体的一比率是根据对所述变体集合142中的所述第二变体的所述支持146对上针对在所述参考集合中的所述第二变体的所述参考频率132来计算出。例如,假如对所述第二变体的所述支持146为所述变体集合142中的85个序列读值中的5个,且所述第一变体的所述参考频率132在所述参考集合128中为0.12,则对于所述第二变体的所述比率为(5/85)/(0.12)或0.49。
在一些实施例中,在方块256的所述评估步骤中比较一个以上的变体,并且对于每个这样的变体计算在对所述生物样本中的每个变体的所述观察到的支持与所述变体集合中的相同变体的所述频率之间的一比率。例如,在一些实施例中,在方块256的所述评估步骤中比较两个以上的变体。在多个这样的实施例中,在对于每个这样的变体计算在对所述生物样本中的每个变体的所述观察到的支持与所述变体集合中的相同变体的所述频率之间的一比率的意义上扩展了上文的多个示例。实际上,在一些实施例中,在方块228的所述比较步骤中比较介于2至200个之间的变体。在一些实施例中,在方块256的所述评估步骤中比较25、50、100、200、300、400、500、1000、2000或5000个以上的变体。
因此,从所述第一异常组织样本观察到多个体细胞变体k的一数量,其中k为一正整数(例如,2、3、大于20、大于100、大于200等)。这可被表示为对于所述参考集合中的每个变体的多个变体频率(支持所述变体a1i的多个序列读值126的数量在映射至对应于所述变体的所述基因组位置的多个序列读值126d1i的总数量上的占比)的一k-长度向量f1=(f11,f12,…,f1k),其中f1的每个组成f1i采取0至1之间的一数值。这形成所述参考变体128。
进一步地,从包括有来自所述受试者的多个无细胞核酸的所述生物样本扫描与由所述向量f1代表的所述k个变体重叠的多个序列读值。对于在所述k个变体位置中的每个个别的变体位置i,确定映射至对应于所述变体位置i的所述基因组位置的多个序列读值140(d2i)的总数量及与所述变体(a2i)配对的这些序列读值140的总数量。所述多个量测值d2i及a2i为非负整数值,从中取得a2i除以d2i的一商数f2i。对于使用从包括有来自所述受试者的多个无细胞核酸模块的所述生物样本量测到的所述多个序列读值140的整个所述参考集合的所述多个变体的所述个别的商数f2i可被表示为,对于所述参考集合中的每个变体的多个变体频率(与所述特定变体配对的映射至由所述特定变体代表的所述基因组位置的多个序列读值140的数量在映射至由一特定变体代表的所述基因组位置的多个序列读值140的总数量上的占比)的所述k-长度向量f2=(f21,f22,…,f2k)。
目的是根据方块256,从所述多个第一序列读值的所述第一变体集合142中的每个变体144的所述观察到的频率(支持146)来确定所述受试者的一单一的估计出的ctdna。换言之,目标是使用从所述第一异常组织样本(例如肿瘤)提供给包括无细胞核酸的所述生物样本(例如血液)的多个突变读值的分数,来确定所述单一的估计出的ctdna。所述多个向量f1及f2总结了从所述多个个别的组织(第一异常组织及含有无细胞核酸的生物样本)量测到的多个序列读值计数,从中将推断出一潜在率(underlyingrate)。在一些实施例中,将明显与癌症不相关的多个变体排除在所述分析之外。换言之,将他们排除在考虑到的所述k个变体之外。
在一些实施例中,假定根据一泊松(poisson)过程产生来自所述异常组织样本的所述多个序列读值126。对于在k中的每个变体i,有观察到的a2i个实际的支持序列读值计数,及f1i乘以d2i个预期的支持读值计数。例如,对于变体1,考虑到一例子,其中a21为100及d21为1000,意味着在从含有无细胞核酸的所述生物样本量测到的与对应于变体1的所述基因组位置重叠的所述1000个序列读值140中,所述多个序列读值140中100个的支持所述变体。进一步假设,从所述第一异常组织,确定了所述第一异常组织中的此变体的所述频率(f11)为0.25。因此,预期会有f11(0.25)乘以d21(1000)或250个读值计数。因此,在一些实施例中,通过以t(所述多个比例突变序列读值是从所述第一异常组织样本提供给含有所述无细胞核酸的所述生物样本)、d(t)为条件的数据来估计一累积分布函数(二项式累积概率函数),从而估计对应于第5个、第50个(中位数)及第95个百分位数,或任何其他所需的百分位数的多个单一的估计出的ctdna分数。在所述无细胞dna生物样本中观察到的是对在所考虑的所述k个变体中的一个别的变体i的a2i个支持读值。进一步地,可根据将对于在所述第一异常组织样本中的所述个别的变体的所述第一异常组织的所述变体频率f1i与d2i(映射到覆盖了在含有所述无细胞核酸的所述生物样本中观察到的变体i的所述基因组位置的多个序列读值的数量)相乘,来计算从含有所述无细胞核酸的所述生物样品中预期在所述k个变体中有多少个序列读值支持所述个别的变体i的一计算值,其中假定为百分之百的脱离率(意味着提供给含有无细胞核酸的所述样本(例如,血液样本)的唯一来源是来自于所述异常组织)。因此,由此可知,t可被计算并引入至一泊松模型中,所述t可被认为是将(i)支持所述变体i的多个读值的所述预期的数量(基于所述第一异常组织分数f1i的分析)转化至(ii)在含有无细胞dna的所述组织中的支持所述变体i的多个读值的所述实际观察到的数量(a2i)的所述分数,并且这可用于估计一累积分布函数(一概率分布),所述累积分布函数提供每个t的试验值的一估计值(其中在一些实施例中,t是从介于百分之0与百分之110之间的任一处被采样)。因此,参考图16,使用在对于t的一数值范围内的所述累积分布函数来计算对于一特定等位基因i的所述个别的t的试验值的一可能性。
在一些实施例中,对于一单一的变体,所述累积分布函数具有范围在0(零概率)与1(百分之百概率)之间的一数值,并具有以下形式
其中,x=a2i,与对应于所述变体i的所述基因组位置配对,且支持位在此位置的所述变体等位基因的来自所述生物样本的多个序列读值的数量;p=t*f1i,其中t为所述单一的估计出的ctdna分数,且f1i为(a)与对应于所述变体i的所述基因组位置配对,且支持位在此位置的所述变体等位基因的来自所述第一异常组织的多个序列读值的数量的比率;及n=d2i,映射至对应于所述变体位置i的所述基因组位置的来自所述生物样本的多个序列读值的总数量。
由此可知,参考图16,可计算基于对于t的在0%至110%之间的所述数值范围内的对于t的可能性分布的对于t的中位数值(对于t的最可能的数值)(1602)、基于对于t的在0至110%之间的所述数值范围内的对于t的可能性分布的对于t的第5个百分位数值(对于t的最低数值、对于t的最低临界值)(1604),及基于对于t的在0至110%之间的所述数值范围内的对于t的可能性分布的对于t的第95个百分位(对于t的最高数值、对于t的最高临界值)数值(1606)。在图16中,实线1610代表一累积密度函数,而线条1608代表所述累积分布函数。在一些实施例中,所述累积分布函数用于计算对于t的多个百分位数值。所述第95个百分位数值意味着在与所述等位位置重叠的多个序列读值的总数量上支持一变体等位基因的多个序列读值的一观察到的分数要超过所述对于t的第95个百分位是极为罕见的,并且预期对于t的一乘积(time)值的95%小于所述对于t的第95个百分位(在图16中约28%)。
可使用其他临界值,例如,第2个百分位及第98个百分位。
上文的讨论有关于如何从一单一的变体计算出t。然而,如本文所讨论,在多个更常见的实施例中,采样多个变体,因此,每个变体产生在对于t所考虑到的所述数值范围(例如,0至100%)内的一独立的可能性(对于t的概率)。因此,所述累计分布函数提供:在一特定的t的试验值下的基于针对变体1的所述观察到的及预期的数值的对于t的一第一概率、在所述特定的t的试验值下的基于针对变体2的所述观察到的及预期的数值的对于t的一第二概率,以此类推。为了达到在所述特定的t的试验值下的对于t的累积可能性,将多个组成概率的每一个(在所述特定的t的试验值下的基于针对变体1的所述观察到的及预期的数值的对于t的所述第一概率、在所述特定的t的试验值下的基于针对变体2的所述观察到的及预期的数值的对于t的所述第二概率,以此类推)合并,并用于计算所述累积分布函数。换言之,使用从任何数量的变体而来的数据可画出图16的所述累积分布函数1608,这是在假设它们为相同潜在的单个估计出的ctdna分数的多个独立观察值的基础上。在一些实施例中,当在一对数空间中表示所述多个概率,以达到对于t的所述试验值的所述计算出的概率时,将针对一特定的t的试验值的所述多个k变体的集合中的每个个别的变体所提供的所述多个概率通过相加在一起来进行合并。例如:
其中k指的是第k个等位基因,总和为所有的k个变体。可替代地,在一些实施例中,当以一自然尺度表示所述多个概率,以达到对于t的所述试验值的所述计算出的概率时,将针对一特定的t的试验值的所述多个k变体的集合中的每个个别的变体所提供的所述多个概率通过相乘在一起来进行合并。
在一些实施例中,对于每个变体k,个别计算在所述t的试验范围内的t的可能性的所述泊松模型,从而计算出每个变体有一个的多个泊松模型。接着,对于所采样的每个t的试验值,将所述多个泊松模型(例如,在对数空间中加总,或假如在所述自然尺度上,就进行相乘)合并,以便获得对于所采样的每个t的试验值的一t的试验值的可能性。因此,在线1608上的每一点被汇总到所述k个变体中,其中k为一正整数(例如,2个或更多个、20个或更多个、1000个或更多个)。以此种方式,为肿瘤分数提供最精简的解释。
在一些实施例中,根据针对t的中位数来取得所述单一的估计出的ctdna分数,所述针对t的中位数是通过使用所述累积密度函数从对于在所述采样到的t的数值范围内的t的多个可能性的分布来取得。
重要的是,在其中在关于所述k个变体的所述生物样本中观察到零个支持读值140的多个例子中,此架构能够基于单一的估计出的ctdna分数来估计多个置信区间。
藉此,以对于在(i)含有所述无细胞核酸的所述生物样本与(ii)所述第一异常组织样本之间的所述多个变体的集合的所述读值信息为条件来估计所述无细胞dna的肿瘤分数。因此,在此实施例中,仅有在所述多个变体的参考集合128及对于所述生物样本的所述变体集合142中表示的那些变体被用于计算所述受试者的所述单一的估计出的ctdna分数。
在多个可替代的实施例中,假定一负二项分布假设,而不是一泊松分布,以便计算出图16的所述累积分布函数1608。
在一些实施例中,对多个观察到的序列读值进行背景拷贝数的校正。例如,将支持源自于在所述受试者中进行重复的多个染色体或多个染色体的多个部分的多个变体的多个序列读值针对此重复进行校正。这可以通过在运行此推断之前进行归一化,或允许一个以上的ctdna分数的数值来完成。允许一个以上的ctdna分数也能够评估肿瘤内/跨肿瘤的异质性。因此,在一些实施例中,将每个变体代表所述单一的估计出的ctdna分数的一独立的观察值的假设针对背景拷贝数进行校正。
作为参考图3的另一示例,如上文所讨论,在一些实施例中,在所述cfdna中的所述单一的预期的ctdna分数介于0.5x10-4至1.5x10-4之间,并且所述第一病况为黑色素瘤。在一些实施例中,在所述cfdna中的所述单一的预期的ctdna分数介于0.5x10-3至1x10-2之间,并且所述第一病况为肾癌、子宫癌、甲状腺癌、前列腺癌、乳腺癌、膀胱癌、胃癌、子宫颈癌或其组合。在一些实施例中,在所述cfdna中的所述单一的预期的ctdna分数介于1x10-2至0.8之间,并且所述第一病况为肺癌、食道癌、头颈癌、结肠直肠癌、肛门直肠癌、卵巢癌、肝胆癌、胰腺癌或淋巴瘤。
在一些实施例中,当所述第一变体集合142中的每个变体144的所述观察到的频率(支持14)满足一第一阈值时,通过将一受试者视为患有一第一病况来对所述受试者进行分类。在一些实施例中,基于对所述变体集合中的所述多个变体的所述参考频率的量化来确定所述第一阈值。在一些实施例中,例如,通过如参考方块258的上文所讨论的对于在所述变体集合中的所述多个对应的变体的所述参考频率来对所述第一变体集合142中的每个变体144的所述观察到的频率(支持14)进行归一化,以实现对于所述受试者的一循环肿瘤核酸分数。例如,在一些实施例中,通过如参考方块258的上文所讨论的对于在所述变体集合中的所述多个对应的变体的所述参考频率来将所述第一变体集合142中的每个变体144的所述观察到的频率(支持14)划分开,以实现对于所述受试者的所述循环肿瘤核酸分数。以此种方式,通过所述第一异常组织样本的所述第一参考集合128中的每个变体130的一频率来确定所述第一阈值。
在一些实施例中,患有一相似病况的一受试者群体被用于改善与一病况相关的所述第一阈值。例如,考虑到一例子,其中所述第一病况为癌症的一阶段,且与癌症类型无关。图4说明在一受试者群体内的脱离率(ctdna分数)。图4中的每个点代表被划分成四个癌症阶段(i、ii、iii及iv)的其中之一的一受试者群体中的一不同受试者的ctdna分数。对于每个个别的受试者,根据针对从所述受试者的一生物样本收集到的所述集合变体142中的所述多个变体的集合的所述支持(例如,根据图2的方块202及210)与针对所述个别的受试者的从来自相同受试者的一肿瘤获得的所述参考集合128中的这些相同的变体的所述参考频率132(例如,根据图2的方块228所概述的公开内容)的所述比率,绘制出所述ctdna分数(肿瘤分数)。图4说明了对于每个癌症受试者有一ctdna分数的数值范围,但是ctdna分数的中位数值通常随着癌症阶段的增加而增加。因此,图4提供了基于在所述变体集合中的所述多个变体的所述参考频率的量化来确定所述第一阈值的动机。意即,图4说明了使用在一特定受试者的所述异常组织中的多个变体的多个观察到的频率的可能性,以及可选择地关于患有一特定的癌症阶段或类型的多个受试者的预期的ctdna的信息,以便确定可对照在对于所述特定受试者的一生物样本的变体集合中的所述多个变体的所述观察到的频率来评估的针对所述特定癌症受试者的一第一阈值,从而将所述受试者分类成具有或不具有所述病况(例如,一特定癌症的一临床阶段)。因此,参考图4,可使用一第一阈值0.05来分析一受试者是否具有一特定癌症的第i期。在这样的例子中,从一受试者获得诸如一肿瘤的一异常组织,并将其用于确定对于一第一参考集合中的每个个别的变体的一参考频率(例如,根据图2的方块228)。事实上,在一些实施例中,将各种可能的变体的所述频率用于辨识对于所述参考集合的所述多个变体。进一步地,从相同受试者的除了所述异常组织之外的一生物样本获得无细胞核酸(例如,参考方块202),并且从所述生物样本中的所述无细胞核酸的多个序列读值确定在所述参考集合中的多个相同变体的所述变体频率(例如,根据方块210)。在所述生物样本中的这些变体的所述变体频率(支持146)是通过在所述异常组织中的多个相同变体的所述参考频率(例如,通过取得一比率等)来进行归一化,从而形成所述生物样本的所述观察到的ctdna分数(例如,根据图2的方块258的公开内容)。在此,通过在所述第一异常组织样本的所述第一参考集合128中的每个变体130的一频率来确定所述第一阈值,因为这些频率形成如上文结合图2的方块258所讨论的所述比率的分母的基础。使用图4的所述群体作为一指导,对一特定生物样本的所述ctdna是否满足所述0.05的阈值状况进行确定为确定所述受试者是否患有此示例中的第一期癌症提供了基础。例如,从图4,当在所述变体集合中的每个变体144的所述观察到的频率(支持146)与在所述参考集合128中的多个相同变体的所述参考频率的所述比较指出所述ctdna分数高于0.05时,将所述受试者视为患有一更晚期的癌症,这是因为在图4的所述群体中很少有第i期患者具有高于0.05的一ctdna分数。另一方面,观察到低于0.001的一ctdna分数与发现所述受试者患有一特定癌症的第i期具有一致性,这是因为在图4的所述群体中相对少有第ii、iii或iv期的受试者具有低于0.001的一ctdna分数。这仅仅是一示例,并且在下文示例1中更详细讨论的图3显示出,当已经知道一受试者患有的所述癌症类型时,可定义多个更精确的阈值。
方块260提供一具体的实施例,其中所述方块256的评估步骤包括:从所述第一变体集合142中的每个变体144的所述观察到的频率(支持146)计算出在所述受试者的所述无细胞dna(cfdna)中的一单一的估计出的循环肿瘤dna(ctdna)分数,其中当所述单一的估计出的循环肿瘤dna(ctdna)分数超过1x10-3,且所述第一病况为乳腺癌的第ii期、第iii期或第iv期时,所述第一变体集合142中的每个第一变体144的所述观察的频率满足一阈值。此阈值的限制受到图5的支持,如下文示例2所讨论。在图5中,每个点为患有乳腺癌的一单独的受试者的所述ctdna分数。在一些实施例中用于计算对于每个受试者的所述cfdna分数的所述方法包括:从一群体中的每个受试者的一生物样本获得电子形式的多个第一序列读值140,其中所述生物样本包括多个无细胞核酸分子。将所述多个第一序列读值140用于辨识对所述生物样本的一变体集合142中的每个变体的支持,从而确定在所述变体集合142中的每个变体144的一观察到的频率(支持146)。在一些实施例中,将所述变体集合142中的每个个别的变体144的所述观察到的频率(支持146)与一参考集合128中对于所述个别的变体的一对应的参考频率132进行比较。所述参考集合128中的每个对应的参考频率132为从所述受试者获得的一第一异常组织样本中的一个别的变体的一频率。以此种方式,在一些实施例中确定每个受试者的所述ctdna分数。除了绘制每个受试者的所述ctdna分数之外,图5通过乳腺癌的阶段将所述多个受试者划分开。对于在每个肿瘤阶段中所观察到的肿瘤分数,图5指出一很大的动态范围。图5指出,假如所述循环肿瘤dna(ctdna)分数超过1x10-3,则所述受试者可能患有乳腺癌第ii、iii或iv期,因为在图5中很少有乳腺癌第0期或第i期的受试者具有超过1x10-3的一ctdna分数。当然,由于图5也显示出大量的第iii期的受试者具有低于1x10-3的ctdna分数,因此可能需要多个额外的测试来确定一乳腺癌受试者的一确切的分类。藉此,所述多个公开的方法支持多个例子,其中所述受试者患有第ii期、第iii期或第iv期乳腺癌,且所述方块256的评估步骤确定所述无细胞核酸的所述第一肿瘤分数低于1x10-3。
参考方块262,在一些实施例中,所述多个公开的方法用于评估患有来自一共同原发部位的癌症的一受试者的一肿瘤分数。例如,参考方块264,在一些实施例中,所述多个公开的方法用于评估患有乳腺癌、肺癌、前列腺癌、结肠直肠癌、肾癌、子宫癌、胰腺癌、食道癌、淋巴瘤、头颈癌、卵巢癌、肝胆癌、黑色素瘤、子宫颈癌、多发性骨髓瘤、白血病、甲状腺癌、膀胱癌、胃癌,或其组合的一受试者的一肿瘤分数。
参考图2e的方块268,在一些实施例中,所述多个公开的方法用于评估患有乳腺癌、肺癌、前列腺癌、结肠直肠癌、肾癌、子宫癌、胰腺癌、食道癌、淋巴瘤、头颈癌、卵巢癌、肝胆癌、黑色素瘤、子宫颈癌、多发性骨髓瘤、白血病、甲状腺癌、膀胱癌或胃癌的一预定阶段的一受试者的一肿瘤分数。
参考图2e的方块270,在一些实施例中,所述多个公开的方法用于评估患有一癌症的一预定亚型的一受试者的一肿瘤分数。在一些这样的实施例中,参考图2e的方块272,所述癌症为乳腺癌、肺癌、前列腺癌、结肠直肠癌、肾癌、子宫癌、胰腺癌、食道癌、淋巴瘤、头颈癌、卵巢癌、肝胆癌、黑色素瘤、子宫颈癌、多发性骨髓瘤、白血病、甲状腺癌、膀胱癌或胃癌。
参考图2e及2f的方块274至286,所述多个公开的方法不限于分析一单一的异常组织,或在一单一的时间点分析一单一的异常组织。在一些实施例中,将方块202至272的所述公开内容扩展至对应于肿瘤间/内异质性的一患者的多个肿瘤样本及多个肿瘤分数。换言之,所述多个公开的方法可用于计算相对于一第二异常组织的一第二生物样本的一额外的ctdna分数。例如,在一些实施例中,上文结合图2的方块202至272所讨论的所述第一异常组织样本为一第一癌症类型,而所述第二异常组织样本为一第二癌症类型(方块278)。在其他实施例中,上文结合图2的方块202至272所讨论的所述第一异常组织样本来自于一第一时间点的一肿瘤,而所述第二异常组织样本来自于一第二时间点的相同肿瘤。在另外的其他实施例中,所述受试者的所述异常组织为异质性的,且所述第一异常组织样本为此异常组织的一第一切片,而所述第二异常组织样本为与所述第一切片同时收集到的此相同异常组织的一第二切片。
更详细地,参考方块274,所述多个第一序列读值140用于辨识对一第二变体142中的每个变体144的支持,从而确定所述第二变体集合中的每个变体144的一观察到的频率。对于在所述第二变体集合142中的每个个别的变体144,在一第二参考集合128中获得对于所述个别的变体的一对应的参考频率132,其中所述第二参考集合中的每个对应的参考频率是针对从所述受试者获得的一第二异常实体组织样本中的一个别的变体。在一些这样的实施例中,所述方块256的评估步骤进一步包括:对照在所述第二参考集合中的所述个别的变体的所述观察到的频率,来使用所述第二变体集合中的每个个别的变体的所述观察到的频率,从而确定在所述受试者的所述液体生物样本的无细胞核酸中的一第二肿瘤分数。以此种方式,可首先相对于所述第一异常组织计算一生物样本的所述ctdna分数(例如,以便确定所述受试者是否患有一第一病况、以便监控所述第一异常组织在一段时间内的进展、以便监控肿瘤异质性等),并且可相对于所述第二异常组织计算所述生物样本的一不同的ctdna分数(例如,以便确定所述受试者是否具有一第二病况、以便监控所述第二异常组织在一段时间内的进展、以便监控肿瘤异质性等)。
参考图2f的方块276,在一些实施例中,将所述多个第一序列读值中的一个别的序列读值140视为支持在所述第二变体142中的一变体144,这是在所述个别的序列读值140(i)映射至对应于所述变体的一基因组位置上,并且(ii)含有所述变体的全部或一部分时。将所述多个第一序列读值中的一个别的序列读值140视为不支持在所述第二变体142中的一变体144,这是在所述个别的序列读值140(i)映射至对应于所述变体的一基因组位置上,并且(ii)不含有所述变体的全部或一部分时。
参考图2f的方块278,在一些实施例中,所述第一异常组织样本是由一第一肿瘤分数组成,而所述第二异常组织样本是由来自所述受试者的一共同(相同)肿瘤的一第二肿瘤分数所组成。
参考图2f的方块280,在一些实施例中,所述第一异常组织样本为一第一癌症类型,而所述第二异常组织样本为一第二癌症类型。所述第一癌症类型可与所述第二癌症类型相同(方块282)。可替代地,所述第一癌症类型可与所述第二癌症类型不同(方块284)。在一些实施例中,所述第一癌症类型跟所述第二癌症类型分别是选自于由乳腺癌、肺癌、前列腺癌、结肠直肠癌、肾癌、子宫癌、胰腺癌、食道癌、淋巴瘤、头颈癌、卵巢癌、肝胆癌、黑色素瘤、子宫颈癌、多发性骨髓瘤、白血病、甲状腺癌、膀胱癌及胃癌所组成的群组(方块286)。
示例性方法实施例-基于cfdna中的ctdna分数随着时间的变体来评估一受试者的一已知病况的侵袭性(aggressiveness)。
本公开的另一方面提供一种评估一受试者的一病况的一状态的方法。所述方法包括:在一计算机系统100中具有一个或多个处理器102及一记忆体111/112,所述记忆体111/112存储通过所述一个或多个处理器执行的一个或多个程序,在所述计算机系统中进行步骤:对于一时期内的多个时间点中的每个个别的时间点,从在每个个别的时间点取得的所述受试者的一个别的生物样本获得电子形式的一对应的数据集138,所述数据集138包括所述个别的生物样本的多个对应的第一序列读值140,从而获得所述受试者的多个数据集(例如,如方块202所阐述)。每个个别的生物样本包括多个无细胞核酸分子。在一些实施例中,从一特定的生物样本获得所述多个无细胞核酸分子,如上文结合图2的方块202至208中的任何一个所讨论。在一些实施例中,获得对于一特定的生物样本的所述多个无细胞核酸分子的所述多个序列读值,如上文结合图2的方块202至208中的任何一个所讨论。
所述方法进一步包括:对于在所述多个个别的数据集中的每个个别的数据集(例如,数据构筑体138),确定对所述变体集合142中的每个变体144的支持(例如,如图2的方块210至226所公开)。将在所述个别的数据集的所述多个第一序列读值中的一个别的序列读值140视为支持所述变体集合142中的一变体144,这是在所述个别的序列读值140(i)映射至对应于所述变体的一基因组位置,并且(ii)含有所述变体144的全部或一部分时。将在所述个别的数据集的所述多个第一序列读值中的一个别的序列读值140视为不支持所述变体集合142中的一变体,这是在所述个别的序列读值(i)映射至对应于所述变体的一基因组位置,并且(ii)不含有所述变体144的全部或一部分时。以此种方式,在所述多个时间点中的每个时间点,使用在所述个别的数据集138的所述多个第一序列读值中的支持或不支持所述变体集合142中的每个变体的所述多个序列读值,来确定所述变体集合142中的每个变体144的一观察到的频率。
在一些实施例中,通过利用b得分分类器来使用所述多个序列读值140识别多个变异,将所多个序列读值140用于找寻对所述变体集合142中的多个变体144的支持。所述b得分分类器在2018年3月13日提交的标题为“用于选择、管理及分析高维度数据的方法及系统”的美国专利公开第62/642,461号中进行描述,其通过引用并入本文中,并且在示例3中进一步详细描述。
在一些实施例中,通过利用m得分分类器来使用所述多个序列读值140识别多个变异,将所多个序列读值140用于找寻对所述变体集合142中的多个变体144的支持。所述m得分分类器在2018年3月13日提交的标题为“甲基化片段异常检测”的美国专利申请第62/642,480号中进行描述,其通过引用并入本文中。
在一些实施例中,通过利用上文结合图2所描述的方块210至216中的任何一个所公开的多个技术来使用所述多个序列读值140识别多个变异,将所多个序列读值140用于找寻对所述变体集合142中的多个变体144的支持。
所述方法进一步包括:在所述多个时间点的每个时间点,对照所述第一异常实体组织中的所述个别的变体的所述观察到的频率(例如,如在方块210的所述第一例子中所确定)来评估所述变体集合142中的每个变体144的所述观察到的频率(例如支持146),从而以所述时期内的所述第一肿瘤分数的增加或减少的形式来确定所述受试者的一疾病状况在所述时期期间的所述状态或进展。
在一些实施例中,为了能够量测在大约数小时(例如,为了测量从一受试者移除异常组织的手术成功率)、数周/数月(例如,为了监控对于一受试者的治疗成功率)或数年(例如,为了监控一受试者的疾病缓解情况)内的ctdna的改变,对所述时期进行校准。因此,在一些实施例中,所述时期为数个月的一时间段,并且在所述多个时间点中的每个时间点为所述数个月的时间段中的一不同的时间点。在一些这样的实施例中,所述数个月的时间段少于四个月。在一些实施例中,所述时期为数个年的一时间段,并且在所述多个时间点中的每个时间点为所述数个年的时间段中的一不同的时间点。在一些这样的实施例中,所述数个年的时间段介于2至10年之间。在一些实施例中,所述时期为数小时的一时间段,并且在所述多个时间点中的每个时间点为所述数小时的时间段中的一不同的时间点。在一些这样的实施例中,所述数小时的时间段介于1小时至6小时之间。
在一些实施例中,所述对照所述第一异常实体组织中的所述个别的变体的所述观察到的频率,来在所述多个时间点的每个时间点评估所述变体集合142中的每个变体144的所述观察到的频率的步骤包括:以上文结合方块256所阐述的方式,从在所述多个时间点的集合中的每个时间点的所述变体集合142中的每个变体144的所述观察到的频率,计算出所述受试者的所述无细胞dna(cfdna)中的各个单一的估计出的循环肿瘤dna(ctdna)分数。在一些这样的实施例中,所述方法进一步包括:当观察到所述受试者的所述cfdna中的所述各个单一的估计出的循环肿瘤ctdna分数在所述时期内以一阈值量变化时,改变所述受试者的一诊断。例如,在一些实施例中,在所述时期内的每个时间点的所述ctdna分数为介于0至1之间的数字,当所述ctdna分数在所述时期期间以一预定值变化时,改变所述受试者的所述诊断。在一示例中,当所述ctdna分数在所述时期内增加超过百分之二、超过百分之三、超过百分之四、超过百分之五、超过百分之十,或超过百分之二十时,将所述受试者的所述诊断降级,这指示出相较于初始诊断,所述受试者患有所述疾病状况的一侵袭性较高的形式及/或所述疾病状况的一较后期阶段。在另一示例中,当所述ctdna分数在所述时期内降低超过百分之二、超过百分之三、超过百分之四、超过百分之五、超过百分之十,或超过百分之二十时,将所述受试者的所述诊断升级,这指示出相较于初始诊断,所述受试者患有所述疾病状况的一侵袭性较低的形式及/或所述疾病状况的一较早期阶段。
在一些实施例中,所述方法进一步包括:当观察到所述受试者的所述cfdna中的所述各个单一的估计出的ctdna分数在所述时期内以一阈值量变化时,改变所述受试者的一预后情形。例如,在一些实施例中,在所述时期内的每个时间点的所述ctdna分数为介于0至1之间的数字,当所述ctdna分数在所述时期期间以一预定值变化时,改变所述受试者的所述预后情形。在一示例中,当所述ctdna分数在所述时期内增加超过百分之二、超过百分之三、超过百分之四、超过百分之五、超过百分之十,或超过百分之二十时,将所述受试者的所述预后情形降级,这指示出所述受试者从所述疾病状况恢复的可能性降低。在另一示例中,当所述ctdna分数在所述时期内降低超过百分之二、超过百分之三、超过百分之四、超过百分之五、超过百分之十,或超过百分之二十时,将所述受试者的所述预后情形升级,这指示出所述受试者从所述疾病状况恢复的可能性提高。
在一些实施例中,所述方法进一步包括:当观察到所述受试者的所述cfdna中的所述各个单一的估计出的ctdna分数在所述时期内以一阈值量变化时,改变所述受试者的一治疗。例如,在一示例中,当所述ctdna分数在所述时期内增加超过百分之二、超过百分之三、超过百分之四、超过百分之五、超过百分之十,或超过百分之二十时,将所述受试者的所述治疗方案改变成一较积极的治疗。在另一示例中,当所述ctdna分数在所述时期内降低超过百分之二、超过百分之三、超过百分之四、超过百分之五、超过百分之十,或超过百分之二十时,将所述受试者的所述治疗方案改变成一较不积极的治疗。
在一些实施例中,所述病况为一疾病,例如癌症。例如,在一些实施例中,所述疾病为一癌症,且所述癌症为乳腺癌、肺癌、前列腺癌、结肠直肠癌、肾癌、子宫癌、胰腺癌、食道癌、淋巴瘤、头颈癌、卵巢癌、肝胆癌、黑色素瘤、子宫颈癌、多发性骨髓瘤、白血病、甲状腺癌、膀胱癌、胃癌或其组合。
在一些实施例中,所述病况为乳腺癌、肺癌、前列腺癌、结肠直肠癌、肾癌、子宫癌、胰腺癌、食道癌、淋巴瘤、头颈癌、卵巢癌、肝胆癌、黑色素瘤、子宫颈癌、多发性骨髓瘤、白血病、甲状腺癌、膀胱癌或胃癌的一阶段。
在一些实施例中,所述疾病状况为一癌症的一预定的亚型,其中所述癌症为乳腺癌、肺癌、前列腺癌、结肠直肠癌、肾癌、子宫癌、胰腺癌、食道癌,淋巴瘤、头颈癌、卵巢癌、肝胆癌、黑色素瘤、子宫颈癌、多发性骨髓瘤、白血病、甲状腺癌、膀胱癌或胃癌。
在一些实施例中,所述变体集合中的每个个别的变体为与一预定的基因组位置相关的一单核苷酸变体、与一预定的基因组位置相关的一插入突变、与一预定的基因组位置相关的一缺失突变、一体细胞拷贝数的改变、与一预定的基因组位点相关的一核酸重组,或与一预定的基因组位置相关的一异常的甲基化模式。
在一些实施例中,所述异常的组织为一肿瘤。在一些实施例中,所述第一异常组织样本为上文参考图2的方块230所描述的多个异常组织的其中一个。
在一些实施例中,所述变体集合142是由一单一变体144组成,所述单一变体144为位于所述受试者的基因组中的一单一位点处的一单一的遗传变异。在一些实施例中,所述变体集合142是由一第一变体及一第二变体组成,所述第一变体为位于所述受试者的基因组中的一第一位点处的一第一遗传变异,且所述第二变体为位于所述受试者的基因组中的一第二位点处的一第二遗传变异。
在一些实施例中,所述变体集合142是由一第一变体、一第二变体及一第三变体组成,所述第一变体为位于所述受试者的基因组中的一第一位点处的一第一遗传变异,所述第二变体为位于所述受试者的基因组中的一第二位点处的一第二遗传变异,且所述第三变体为位于所述受试者的基因组中的一第三位点处的一第三遗传变异。
在一些实施例中,所述变体集合142是由介于2至20个之间的变体144组成,其中所述变体集合中的每个变体144为所述受试者的所述基因组中的(可选择地位于一不同的位点处的)一不同的遗传变异。在一些实施例中,所述变体集合142包括30个变体144、50个变体144、75个变体144、100个变体144、125个变体144、250个变体144、500个变体144、750个变体144、1000个变体144、2500个变体144,或5000个变体144,其中所述变体集合中的每个变体144为所述受试者的所述基因组中的(可选择地位于一不同的位点处的)一不同的遗传变异。
在一些实施例中,对于在所述多个个别的数据集中的每个个别的数据集,确定对一变体集合142中的每个变体144的支持的步骤包括:将在一个别的数据集的所述多个第一序列读值中的一序列读值140与一参考基因组中的一区域进行比对,以便确定所述序列读值是否含有所述变体集合中的一变体的全部或一部分。例如,参见图2a的方块212及上文呈现出的相同公开内容。
在一些实施例中,对于在所述多个个别的数据集中的每个个别的数据集,确定对一变体集合142中的每个变体144的支持的步骤包括:将在一个别的数据集的所述多个第一序列读值中的一序列读值140与多个变体的一查找表进行比对,以便确定所述序列读值是否含有所述变体数据集中的一变体的全部或一部分。例如,参见图2a的方块214及上文呈现出的相同公开内容。
在一些实施例中,对于在所述多个个别的数据集中的每个个别的数据集,确定对一变体集合142中的每个变体144的支持的步骤包括:将在一个别的数据集的所述多个第一序列读值中的一序列读值140与一查找表中的每个条目进行比对,其中所述查找表中的每个条目代表一参考基因组的一不同部分。例如,参见图2a的方块216及上文呈现出的相同公开内容。
在一些实施例中,所述受试者为一人类受试者。在一些实施例中,所述受试者为哺乳动物。在一些实施例中,所述受试者为上文结合图2的方块204所公开的任何一个物种。
在一些实施例中,所述个别的生物样本包括所述受试者的血液、全血、血浆、血清、尿液、脑脊髓液、粪便、唾液、汗水、眼泪、胸水、心包液,及/或腹膜液。意即,所述生物样本为所述受试者的血液、全血、血浆、血清、尿液、脑脊髓液、粪便、唾液、汗水、眼泪、胸水、心包液及/或腹膜液,与所述受试者的其他组成物的一混合物。
在一些实施例中,所述个别的生物样本是由所述受试者的血液、全血、血浆、血清、尿液、脑脊髓液、粪便、唾液、汗水、眼泪、胸水、心包液,及/或腹膜液组成。意即,所述生物样本为所述受试者的血液、全血、血浆、血清、尿液、脑脊髓液、粪便、唾液、汗水、眼泪、胸水、心包液,及/或腹膜液,并且没有所述受试者的其他组成物。
示例性方法实施例-使用肿瘤分数把关一分类器的多个结果的使用
本公开的另一方面提供一种用于分类一受试者的方法。所述方法包括:在一计算机系统100中具有一个或多个处理器102及一记忆体111/112,所述记忆体111/112存储通过所述一个或多个处理器执行的一个或多个程序(例如,病况监控模块120),在所述计算机系统中获得电子形式的一数据集(例如,数据构筑体138),所述数据集包括从所述受试者的一生物样本而来的多个第一序列读值140。在此,所述生物样本包括多个无细胞核酸分子。在一些实施例中,以任何结合方块202至208的方式来获得所述多个第一序列读值。并且,在多个这样的实施例中,将所述多个第一序列读值140用于辨识对一第一变体集合142中的每个变体144的支持,从而参考上文结合图2所公开的方块210至226中的任何一个,以上文公开的方式确定所述第一变体集合中的每个变体的一观察到的频率。并且,针对在所述第一变体集合132中的每个个别的变体144,以上文参考图2的方块228至248所公开的方式获得在一第一参考集合128中对于所述个别的变体的一对应的参考频率,其中所述第一参考集合中的每个对应的参考频率是针对从所述受试者获得的一第一异常实体组织样本中的一个别的变体。所述方法进一步公开:对照在所述第一异常实体组织的所述第一参考集合128中的所述个别的变体的所述观察到的频率,来评估在所述第一变体集合142中的每个个别的变体的所述观察到的频率,从而以上文参考图2的方块256至272所公开的任何方式确定在所述受试者的所述液体生物样本的无细胞核酸中的一第一肿瘤分数。
所述方法进一步包括:将所述多个第一序列读值(或来自所述多个序列读值的维度缩减数据,例如多个主成分)应用至一分类器,从而获得一分类器的结果。所述分类器的结果指示出所述受试者是否患有一第一癌症病况。进一步地,在执行所述即时方法之前,透过在多个受试者的无细胞dna(cfdna)中所观察到的肿瘤分数以外的数据来训练所述分类器。在一些实施例中,当所述第一肿瘤分数介于0.003至1.0之间,且所述训练过的分类器的结果指出所述受试者患有所述第一癌症病况时,使用所述训练过的分类器的结果作为所述受试者对于所述第一癌症病况的诊断或预后情形的一基础。如本文所使用,术语“训练过的分类器”指的是具有多个固定(锁定)参数(权重)及阈值的一模型(例如,一机器学习算法,如逻辑回归、神经网络、回归、支持向量机、聚类算法、决策树等),可准备将其应用于先前未见的多个样本中。
在一些实施例中,使用参考图2的上文及下面的多个示例所公开的多个技术来确定在所述受试者的所述cfdna中的所述估计出的肿瘤分数。
在一些实施例中,所述第一病况为一癌症(例如,乳腺癌、肺癌、前列腺癌、结肠直肠癌、肾癌、子宫癌、胰腺癌、食道癌、淋巴瘤、头颈癌、卵巢癌、肝胆癌、黑色素瘤、子宫颈癌、多发性骨髓瘤、白血病、甲状腺癌、膀胱癌、胃癌或其组合)。
在一些实施例中,所述第一病况为一癌症的一亚型。在一些这样的实施立中,所述癌症为乳腺癌、肺癌、前列腺癌、结肠直肠癌、肾癌、子宫癌、胰腺癌、食道癌、淋巴瘤、头颈癌、卵巢癌、肝胆癌、黑色素瘤、子宫颈癌、多发性骨髓瘤、白血病、甲状腺癌、膀胱癌或胃癌。
在一些实施例中,所述估计出的肿瘤分数介于0.003至1.0之间,并且所述第一癌症病况为一癌症的一起源组织。
在一些实施例中,计算在所述cfdna中的所述估计出的肿瘤分数的步骤包括:使用所述数据集来辨识对一变体集合142中的每个变体144的支持,其中将所述多个第一序列读值中的一个别的序列读值140视为支持所述变体集合142中的一变体144,这是在所述个别的序列读值140(i)映射到所述基因组的与所述变体对应的部分上,并且(ii)含有所述变体144的全部或一部分时;以及将所述多个第一序列读值中的一个别的序列读值140视为不支持所述变体集合142中的一变体144,这是在所述个别的序列读值140(i)映射到所述基因组的与所述变体对应的部分上,并且(ii)不含有所述个别的变体144时。以此种方式,从所述多个第一序列读值中的支持或不支持所述变体集合中的每个变体的所述多个序列读值中,确定所述变体集合142中的每个变体144的一观察到的频率。
在一些实施例中,通过利用b得分分类器来使用所述多个序列读值140识别多个变异,将所多个序列读值140用于找寻对所述变体集合142中的多个变体144的支持。所述b得分分类器在以62/642,461提交的标题为“用于选择、管理及分析高维度数据的方法及系统”的美国专利公开第62/642,461号中进行描述,其通过引用并入本文中,并且在示例3中进一步详细描述。
在一些实施例中,通过利用m得分分类器来使用所述多个序列读值140识别多个变异,将所多个序列读值140用于找寻对所述变体集合142中的多个变体144的支持。所述m得分分类器在2018年3月13日提交的标题为“甲基化片段异常检测”的美国专利申请第62/642,480号中进行描述,其通过引用并入本文中。
在一些实施例中,通过利用上文结合图2所描述的方块210至216中的任何一个所公开的多个技术来使用所述多个序列读值140识别多个变异,将所多个序列读值140用于找寻对所述变体集合142中的多个变体144的支持。
进一步地,在多个这样的实施例中,从在所述变体集合中的每个变体的所述观察到的频率计算出所述受试者的所述cfdna中的一单一的估计出的肿瘤分数。例如,关于计算出所述cfdna中的所述单一的估计出的肿瘤分数的公开,请参见图2的方块258的公开内容。
在一些这样的实施例中,所述变体集合中的一变体为与一预定的基因组位置相关的一单核苷酸变体、与一预定的基因组位置相关的一插入突变、与一预定的基因组位置相关的一缺失突变、一体细胞拷贝数的改变、与一预定的基因组位点相关的一核酸重组,或与一预定的基因组位置相关的一异常的甲基化模式。
在一些实施例中,所述异常的组织为一肿瘤的全部或一部分。在一些实施例中,所述异常的组织样本为上文结合方块230所描述的任何一种异常组织。
在一些实施例中,所述变体集合142是由一单一变体144组成,所述单一变体为位在所述受试者的所述基因组中的一单一位点处的一单一的遗传变异。
在一些实施例中,所述变体集合142是由一第一变体144及一第二变体144组成,所述第一变体144为位于所述受试者的基因组中的一第一位点处的一第一遗传变异,且所述第二变体144为位于所述受试者的基因组中的一第二位点处的一第二遗传变异。
在一些实施例中,所述变体集合142是由一第一变体144、一第二变体144及一第三变体144组成,所述第一变体144为位于所述受试者的基因组中的一第一位点处的一第一遗传变异,所述第二变体144为位于所述受试者的基因组中的一第二位点处的一第二遗传变异,且所述第三变体为位于所述受试者的基因组中的一第三位点处的一第三遗传变异。
在一些实施例中,所述变体集合142是由介于2至20个之间的变体组成,其中所述变体集合142中的每个变体144为所述受试者的所述基因组中的(可选择地位于一不同的位点处的)一不同的遗传变异。在一些实施例中,所述变体集合包括40个变体、50个变体、75个变体、100个变体、200个变体、500个变体、1000个变体、2000个变体,或5000个变体,并且所述变体集合中的每个变体为所述受试者的所述基因组中的(可选择地位于一不同的位点处的)一不同的遗传变异。
在一些实施例中,在所述cfdna中的所述单一的估计出的肿瘤分数介于0.5x10-4至1.5x10-4之间,并且所述第一病况为黑色素瘤。在一些实施例中,在所述cfdna中的所述单一的估计出的肿瘤分数介于0.5x10-3至1x10-2之间,并且所述第一病况为肾癌、子宫癌、甲状腺癌、前列腺癌、乳腺癌、膀胱癌、胃癌、子宫颈癌或其组合。在一些实施例中,在所述cfdna中的所述单一的估计出的肿瘤分数介于1x10-2至0.8之间,并且所述第一病况为肺癌、食道癌、头颈癌、结肠直肠癌、肛门直肠癌、卵巢癌、肝胆癌、胰腺癌、淋巴瘤或其组合。
在一些实施例中,使用所述多个第一序列读值来辨识对一变体集合中的每个变体的支持的步骤包括:将所述多个第一序列读值中的一个别的序列读值140与一参考基因组中的一区域进行比对,以确定所述个别的序列读值140是否含有所述变体集合中的一变体的全部或一部分。例如,参见图2a的方块212及上文呈现出的相同公开内容。
在一些实施例中,使用所述多个第一序列读值来辨识对一变体集合142中的每个变体144的支持的步骤包括:将所述多个第一序列读值中的一个别的序列读值140与多个变体的一查找表进行比对,以确定所述序列读值是否含有所述变体集合中的一变体的全部或一部分。例如,参见图2a的方块214及上文呈现出的相同公开内容。
在一些实施例中,使用所述多个第一序列读值来辨识对一变体集合142中的每个变体144的支持的步骤包括:将所述多个第一序列读值中的一个别的序列读值140与一查找表中的每个条目进行比对,其中所述查找表的每个条目代表一基因组的一不同部分。例如,参见图2a的方块216及上文呈现出的相同公开内容。
在一些实施例中,所述受试者为一人类受试者。在一些实施例中,所述受试者为哺乳动物。在一些实施例中,所述受试者为上文结合图2的方块204所公开的任何一个物种。
在一些实施例中,所述生物样本包括所述受试者的血液、全血、血浆、血清、尿液、脑脊髓液、粪便、唾液、汗水、眼泪、胸水、心包液,或腹膜液。意即,所述生物样本为所述受试者的血液、全血、血浆、血清、尿液、脑脊髓液、粪便、唾液、汗水、眼泪、胸水、心包液及/或腹膜液,与所述受试者的一个或多个其他组成物的一混合物。
在一些实施例中,所述个别的生物样本是由所述受试者的血液、全血、血浆、血清、尿液、脑脊髓液、粪便、唾液、汗水、眼泪、胸水、心包液,或腹膜液组成。意即,所述生物样本为所述受试者的血液、全血、血浆、血清、尿液、脑脊髓液、粪便、唾液、汗水、眼泪、胸水、心包液,及/或腹膜液,并且没有所述受试者的其他组成物。
在一些实施例中,所述分类器使用以62/642,461提交的标题为“用于选择、管理及分析高维度数据的方法及系统”的美国专利公开第62/642,461号所描述的b得分分类器,其通过引用并入本文中。
在一些实施例中,所述分类器使用在2018年3月13日提交的标题为“甲基化片段异常检测”的美国专利申请第62/642,480号所描述的m得分分类器,其通过引用并入本文中。
在一些实施例中,所述分类器为一神经网络或一常规的神经网络。参见vincent等人,2010,“堆迭式降噪自动编码器:利用本地降噪标准在深度网络中学习有用的表征”,jmachlearnres11,pp.3371-3408;larochelle等人,2009,“探索用于训练深度神经网络的策略”,jmachlearnres10,pp.1-40;及hassoun,1995,人工神经网络基础,麻省理工学院,其中的每一个通过引用并入本文中。
在一些实施例中,所述分类器为一支持向量机(svm)。多个svm在cristianini及shawe-taylor,2000,“对于支持向量机的介绍”,剑桥大学出版社,剑桥;boser等人,1992,“一种用于最佳边际分类器(marginclassifier)的训练算法”,第五届acm计算学习理论年度研讨会论文集,acm出版社,匹兹堡,宾夕法尼亚州,pp.142-152;vapnik,1998,统计学习理论,威立,纽约州;mount,2001,生物信息学:序列及基因组分析,冷泉港实验室出版社,冷泉港,纽约州;duda,模式分类,第二版,2001,约翰威立公司,pp.259,262-265;及hastie,2001,统计学习的要素,施普林格,纽约州;及furey等人,2000,bioinformatics16,906-914进行描述,其中的每一个通过引用整体并入本文中。当用于分类时,多个svm将具有一超平面的一特定的二进制标记数据集分离,所述超平面与所述标记数据的距离最大。对于无法进行线性分离的情况,多个svm可与‘核函数(kernels)’技术结合运作,所述核函数技术可自动实现一种与一特征空间的非线性映射。所述特征空间中的通过所述svm发现的所述超平面对应于所述输入空间中的一非线性决策边界。
在一些实施例中,所述分类器为一决策树。多个决策树一般通过duda,2001,模式分类,约翰威立公司,纽约州,pp.395-396进行描述,其通过引用并入本文中。多个基于树的方法将所述特征空间划分为一组矩形,然后在每个矩形中拟合一模型(如一常量)。在一些实施例中,所述决策树为一随机森林回归。可使用的一特定的运算法为一分类及回归树(cart)。其他特定的决策树算法包括,但不限于,id3、c4.5、mart及多种随机森林。cart、id3及c4.5在duda,2001,模式分类,约翰威立公司,纽约州,pp.396-408及pp.411-412中进行描述,其通过引用并入本文中。cart、mart及c4.5在hastie等人,2001,统计学习的要素,施普林格出版社,纽约州,第9章中进行描述,其通过引用整体并入本文中。多种随机森林在breiman,1999,“随机森林--随机的特征”,技术报告567,统计学系,柏克莱加利福尼亚大学,1999年9月中进行描述,其通过引用整体并入本文中。
在一些实施例中,所述分类器为一非监督式聚类模型。在一些实施例中,所述分类器为一监督式聚类模型。在duda及hart,模式分类及事件(scene)分析,1973,约翰威立公司,纽约州(下文的“duda1973”)的页数211至216中描述聚类的进行,其通过引用整体并入本文中。如duda1973的第6.7节所描述,所述聚类的问题被描述为在一数据集中找寻自然编组的一种。为了辨识多个自然编组,要解决两个问题。第一,确定一种用于量测两个样本之间的相似度(或相异度)的方法。将此度量(相似度的量测)用于确保一个群集中的多个样本相较于它们与其他群集中的多个样本彼此间更相似。第二,确定一种用于利用所述相似度量测来将所述数据划分为多个群集的机构。多个相似度的量测在duda1973的第6.7节中进行讨论,其中说明了开始一聚类调查的一种方法是定义一距离函数,并计算在一训练集合中所有样本对之间的距离矩阵。假如所述距离为一良好的相似度量测,则在相同群集中的多个参考实体(entities)之间的所述距离将显着地小于在不同群集中的多个参考实体之间的所述距离。然而,如duda1973的第215页所陈述,聚类的进行不需要使用一距离度量。例如,一非度量性相似度函数s(x,x')可被用于比较两个向量x及x'。按照常规,当x及x'某种程度上为“相似的”时,s(x,x')为数值很大的一对称函数。在duda1973的第218页提供了一非度量性相似度函数s(x,x')的一示例。一旦已经选择一种用于量测一数据集中的多个点之间的“相似度”或“相异度”的方法,聚类的进行就需要一标准函数,所述标准函数量测所述数据的任何分区的一聚类质量。将所述标准函数极化的所述数据的多个分区被用于聚集所述数据。参见duda1973的第217页。多个标准函数在duda1973的第6.8节中进行讨论。近期,已经出版duda等人,模式分类,第二版,约翰威立公司,纽约州。页数537至563详细描述了聚类的进行。更多关于多个聚类技术的信息可在kaufman及rousseeuw,1990,在一数据中找寻群组:对于聚类分析的介绍,威立,纽约,纽约州;everitt,1993,聚类分析(3d版),威立,纽约,纽约州;及backer,1995,聚类分析中的计算机辅助推理,普林迪斯豪尔,上萨德尔里弗,新泽西州中发现,其中的每一个通过引用并入本文中。可用于本公开中的多个特定的示例性聚类技术包括,但不限于,层次聚类(使用最近邻算法、最远邻算法、平均链接算法、质心算法或平方和算法进行的聚合式聚类)、k均值聚类、模糊k均值聚类算法,及贾维斯-派翠克(jarvis-patrick)聚类。在一些实施例中,所述聚类包括非监督式聚类,其中没有对所述训练集进行聚类时应形成什么聚类的先入为主的概念。
在一些实施例中,所述分类器为一回归模型,例如,在agresti,对于分类数据分析的介绍,1996,约翰威立公司,纽约州,第8章中所描述的多个多类别罗吉斯(logit)模型,其通过引用整体并入本文中。在一些实施例中,所述分类器使用在hastie等人,2001,统计学习的要素,施普林格出版社,纽约州所公开的一回归模型。
一种不需要肿瘤配对的用于确定肿瘤分数的可替代的方法上文结合图2所公开的多个方法需要使用来自所述受试者的一异常组织,例如一肿瘤组织,的一参考集合128。本公开的另一方面提供一种用于确定在一受试者的一液体生物样本的无细胞核酸中肿瘤分数,其不需要将多个等位基因的频率与一对应的肿瘤样本进行配对。这种无参考的方法包括:在一计算机系统中具有一个或多个处理器及一记忆体,所述记忆体存储通过所述一个或多个处理器执行的一个或多个程序,在所述计算机系统中获得来自所述受试者的所述液体生物样本的电子形式的多个序列读值,其中所述液体生物样本包括多个无细胞核酸分子。在一些实施例中,使用任何用于获得上文结合图2的方块202至208所公开的这类的序列读值的方法。
所述方法进一步包括:使用所述多个序列读值来辨识对一变体集合中的每个变体的支持,从而确定所述第一变体集合中的每个变体的一观察到的频率。在一些实施例中,使用上文结合方块210至226所公开的任何使用多个序列读值来辨识对一变体集合中的每个变体的支持,从而确定所述变体集合中的每个变体的一观察到的频率的方法。
所述方法进一步包括:将在所述变体集合中具有第n个最高等位基因频率的所述变体的所述观察到的频率视为所述受试者的所述液体生物样本的无细胞核酸中的所述肿瘤分数,其中n为1以外的正整数(例如,1、2、3、4、5等)。图17提供从肿瘤变体覆盖率估计出的肿瘤分数与单独从cfdna估计出的无参考肿瘤分数的一比较。因此,图17将从在本公开的一方面的cfdna中重新识别的多个小变体估计出的所述无参考tf,其中n被设置为2(y轴)与从通过使用上文结合图2所描述的配对方法所评估的cfdna中的肿瘤突变覆盖率估计出的tf(x轴)进行比较。为了估计所述无参考tf,重新从示例12所描述的ccga群体的多个art测定序列读值识别多个体细胞变体。在进行噪声建模、利用白细胞(wbc)的接合建模,及边缘变体的伪影建模之后,将多个变体过滤,如在2018年11月27日提交的标题为“用于靶向定序的模型”的美国专利申请第16/201,912号所公开,其通过引用并入本文中。再者,对多个变体进行变体归因。例如,参见2018年11月27日提交的标题为“用于靶向定序的模型”的美国专利申请第16/201,912号,其通过引用并入本文中。在被辨识为体细胞且不归因于wbc起源的多个变体中,肿瘤分数被估计为排名第二的变体等位基因频率(af_max2)。在图17中,透过肿瘤证据(至少在cfdna中的至少一肿瘤突变读值,真实)是否对上在cfdna中没有肿瘤证据(错误)对多个结果进行分面(faceted)。图17显示出在对于具有阳性读值证据的多个样本的多个估计值中,本公开的即时方面的所述无参考方法与图2的所述配对方法的一致性下降至大约1/1000的肿瘤分数。
在一些实施例中,所述变体集合中的一变体为与一预定的基因组位置相关的一单核苷酸变体、与一预定的基因组位置相关的一插入突变、与一预定的基因组位置相关的一缺失突变、一体细胞拷贝数的改变、与一预定的基因组位点相关的一核酸重组,或与一预定的基因组位置相关的一异常的甲基化模式。
在一些实施例中,将所述多个序列读值中的一个别的序列读值视为支持所述变体集合中的一第一变体,这是在所述个别的序列读值含有所述第一变体的全部或一部分时,以及将所述多个序列读值中的一个别的序列读值视为不支持所述变体集合中的一第一变体,这是在所述个别的序列读值不含有所述第一变体时,并且所述多个序列读值中的支持所述第一变体的多个序列读值的一数量对上所述多个序列读值中的不支持所述第一变体的多个序列读值的一数量用以确定所述第一变体的所述观察到的频率,所述第一变体的所述观察到的频率可估计在所述液体生物样本中的所述第一变体的所述变体频率。
在一些实施例中,所述受试者患有来自一单一原发部位的一癌症。在一些实施例中,所述受试者患有源自于两个或更多个不同器官的一癌症。在一些实施例中,所述受试者患有乳腺癌、肺癌、前列腺癌、结肠直肠癌、肾癌、子宫癌、胰腺癌、食道癌、淋巴瘤、头颈癌、卵巢癌、肝胆癌、黑色素瘤、子宫颈癌、多发性骨髓瘤、白血病、甲状腺癌、膀胱癌、胃癌或其组合。
在一些实施例中,所述变体集合包括五个或更多个变体,并且在所述变体集合中的每个个别的变体位在所述受试者的所述基因组的一不同的位点上。在一些实施例中,所述变体集合是由介于3至20个之间的变体组成,并且在所述变体集合中的每个变体为所述受试者的所述基因组中的一不同的遗传变异。
在一些实施例中,所述变体集合是由介于2至200个之间的变体组成,并且在所述变体集合中的每个变体为所述受试者的所述基因组中的一不同的遗传变异。在一些实施例中,所述变体集合包括1000个变体,并且在所述变体集合中的每个变体为所述受试者的所述基因组中的一不同的遗传变异。
在一些实施例中,使用所述多个序列读值来辨识对一变体集合中的每个变体的支持的步骤包括:将所述多个序列读值中的一序列读值与一参考基因组中的一区域进行比对,以确定所述序列读值是否含有一第一变体的全部或一部分。
在一些实施例中,使用所述多个序列读值来辨识对一变体集合中的每个变体的支持的步骤包括:将所述多个序列读值中的一序列读值与多个变体的一查找表进行比对,以确定所述序列读值是否含有一第一变体的全部或一部分。
在一些实施例中,使用所述多个序列读值来辨识对一变体集合中的每个变体的支持的步骤包括:将所述多个第一序列读值中的一序列读值与一查找表中的每个条目比对,其中所述查找表中的每个条目代表一基因组的一不同部分。
在一些实施例中,所述液体生物样本包括或由以下组成:所述受试者的血液、全血、血浆、血清、尿液、脑脊髓液、粪便、唾液、汗水、眼泪、胸水、心包液或腹膜液。
在一些实施例中,所述方法进一步包括:从在每个个别的时间点取得所述受试者的一个别的生物样本中,于一时期内的多个时间点中的每个个别的时间点重复所述获得多个序列读值的步骤,其中所述个别的生物样本包括多个无细胞核酸分子,从而获得在每个个别的时间点对于所述受试者的对应的多个序列读值;及对于在所述多个时间点中的每个个别的时间点,确定对所述原先的认定步骤的所述变体集合中具有所述第n个最高等位基因频率的所述变体的支持,从而以在所述时期内的所述变体的所述等位基因频率的增加或减少的形式来确定所述时期期间的所述受试者的一病况的状态或进展。
在一些实施例中,所述时期为数个月的一时间段(例如,介于1个月至4个月之间),幷且在所述多个时间点中的每个时间点为所述数个月的时间段中的一不同的时间点。在一些实施例中,所述时期为数个年的一时间段(例如,介于2至10年之间),幷且在所述多个时间点中的每个时间点为所述数个年的时间段中的一不同的时间点。在一些实施例中,所述时期为数小时的一时间段(例如,介于1小时至6小时之间),幷且在所述多个时间点中的每个时间点为所述数小时的时间段中的一不同的时间点。
在一些实施例中,所述方法进一步包括:当观察到所述变体的所述等位基因频率在所述时期内以一阈值量变化时(例如,如在第一量测时间点,相对于一参考量改变百分之十、百分之二十、百分之三十),改变所述受试者的一诊断。
在一些实施例中,所述方法进一步包括:当观察到所述变体的所述等位基因频率在所述时期内以一阈值量变化时(例如,如在第一量测时间点,相对于一参考量改变百分之十、百分之二十、百分之三十),改变所述受试者的一预后情形。
在一些实施例中,所述方法进一步包括:当观察到所述变体的所述等位基因频率在所述时期内以一阈值量变化时(例如,如在第一量测时间点,相对于一参考量改变百分之十、百分之二十、百分之三十),改变所述受试者的一治疗。
在一些实施例中,所述病况为一癌症(例如,乳腺癌、肺癌、前列腺癌、结肠直肠癌、肾癌、子宫癌、胰腺癌、食道癌、淋巴瘤、头颈癌、卵巢癌、肝胆癌、黑色素瘤、子宫颈癌、多发性骨髓瘤、白血病、甲状腺癌、膀胱癌、胃癌或其组合)。在一些实施例中,所述病况为一癌症的一阶段(例如,一乳腺癌的一阶段、一肺癌的一阶段、一前列腺癌的一阶段、一结肠直肠癌的一阶段、一肾癌的一阶段、一子宫癌的一阶段、一胰腺癌的一阶段、一食道癌的一阶段、一淋巴瘤的一阶段、一头颈癌的一阶段、一卵巢癌的一阶段、一肝胆癌的一阶段、一黑色素瘤的一阶段、一子宫颈癌的一阶段、一多发性骨髓瘤的一阶段、一白血病的一阶段、一甲状腺癌的一阶段、一膀胱癌的一阶段或一胃癌的一阶段)。在一些实施例中,所述病况为一癌症的一预定亚型。
在一些实施例中,所述方法进一步包括:将所述多个序列读值应用于一训练过的分类器,从而获得一分类器的结果,其中所述训练过的分类器的结果指示出所述受试者是否患有一第一癌症病况;及当所述肿瘤分数介于0.003至1.0之间,且所述训练过的分类器的结果指出所述受试者具有所述第一癌症病况时,使用所述训练过的分类器的结果作为所述受试者对于所述第一癌症病况的诊断的一基础。在一些这样的实施例中,所述第一癌症病况为一癌症(例如,乳腺癌、肺癌、前列腺癌、结肠直肠癌、肾癌、子宫癌、胰腺癌、食道癌、淋巴瘤、头颈癌、卵巢癌、肝胆癌、黑色素瘤、子宫颈癌、多发性骨髓瘤、白血病、甲状腺癌、膀胱癌、胃癌或其组合)。在一些这样的实施例中,所述第一癌症病况为一癌症的一亚型(例如,乳腺癌、肺癌、前列腺癌、结肠直肠癌、肾癌、子宫癌、胰腺癌、食道癌、淋巴瘤、头颈癌、卵巢癌、肝胆癌、黑色素瘤、子宫颈癌、多发性骨髓瘤、白血病、甲状腺癌、膀胱癌或胃癌的一亚型)。在一些这样的实施例中,所述第一肿瘤分数介于0.003至1.0之间,且所述第一癌症病况为一癌症的一起源组织。在一些实施例中,所述训练过的分类器为一神经网络、一支持向量机、一决策树、一非监督式聚类模型、一监督式聚类模型或一回归模型。
示例1
根据癌症阶段增加的ctdna分数的中位数
参考图4,不管多个受试者患有的癌症类型,将所述多个受试者通过癌症阶段i、ii、iii及iv进行分组。在图4中,x轴指出每个受试者患有的癌症阶段为何,而y轴指出对于每个受试者的所述观察到的ctdna分数。用于计算对于每个受试者的所述cfdna分数的所述方法包括:获得从一群体中的每个受试者的一生物样本而来的电子形式的多个第一序列读值140,其中所述生物样本包括多个无细胞核酸分子。所述多个第一序列读值140用于辨识对所述生物样本的一变体集合142中的每个变体144的支持,从而确定在所述变体集合142中的每个变体144的一观察到的频率(支持146)。将所述变体集合142中的每个个别的变体144的所述观察到的频率(支持146)与一参考集合128中对于所述个别的变体的一对应的参考频率132进行比较。在所述参考集合128中的每个这样的对应参考频率132为在从所述受试者获得的一第一异常组织样本中的一个别的变体的一频率。图4未包括不具有多个阳性读值的多个受试者,其意思为所述多个受试者不具有支持在这类受试者的所述配对参考集合中观察到的所述多个变体的多个序列读值140。
在所述变体集合是由一单一变体组成的例子中,将所述变体集合142中的每个个别的变体144的所述观察到的频率与一参考集合128中对于所述个别的变体的一对应的参考频率132进行比较的步骤包括:取得在所述变体集合中的所述变体的所述频率(从所述生物样本的cfdna的多个序列读值获得)与在所述参考集合中的相同变体的所述频率(从所述异常组织中的dna的多个序列读值获得)的一比率。
在所述变体集合是由两个或更多个变体组成的例子中,将所述变体集合142中的每个个别的变体144的所述观察到的频率与一参考集合128中对于所述个别的变体的一对应的参考频率132进行比较的步骤包括:取得在所述变体集合中的每个个别的变体的所述频率(从所述生物样本的cfdna的多个序列读值获得)与在所述参考集合中的与所述个别的变体相同的变体的所述频率(从所述异常组织中的dna的多个序列读值获得)的一比率。意即,在所述参考集合中的多个变体与所述变体集合中的多个变体之间具有一种一对一的对应性。
因此,图4提供一种不管癌症类型为何,在多个受试者中的ctdna分数如何随着癌症阶段而变化的分析,所述多个受试者具有支持它们的潜在癌症的多个无细胞序列读值。因此,图4显示出由于根据临床分期(第1至4期)确定所述疾病更为严重,因此在所述cfdna中发现更高的肿瘤分数(更大的ctdna分数)的证据。虽然图4显示出这是整个所述ccga群体的一般情况(对于ccga群体的的多个细节,请参见示例12),但针对此趋势存在有多个违背值(violations)(离群值)。图4中的多个这样的离群值是暗示性的,最好通过临床的错误分类来解释。因此,图4显示出所述潜在疾病的一基础组成,其为所述cfdna中的通常所预期的肿瘤分数比率。图4还显示第4期具有一些拥有非常低的脱离率的个体,这指示出在第4期中存在有不同的子状态。
图4说明从所述参考集合中的所述多个变体的多个观察到的频率,可使用多个脱离率(ctdna分数)作为建立多个有意义且有益的阈值的一基础。意即,例如,在一特定的受试者的所述异常组织中的多个变体的多个特定的频率,以及可选择地关于患有癌症的一特定时期的多个受试者的预期ctdna分数的信息、对于所述特定受试者的一阈值可被确定,并且对照针对所述特定受试者的一变体集合中的所述多个变体的所述观察到的频率来进行评估,以便将所述受试者分类成患有或未患有所述病况(例如,一特定癌症的一临床阶段)。例如,参考图4,可使用一阈值0.05来分析一受试者是否患有一特定癌症的第i期。在此示例中,诸如一肿瘤的一异常组织可从一患者获得,并用于确定一第一参考集合中对于每个个别的变体的一参考频率。事实上,在一些实施例中,各种可能的变体的所述频率可用于定义所述变体集合的所述多个变体。接着,从相同受试者的除了所述异常组织以外的一生物样本获得无细胞核酸,并且从所述无细胞核酸的多个序列读值确定在所述参考集合中的多个相同变体的所述变体频率,从而形成在所述第一变体集合中的每个个别的变体的所述观察到的ctdna频率。然后,用于确定是否满足所述0.05的阈值状况的所述ctdna分数与所述多个参考频率的比较提供了用于确定所述受试者是否患有第i期癌症的一基础。例如,假如所述比较指出所述ctdna高于0.05,则其指出所述受试者患有一更晚期的癌症。另一方面,观察到低于0.001的从所述第一变体集合中的每个个别的变体的所述观察到的频率形成的一ctdna分数与发现所述受试者患有一特定癌症的第i期具有一致性。
示例2
根据一乳腺癌阶段函数来侦测ctdna的能力
在图5中,每个点为在下文示例12所描述的ccga群体中的患有乳腺癌的一单独的受试者的所述ctdna分数,其中使用wgs定序。用于计算对于每个受试者的所述cfdna分数的所述方法包括:从一群体中的每个受试者的一生物样本获得电子形式的多个第一序列读值140,其中所述生物样本包括多个无细胞核酸分子。所述多个第一序列读值140用于辨识对所述生物样本的一变体集合142中的每个变体144的支持,从而确定所述变体集合142中的每个变体144的一观察到的频率(支持146)。将所述变体集合142中的每个个别的变体144的所述观察到的频率(支持146)与一参考集合128中对于所述个别的变体的一对应的参考频率132进行比较。在所述参考集合128中的每个这样的对应参考频率132为在从所述受试者获得的一第一异常组织样本中的一个别的变体的一频率。
除了绘制出每个受试者的所述ctdna分数,图5依据乳腺癌的阶段划分所述多个受试者,并依据三种不同类别的其中一个对每个受试者进行注释。第一类别(红色三角形)为所述受试者的一生物样本的所述多个序列读值140提供来足够的基础来独立地识别与所述参考集合中的所述多个变体的其中一个配对的至少一变体144的情况。因此,在多个这样的实施例中,所述异常组织样本(例如肿瘤)没有使用所述多个无细胞dna变体,反之亦然;而是在不需要依靠来自所数肿瘤的定序数据的情况下,基于来自所述生物样本(例如血液)的所述多个序列读值的所述靶向测定法独立地辨识所述变体。第二类别(蓝色三角形)代表对于一肿瘤变体的基于读值证据的分析,其中cfdna被观察到具有支持通过所述肿瘤的直接肿瘤定序所识别到的至少一变体的多个序列读值。第三类别(黑色圆形)指出没有证据表明所述多个cfdna序列读值具有与直接在所述异常组织(乳腺癌组织)中观察到的所述多个变体配对的多个变体。
图5指出对于在每个肿瘤阶段内观察到的肿瘤分数的一非常大的动态范围。图5进一步指示当所述肿瘤分数为百分之一或更高时,所述测定法利用一可评估的置信区间来侦测乳腺癌。在1.0%至0.1%之间,所述测定法的成效会下降。对于多个黑点,所述置信区间一直为零,意味着对于多个这样的个体,可确信这些个体样本不会超过所述肿瘤分数。因此,对图5中的第ii期进行分析,可看到大量的患有乳腺癌第ii期的受试者群体具有低于所述测定侦测的多个界限的一肿瘤分数。换言之,有大量的患有第ii期乳腺癌的受试者具有低脱离率,其指出对于多个这样的受试者的所述cfdna中的ctdna的辨识低于所述多个侦测界限。
示例3
根据一cfdna分数的函数来侦测ctdna的能力
图6提供了在下文使用wgs定序的示例12所描述的ccga群体中,有多少个体被分类成患有癌症的一估计值,其中使用三种不同分类器的其中一种(y轴)作为所述cfdna分数的一函数(x轴)。意即,基于cfdna分数将多个受试者分成所述x轴上的八个数据箱的其中一个,接着,对于三种不同的分类器的每一个,在所述y轴上绘制出每个这类的数据箱在95%特异性下的敏感度的平均值集范围。对于图6中的每个cfdna数据箱,所述三种不同的分类器从左到右(使用所述数据箱(0,0.000316]来说明)为“a得分”602、“b得分”604及“m得分”606。
本文所描述的a得分分类器是基于多个非同义性突变的靶向定序分析的肿瘤突变负荷的一分类器。例如,可使用针对肿瘤突变负荷数据的逻辑回归来计算一分类得分(例如“a得分”),其中从所述靶向cfdna测定法获得对于每个个体的肿瘤突变负荷的一估计值。在一些实施例中,根据每个个体的多个变体的总数量可估计一肿瘤突变负荷,所述个体:被识别为所述cfdna中的多个候选变体、通过噪声建模及接合识别,及/或在与所述多个变体重叠的任何基因注释中被发现为非同义的。一训练集合的所述肿瘤突变负荷的数量可被输入至一惩罚型逻辑回归分类器,以使用交叉验证来确定达到95%特异性的临界值(cutoff)。图6显示出所述交叉验证成效的一示例。例如,在r.chaudhary等人,2017,“使用次世代定序测定法来估计肿瘤突变负荷”,journalofclinicaloncology,35(5),suppl.e14529,线上公开预印本中可找到a得分的多个额外的细节,其通过引用整体并入本文中。
所述b得分分类器在以62/642,461提交的标题为“用于选择、管理及分析高维度数据的方法及系统”的美国专利公开第62/642,461号中进行描述,其通过引用并入本文中。根据所述b得分方法,对于多个低变异度的区域,分析来自在多个健康受试者的一参考群组中的多个健康受试者的多个核酸样本的一第一序列读值集合。因此,将在来自每个健康受试者的多个核酸样本的所述第一序列读值集合中的每个序列读值与所述参考基因组中的一区域进行比对。由此,选择从来自一训练群组中的多个受试者的多个核酸分子的多个序列读值而来的一训练的序列读值集合。将所述序列集合中的每个训练读值与从所述参考集合辨识到的所述参考基因组中的所述多个低变异的区域中的一区域进行比对。所述训练集合包括来自多个健康受试者的多个核酸样本的多个序列读值,以及来自其已知患有癌症的多个患病受试者的多个核酸样本的多个序列读值。来自所述序列群组的所述多个核酸样本为与来自多个健康受试者的所述参考群组的所述多个核酸样本的类型相同或相似的一类型。由此,使用源自于所述训练集合的多个序列读值的数量,来确定一个或多个参数,所述一个或多个参数反映出在来自所述多个健康受试者的多个核酸样本的多个序列读值与来自所述训练群组中的所述多个患病受试者的多个核酸分子的多个序列读值间的差异。接着,接收与多个核酸分子相关的一测试的序列读值集合,所述测试的序列读值集合包括来自其状态在所述癌症方面是未知的一测试受试者的多个cfna片段,并且基于所述一个或多个参数来确定所述测试患者患有所述癌症的可能性。
所述m得分分类器在2018年3月13日提交的标题为“甲基化片段异常检测”的美国专利申请第62/642,480号中进行描述,其通过引用并入本文中。
图6指出在高于3%的cfdna分数时,三种分类器均检测出患有所述癌症的所述多个个体。对于较低的cfdna分数,所述m得分分类器的敏感度在(0.00316,0.01]区间相对于所述b得分分类器在统计上具有显着的提高。因此,对于中间的所述脱离率,所述m得分分类器似乎更具优势。对于较低的脱离率,在cfdna低于00.316时,似乎没有分类器为合适的。因此,图6为如何精进改善所述癌症检测分类器提供动机。在所述x轴上,在两个数值之间的逗点表示范围,圆括号表示不包括在内,方括号表示“包括在内”。对于3%或更高的cfdna分数,所述多个分类器分别具有95%或更高的一敏感率以及5%的一伪阳率。
示例4
根据一cfdna分数函数、定序实验步骤及乳腺癌亚型来识别乳腺癌的能力
图7a及7b详细描述一乳腺癌识别分类器的敏感度,其使用全基因组亚硫酸氢盐定序(wgbs)(图7a)及全基因组定序(wgs)(图7b)来进行变体识别,从而通过使用下文示例12所描述的ccga群体,来根据对于乳腺癌的四个不同亚型的一cfdna分数函数将多个受试者识别为是否患有乳腺癌,所述乳腺癌的四个不同亚型为her2+(实心圆)、hr+/her2-(空心圆)、其他/遗失(实心方块)及tnbc(空心方块)。图7证明的是,考虑到一乳腺癌亚型(例如,her2+对上激素受体+(hr+),不同类型的变体识别方法在分类器敏感度上存在差异。图7进一步指出,相较于侵袭性较低形式的乳腺癌,侵袭性较高的癌症对于her2+的信号可用性较佳。例如,参见图7a中的所述(0.001,0.00316]区间的敏感度。在图7中,敏感度为一癌症对上非癌症的指派(assignment)。对于图7,不需要使用任何ctdna脱离信息来分别基于wgsb及wgs数据将多个受试者识别为“患有癌症”及“未患有乳腺癌”。图7证明癌症侦测分类器对于这些癌症具有较高的ctdna分数。
示例5
根据一cfdna分数函数的一全基因组亚硫酸氢盐定序的多类别癌症类型分类器的精确度
图8详细描述根据一cdna分数函数的针对已经使用全基因组亚硫酸氢盐定序(wgbs)进行定序的所述ccga受试者群体(下文示例12)的一多类别分类器的精确度,其跨越了图3中所辨识出的不同癌症的范围(spectrum)。对于有关wgbs的多个细节,例如,参见示例13。还可参见在2018年3月13日提交的标题为“甲基化片段异常检测”的美国专利申请第62/642,480号,其通过引用并入本文中。如图8所说明,将所述群体分成八个不同的cfdna分数的数据箱,并且提供对于每个这样的数据箱的所述wgbs分类器的所述精确度,及在每个这样的数据箱的所述群体中的多个受试者的数量,所述精确度被定义为将一特定受试者的正确癌症置于前两个癌症类别概率中的能力。图8表明了使用所述wgbs多类别癌症类型分类器需要一ctdna分数水平的阈值才能实现正确的指派。
示例6
根据一临床阶段函数的显示出一最小ctdna分数的多个受试者的比例
图9说明在所述ccga群体中,显示出跨越由所述群体代表的所有癌症的一最小ctdna分数的多个样本的数量。如同示例1中的例子,对于图9中公开的每个受试者,用于计算所述cfdna分数的方法包括:获得从所述群体中的每个受试者的一生物样本而来的电子形式的多个第一序列读值140,其中所述生物样本包括多个无细胞核酸分子。所述多个第一序列读值140用于辨识对所述生物样本的一变体集合142中的每个变体144的支持,从而确定在所述变体集合142中的每个变体144的一观察到的频率(支持146)。将所述变体集合142中的每个个别的变体144的所述观察到的频率(支持146)与一参考集合128中对于所述个别的变体的一对应的参考频率132进行比较,以确定每个受试者的所述ctdna分数。在所述参考集合128中的每个这样的对应参考频率132为在从所述受试者获得的一第一异常组织样本中的一个别的变体的一频率。
图9公开所述群体中的多个受试者的比例,其显示出0.01的ctdna分数从刚好高于0.00的针对由所述群体代表的所有第i期癌症(在所述群体中患有第i期癌症的受试者n=157)爬升至大约0.75的针对由所述群体代表的所有第ii期癌症(在所述群体中患有第iv期癌症的受试者n=59)。图9说明可使用多个癌症患者的所述ctdna分数中的可用信息,以便根据包括图2所描述的多个方法在内的本公开内容来对多个受试者的病况进行分类。示例1至6共同显示出,本公开的所述多个方法能够分类多个受试者、基于ctdna分数来评估多个分类器的性能,以及鉴于跨越不同癌症类型的一固定的ctdna分数来评估信号的质量。有利的是,示例1至6共同显示出,所公开的多个系统及方法能够侦测更具侵袭性的癌症形式,这是高度需要的。
示例7
将ctdna分数与源自于肿瘤结合的数字病理学的计算机模型
示例1至6指出可将根据本公开的多个方法所确定的所述ctdna分数与从数字病理学获得的信息结合,以提供一种预测一特定癌症的所述侵袭性的模型。因此,本公开证明多个模型的实用性考虑到ctdna分数,并进一步包括数字病理学,以确定一特定受试者的一特定癌症病况的所述侵袭性。通过从一受试者的一生物样本获得电子形式的多个第一序列读值140,来确定对于所述受试者的所述cfdna分数,其中所述生物样本包括多个无细胞核酸分子(例如,来自所述受试者的血液)。根据本公开的多个教示,所述多个第一序列读值140用于辨识对所述生物样本的一变体集合142中的每个变体144的支持,从而确定在所述变体集合142中的每个变体144的一观察到的频率(支持146)。将所述变体集合142中的每个个别的变体144的所述观察到的频率(支持146)与一参考集合128中对于所述个别的变体的一对应的参考频率132进行比较,以确定所述受试者的所述ctdna分数。多个这样的参考频率132是从多个序列读值获得,所述多个序列读值是从所述受试者的一肿瘤或一肿瘤部分取得。并且,使用多个计算机视觉技术来分析所述肿瘤或肿瘤部分的一个或多个切片,从而估计密度、有多少免疫细胞浸润、估计细胞坏死,及/或估计增殖率,或其他与一癌症的侵袭性有关的参数。接着,将此信息与所述ctdna结合,并输入至一分类器中,所述分类器评估所述受试者的所述癌症的所述侵袭性,及/或任何与所述癌症相关的其他状态。
示例8
图10说明了使用示例12所描述的ccga群体的跨越癌症的所有阶段的ctdna分数与肿瘤尺寸的正相关性。由于在许多例子中的肿瘤尺寸与癌症侵袭性具有正相关,因此根据本公开,示例8为使用cfdna分数来分类多个受试者提供额外的支持,包括结合图2来公开的多个方法、下文所公开的多个额外的实施例,及本公开的多个权利要求。
示例9
ctdna分数与针对增殖的ki67标志物的相关性
ki-67为一种与细胞增殖相关的核蛋白。参见gerdes等人,1983,“与有关细胞增殖的人类核抗原反应的小鼠单克隆抗体的产生”,int.j.cancer31(1),13-20,其通过引用并入本文中。一种用于分析一受试者的所述ki-67抗原的方法为免疫组化评估。其已经显示,所述ki-67核抗原在细胞周期的某些时期中表现,即s、g1、g2及m时期,而不存在g0中。例如,参见gerdes等人,1984,“通过单克隆抗体ki-67所限定的与细胞增殖相关的人类核抗原的细胞周期的分析”,jimmunol.133(4),1710–1715;及scholzen及gerdes,2000,“ki-67蛋白:来自已知及未知”,jcellphysiol.182(3),311–322,其中的每一个通过引用并入本文中。在来自正常乳腺组织的多个样本中,已经发现ki-67在er阴性细胞中也以低水平表现(细胞的<3%),而不在er阳性细胞中表现。例如,参见urruticoechea等人,2005,“早期乳腺癌中的增殖标志物ki-67”,jclinoncol.23:7212–7220,其通过引用并入本文中。通过利用所述单克隆抗体ki-67进行的免疫染色,可评估肿瘤细胞群的生长分数。
对于此示例,实施免疫组化染色,并且使用光学显微镜以一定量及视觉的方式评估对于所述核抗原ki-67的染色呈阳性的多个恶性细胞的比例。通过使用所述抗人类ki-67单克隆抗体,根据多个阳性标记的恶性细胞的百分比来获取多个ki-67值。在图11中,所述ki-67的百分比得分被定义为在所评估的恶性细胞的总数量中的多个阳性染色的肿瘤细胞的百分比。参见inwald,2013,“ki-67为乳腺癌患者中的预后参数:大量的基于人群的癌症登记群体的结果”,breastcancerres.treat.139(2):539-552,其通过引用并入本文中。
在图11中,通过获得来自所述受试者的一生物样本的电子形式的多个第一序列读值140,来确定在下文示例12所描述的显示出实体侵袭性癌症的ccga群体中的对于每个特定受试者的所述cfdna,其中所述生物样本包括多个无细胞核酸分子(例如,来自所述受试者的所述血液)。所述多个第一序列读值140用于辨识对所述生物样本的一变体集合142中的每个变体144的支持,从而确定在所述变体集合142中的每个变体144的一观察到的频率(支持146)。将所述变体集合142中的每个个别的变体144的所述观察到的频率(支持146)与一参考集合128中对于所述个别的变体的一对应的参考频率132进行比较,以确定所述受试者的所述ctdna分数。多个这样的参考频率132是从多个序列读值获得,所述多个序列读值是从获得所述多个ki-67数值的所述受试者的一肿瘤或一肿瘤部分取得。
在图11中,在最左边的柱体中,具有大于10的一ki-67得分的多个样本显示出,在具有大于0.1000的脱离率的尾部中有许多样本。这表明ki-67与所述ctdna分数的结合可为诊断一受试者的一病况(例如更具侵袭性的乳腺癌形式)提供一基础。
示例10
获得多个序列读值
图12为根据一实施例的一种制备用于定序的一核酸样本的方法1200的一流程图。所述方法包括,但不限于,以下多个步骤。例如,所述方法1200的任何步骤可包括一个用于质量控制的定量子步骤,或本领域技术人员已知的其他实验室测定流程。
在方块1202中,从一受试者提取一核酸样本(dna或rna)。所述样本可为人类基因组的任何子集合,包括全基因组。所述样本可从已知患有或疑似患有癌症的一受试者提取出。所述样本可包括血液、血浆、血清、尿液、粪便、唾液、其他类型的体液,或其任何组合。在一些实施例中,用于抽取一血液样本的多个方法(例如,注射器或扎手指)可比用于获得一组织活检的多个流程的侵入性小,所述用于获得一组织活检的多个流程可能需要手术。所述提取出的样本可包括cfdna及/或ctdna。对于多个健康个体,人体可自然清除cfdna及其他细胞碎片。假如一受试者患有一癌症或疾病,则一提取出的样本中的ctdna可能以一可侦测的水平存在,以进行诊断。
在方块1204中,制备一定序文库。在文库制备期间,通过衔接子连接可将多个独特分子指标(umi)添加到所述多个核酸分子(例如dna分子)中。所述多个umi为多个短核酸序列(例如,4至10个碱基对),所述多个短核酸序列在衔接子连接期间被添加到多个dna片段的末端。在一些实施例中,多个umi为多个简并(degenerate)碱基对,所述多个简并碱基对用作为一独特标签,所述独特标签可用于辨识源自于一特定的dna片段的多个序列读值。在衔接子连接后的pcr扩增期间,所述多个umi与附着的dna片段一起复制。这提供了一种在下游分析中辨识来自相同原始片段的多个序列读值的方法。
在方块1206中,从所述文库富集多个目标dna序列。在富集期间,使用多个杂交探针(本文也称为“探针”)来靶向及拉下多个核酸片段,所述多个核酸片段提供是否存在癌症(或疾病)、癌症状态,或一癌症分类(例如,癌症类型或起源组织)的信息。对于一特定的工作流程,所述多个探针可被设计用以粘合(或杂交)至dna的一目标(互补)股。所述目标股可为“正”股(例如,此股被转录成mrna,随后被转译成一蛋白质)或“负”股。所述多个探针的长度范围可为数十个、数百个或数千个碱基对。在一些实施例中,基于一基因小组来设计所述多个探针,以便分析对应于某些癌症或其他疾病类型的(例如,人类或其他生物体的)所述基因组的多个特定的突变或多个目标区域。并且,所述多个探针可覆盖一目标区域的多个重叠部分。
图13为根据一实施例的一个用于获得多个序列读值的过程的一图解表示法。图13描绘来自所述样本的一核酸片区段1300的一示例。在此,所述核酸片段1300可为一单股核酸片段,例如一单一股。在一些实施例中,所述核酸片段1300为一双股cfdna片段。此说明示例描绘出可通过不同探针靶向的所述核酸片段的三个区域1305a、1305b及1305c。具体来说,所述三个区域165a、165b及165c中的每一个包括在所述核酸片段160上的一重叠位置。一示例性重叠位置在图13中被描绘成胞嘧啶(“c”)核苷酸碱基1302。所述胞嘧啶核苷酸碱基1302位于区域1305a的一第一边缘附近、位于区域1305b的中间,及位于区域1305c的一第二边缘附近。
在一些实施例中,基于一基因小组来设计所述多个探针的一个或多个(或全部),以便分析疑似对应于某些癌症或其他疾病类型的(例如,人类或其他生物体的)所述基因组的多个特定的突变或多个目标区域。通过使用一目标基因小组,而不是将一基因组的所有表现基因进行定序,也称为“全外显子组定序”,所述方法1200可用于增加所述多个目标区域的定序深度,其中深度指的是在所述样本中的一特定目标序列被定序的次数的计数。定序深度的增加降低了所述核酸样本需要输入的量。
使用一个或多个探针对所述核酸样本1300进行杂交,以产生对一目标序列1370的理解。如图13所示,所述目标序列1370为由一杂交探针靶向的所述区域1305的所述核苷酸碱基序列。所述目标区域1307也可被称为一杂交核酸片段。例如,所述目标1370a对应于由一第一杂交探针靶向的区域1305a,所述目标区域1370b对应于由一第二杂交探针靶向的区域1305b,及所述目标区域1370c对应于由一第三杂交探针靶向的区域1305c。考虑到所述胞嘧啶核苷酸碱基1302位在由一杂交探针靶向的每个区域1305a至c中的不同位置处,每个目标序列1370包括与位在所述目标序列1370上的一特定位置处的所述胞嘧啶核苷酸碱基1302对应的一核苷酸碱基。
在一杂交步骤后,所述多个杂交核酸片段被捕捉,并且还可使用pcr进行扩增。例如,所述多个目标序列1370可被富集,以获得后续可进行定序的多个经富集的序列1380。在一些实施例中,从一目标序列1370复制每个经富集的序列1380。分别从目标序列1370a及1370c扩增的多个经富集的序列1380a及1380c还包括位在每个序列读值180a或180c的边缘附近的胸腺嘧啶核苷酸碱基。如下文所使用,在所述经富集的序列1380中的相对于所述参考等位基因(例如,胞嘧啶核苷酸碱基1302)发生突变的所述突变核苷酸序列(例如,胸腺嘧啶核苷酸碱基)被认为是一替代等位基因。此外,从目标序列1370b扩增的每个经富集的序列1380b包括靠近或位于每个经富集的序列1380b中间的所述胞嘧啶核苷酸碱基。
在方块1208中,从所述多个经富集的dna序列中产生多个序列读值,例如,图13所示的多个经富集的序列180。通过本领域已知的工具可从所述多个经富集的dna序列获得多个定序数据。例如,所述方法1200可包括次世代定序(ngs)技术,其包括合成技术(伊卢米那)、焦磷酸定序(454莱富生命科技)、离子半导体技术(离子倾注定序)、单一分子即时定序法(太平洋生物科学)、连接定序法(solid定序)、纳米孔定序法(牛津纳米孔公司),或成对末端定序。在一些实施例中,使用合成定序与多个可逆染色终止子来进行大规模平行定序。
在一些实施例中,使用本领域已知的多个方法来将所述多个序列读值与一参考基因组进行比对,以确定比对位置的信息。所述比对位置的信息可指示出所述参考基因组中对应于一特定序列读值的一起始核苷酸碱基与一末端核苷酸碱基的一区域的一起始位置与一终点位置。所述比对位置的信息还可包括由所述起始位置与所述末端位置决定的序列读值长度。在所述参考基因组中的一区域可与一基因或一基因的一片段相关。
在各种实施例中,一序列读值包括表示为r1及r2的一读值对。在一些示例中,所述第一读值r1是从一核酸片段的一第一末端定序,而所述第二读值r2是从所述核酸片段的一第二末端定序。因此,在所述示例中,所述第一读值r1及所述第二读值r2的多个核苷酸碱基对与所述参考基因组的多个核苷酸碱基在比对上为一致的(例如,在相反方向上)。源自于所述读值对r1及r2的所述比对位置信息可包括:在所述参考基因组中对应于一第一读值(例如r1)的一末端的一起始位置,及在所述参考基因组中对应于一第二读值(例如r2)的一末端的一终点位置。换言之,所述参考基因组中的所述起始位置及所述终点位置代表在所述参考基因组中与所述核酸片段对应的可能位置。可产生具有sam(序列比对图谱)格式或bam(二进制)格式的一输出文件,并将所述输出用于进一步的分析,例如,上文结合图2及示例11所描述的变体识别。
示例11
辨识变体
图14为根据一实施例的一种用于确定多个序列读值的多个变体的方法1400的流程图。在一些实施例中,如上文结合图2及示例10所讨论,进行基于输入定序数据的变体识别(例如,对于多个snv及/或多个插入缺失(indel))。在步骤1402,将所述输入定序数据的多个比对过的序列读值进行分裂。在一些实施例中,将多个序列读值分裂的步骤包括:使用多个umi及可选择地来自一输出文件(例如,从示例10所描述的方法而来)的定序数据的比对位置信息,来将多个序列读值分裂成一共有序列,以便用于确定一核酸片段或所述核酸片段的一部分的最可能的序列。在一些实施例中,所述独特序列标签的长度为大约4至20个核酸。由于所述多个umi与所述多个经连接的核酸片段通过富集及pcr一起进行复制,因此可确定某些源自于一核酸样本的相同分子的序列读值。在一些实施例中,将具有相同或相似的比对位置信息(例如,一阈值偏移量(offset)的起始及终点位置)及包括一共同umi的多个序列读值分裂,并且产生一分裂过的读值(本文也称为一共有读值)来代表所述核酸片段。假如对应的所述分裂过的读值对具有一共同的umi,则一共有读值被指定为“双链体(duplex)”,这指出可捕捉到所述来源核酸分子的正股跟负股两种;否则,所述分裂过的读值被指定为“非双链体(non-duplex)”。在一些实施例中,作为将多个序列读值进行分裂的一替代方法或补充,可在多个序列读值上进行其他类型的错误校正。
在步骤1405,基于对应的比对位置信息来将所述多个分裂过的读值缝合。在一些实施例中,比较在一第一读值与一第二读值之间的比对位置信息,以便确定所述第一及第二读值的核苷酸碱基对在所述参考基因组中是否重叠。在一使用例子中,响应于确定在所述第一及第二读值之间的一重叠(例如,一特定数量的核苷酸碱基)大于一阈值长度(例如,核苷酸碱基的阈值数量),所述第一及第二读值被指定为“经缝合的”;否则,所述多个分裂过的读值被指定为“未经缝合的”。在一些实施例中,假如所述重叠大于所述阈值长度,且假如所述重叠不是一滑行重叠,则一第一及第二读值被缝合。例如,一滑行重叠可包括一均聚物运行(例如,一单一的重复核苷酸碱基)、一双核苷酸运行(例如,两个核苷酸碱基序列),或一三核苷酸运行(例如,三个核苷酸碱基序列),其中所述均聚物运行、双核苷酸运行或三核苷酸运行皆至少具有多个碱基对的一阈值长度。
在步骤1410,将多个读值组装成多个路径。在一些实施例中,这涉及组装多个读值以产生针对一目标区域(例如一基因)的一有向图(directedgraph),例如一德布鲁因图。所述有向图的多个单向边缘代表在所述目标区域中的k个核苷酸碱基(本文也称为“k聚体(k-mers)”)的多个序列,并且所述多个边缘通过多个顶点(或节点)连接。将多个分裂过的读值与一有向图进行比对,使得所述多个分裂过的读值的任何一个可由所述多个边缘及多个对应的顶点的一子集依序表示。
在一些实施例中,确认描述多个有向图及处理多个有向图的多个参数集。所述参数集可包括将来自多个分裂过的读值的多个k聚体与由所述有向图中的一节点或边缘代表的一k聚体成功比对的一计数。在一些实施例中,所述多个有向图及多个对应的参数集可被存储,以便随后的检索更新多个图示或产生多个新图示。例如,基于所述参数集可产生一有向图的一压缩版本(例如,或修改一现有图式)。在一使用例子中,为了过滤掉具有较低重要性级别的一有向图的数据,将具有小于一阈值的一计数的多个节点或多个边缘移除(例如,“修整”或“修剪”),同时维持具有大于或等于所述阈值的多个计数的多个节点或多个边缘。
在步骤1415,所述变体识别器240产生来自所述多个组装路径的多个候选变体。在一实施例中,通过将一有向图(已经通过步骤1410中的修剪多个边缘或多个节点来被压缩)与一基因组的一目标区域的一参考序列进行比较来产生多个候选变体。所述有向图的多个边缘可与所述参考序列进行比对,并且将多个错误配对边缘的所述多个基因组位置及邻近于所述多个边缘的多个错误配对的核苷酸碱基记录为多个候选变体的位置。在一些实施例中,将所述多个错误配对边缘的多个基因组位置及针对多个边缘的左侧及右侧的多个错误配对的核苷酸碱基记录为多个经识别的变体的位置。另外,基于一目标区域的所述定序深度可产生多个候选变体。特别地,可更有信心地辨识具有更大定序深度的多个目标区域中的多个变体,例如,因为更多的序列读值有助于解决(例如,使用冗余性)多个错误配对或多个序列之间的其他碱基对的变异。
在一些实施例中,使用一模型来产生多个候选变体,以确定对于来自一受试者的多个序列的预期噪声率。虽然在一些实施例中,使用一个或多个不同类型的模型,但所述模型可为贝叶斯层次性模型(bayesianhierarchical)。并且,一贝叶斯层次性模型可为可用于产生多个候选变体且彼此相关的许多可能的模型架构的其中之一,因为它们都对位置特异性的噪声信息进行建模,以提高变体识别的敏感度/特异性。更具体地,可使用来自多个健康个体的多个样本来训练所述模型,以对每个序列读值的位置的所述多个预期噪声率进行建模。
进一步地,多个不同的模型可用于应用程式的后训练。在一示例中,训练一第一模型来对多个snv噪声率进行建模,并且训练一第二模型来对多个插入缺失的噪声率进行建模。进一步地,所述模型的多个参数可用于确定在一序列读值中的一个或多个真实阳性的一可能性。基于所述可能性可确定一质量得分(例如,在一对数尺度上)。在一示例中,所述质量得分为一phred质量得分q=-10log10p,其中p为一错误的候选变体识别(例如伪阳性)的可能性。诸如一接合模型的其他模型使用一个或多个贝叶斯层次性模型的输出来确定在不同样本的多个序列读值中的多个核苷酸突变的预期噪声。
在步骤1420,使用一个或多个类型的模型或过滤器来过滤所述多个候选变体。在一实施例中,使用一接合模型、一边缘变体预测模型,或者对应的真实阳性的可能性或质量得分来评分所述多个候选变体。另外,分别使用一边缘过滤器及/或一非同义过滤器来过滤多个边缘变体及/或多个非同义突变。
在步骤1425,输出所述多个过滤过的候选变体。在一些实施例中,所述多个确定的候选变体的一些或全部与来自所述多个过滤步骤的一对应的得分一起被输出。
示例12
无细胞基因组图谱研究(ccga)群体
将来自所述ccga[nct02889978]的多个受试者用于本公开的多个示例中。ccga为一个带有纵向随访的具前瞻性的、多中心的、病例对照的及观察性的研究。所述研究在141个地点招募了在人口统计上平衡的15,000名参与者中的9,977名。从入选时定义为未针对癌症进行新诊断型治疗(newlydiagnosedtherapy-naivecancer)的受试者(c,病例)及未诊断出癌症的受试者(非癌症[nc],对照组)中收集血液。此预先计划的子研究包括1628个病例及1172个对照组,跨越20个肿瘤类型及所有的临床阶段。在分析之前,将多个样本划分为一训练集合(1,785)及一测试集合(1,015)。选择多个样本以确保在每个群体中跨越地点的多个癌症类型及非癌症的一预先指定的分布,并使癌症与非癌症样本的频率按照性别与年龄匹配。图18提供了在最终分析中的多个参与者的人口统计信息。
从血浆分离出无细胞dna,同时使用多个标准方法从多个白细胞(wbcs)及肿瘤组织分离出基因组dna(gdna)。在cfdna分析中采用三种不同的高强度定序方法:(i)cfdna全基因组亚硫酸氢盐定序(wgbs;30倍深度),其中使用多个异常甲基化的片段来产生多个归一化的得分;(ii)成对cfdna及wbc全基因组定序(wgs;30倍深度),其中一新型机器学习算法产生多个与癌症相关的信号得分,以及接合分析辨识出多个共享事件;及(iii)成对cfdna及wbc靶向定序(507个基因的小组;60,000倍深度,本文指的是所述“art”测定法),其中一接合识别器移除多个源自于wbc的体细胞变体及残留的技术噪声。对wbcgdna进行靶向定序,以辨识出克隆性造血作用(ch)。对肿瘤组织的gdna进行wgs,以辨识多个体细胞变体,其可用于计算cfdna肿瘤分数。
在所述靶向测定中,多个与非肿瘤wbc配对的cfdna体细胞变体(snvs/插入缺失)在nc中占所有变异的76%,及在c中占65%。在与体细胞镶嵌现象(例如克隆性造血作用)一致的情况下,多个与wbc配对的变体随着年龄的增长而增加;一些是先前未报导过的非典型的功能丧失突变。在移除wbc变体之后,多个典型的驱动体细胞变体对c具有高度特异性(例如,在egfr及pik3ca中,0个nc具有变体对上分别为c的11及30)。相似地,利用wgs检测到的8个nc具有多个体细胞拷贝数改变(scnas),其中4个源自于wbc。所述ccga的wgbs数据显示出有用的高片段水平及低片段水平的cpg(1:2比例)的信息;其子集用于计算多个甲基化得分。在所有测定内的<1%的nc参与者中观察到一致的“类癌症”信号(代表潜在地未诊断出癌症)。在nc对上第i至iii期对上第iv期中观察到一增加的趋势(每个mb的nonsyn.snvs/indels[平均值±标准差]nc:1.01±0.86,第i至iii期:2.43±3.98;第iv期:6.45±6.79;wgs得分nc:0.00±0.08,i至iii:0.27±0.98;iv:1.95±2.33,甲基化得分nc:0±0.50;i至iii:1.02±1.77;iv:3.94±1.70)。这些数据证明针对侵袭性癌症实现>99%的特异性的可行性,并且支持cfdna分析有望用于早期癌症侦测。
在klein等人,“对于早期侦测多个肿瘤类型的一综合无细胞dna(cfdna)测定法的开发:循环无细胞基因组图谱(ccga)研究”,2018asco年度会议,6月1-5日,2018,摘要12021#134,芝加哥,伊利诺伊州中公开更多ccga测定法的信息,其通过引用并入本文中。
使用wgbs确定甲基化等位基因。对于每个样本,将所述wgbs片段集合缩减为一极端甲基化状态的异常片段的子集(ufxm)。在未患有癌症的个体中以高频率出现的,或具有不稳定甲基化的多个片段不太可能产生用于分类癌症状态的多个高度区别性的特征。通过利用来自所述ccga研究的108名未患有癌症的未吸烟参与者(年龄:58±14岁,79名[73%]女性)的一独立的参考集合,来使用多个典型片段的一统计模型。将这些样本用于训练一马尔可夫链模型(第3级),所述马尔可夫链模型估计出在一片段中的一特定序列的cpg甲基化状态的可能性。此模型被证明是在一正常的片段范围内进行校准(p值≥0.001),并且被用于利用来自所述马尔可夫模型p≥0.001的一p值来拒绝多个片段,因为其异常不足。
一进一步的数据缩减步骤仅选择具有覆盖至少5个cpg的多个片段,并且每个片段的平均甲基化>0.9(高甲基化)或<0.1(低甲基化)。此流程产生针对在训练中未患有癌症的多个参与者的ufxm片段的一中位数(范围)2,800(1,500-12,000),以及针对在训练中患有癌症的多个参与者的ufxm片段的一中位数(范围)3,000(1,200-220,000)。由于此数据减小流程仅使用参考集合的数据,因此这个阶段仅需要对每个样本应用一次。
在所述基因组内选定的基因座上,针对高甲基化及低甲基化的ufxm独立地构建了对于癌症状态信息的一大概的对数比率得分。首先,对于位在所述基因座的每个样本,产生一个二进制特征:假如没有ufxm片段与此样本内的此基因座重叠,则为0;假如存在有与此基因座重叠的一ufxm片段,则为1。然后,从患有癌症(cc)或未患有癌症(cnc)的多个参与者计算出多个样本中的阳性数值(1s)的数量。接着,将所述对数比率得分构建为:log(cc+1)-log(cnc+1),其中在计数中添加一正则项,并抛弃与每个群组(nc及nnc)中的多个样本的总数量有关的归一化项,因为其为常数(log[nnc+2]-log[nc+2])。在所述基因组中的所有cpg位点的多个位置处构建多个得分,从而产生大约25m个具有多个指定得分的基因座:一个针对ufxm高甲基化片段的得分及一个针对ufxm低甲基化片段的得分。
鉴于一基因座特异性的对数比率得分,通过取得在所述片段内的对于基因座的所有对数比率的最大值,并与高甲基化或低甲基化的甲基化类别配对,来对一样本中的多个ufxm片段进行评分。这使得在一样本中的每个ufxm片段有一得分。
通过单独地针对高甲基化片段及低甲基化片段取得在每个样本中的多个极端排序片段的一子集的所述得分,将一样本中的这些片段级别的得分缩减为每个样本的多个特征的一小集合。以此种方式,使用多个有用特征的一小集合来捕捉对于每个样本中的信息量最多的多个片段的信息。在一低cfdna肿瘤分数样本中,预期仅有少数片段具有异常的信息量。
在每个片段的类别中,针对高甲基化及低甲基化ufxm的每个类别中的多个片段选择所述排序1、2、4、…、64(2i,i在0:6中)的最大分数,从而产生14个特征(7与7)。为了调整样本的定序深度,所述排序流程被视为将多个排序与多个得分进行映射的一函数,并在所述多个观察到的得分之间进行内插,以获得对应于调整后的排序的多个得分。所述多个排序以针对相关样本深度的线性比例来进行调整:假如所述相关样本深度为x,则在所述x乘以所述多个初始排序的情况下取得多个内插分数(例如,对于x=1.1,我们取得在排序floor(1.1)、floor(2.2)、…、floor(x·2i)下计算出的多个得分)。接着,每个样本被指派14个调整过的极端排序得分的一集合,以便用于进一步的分类中。
考虑到所述特征向量,在从所述多个特征预测癌症/非癌症状态时,使用一核函数逻辑回归分类器捕捉多个潜在的非线性。具体地,使用作为具有尺度参数γ的核函数的等项性径向基础函数(幂指数2),及l2正则化参数λ(通过除以m2来进行调整,其中m为样本数量,因此λ自然地随着训练数据量按比例调整),来训练正则化内核逻辑回归分类器(klr)。在指定的训练数据中,使用内部交叉验证使γ及λ最佳化,以便留出(holdout)对数损失,并且使用在7个相乘步骤中的范围1-0.01(γ)、1000-10(λ)内的网格搜寻来进行最佳化,所述7个相乘步骤从最大值开始,且每个步骤将所述参数减半。在内部交叉验证折迭(fold)期间的多个最佳参数的中位数对于γ为0.125,且对于λ为125。
为了评估此极端排序得分分类器的流程对所述ccga子研究数据集的成效,将交叉验证应用于所述训练集合,其中将所述多个样本划分成10个折迭。将每个折迭留出,并且用所述数据剩余的9/10训练所述极端排序得分(ers)分类器(使用这些折迭中的内部交叉验证来将γ及λ最佳化)。在特征化中使用的所述多个对数比率得分仅访问来自多个训练折迭的数据。将来自每个留出折迭的多个输出得分合并,并用于构建对于性能的一roc曲线。
敏感度估计。
图19a及19b提供针对模型敏感度的信息,其中使用图18总结的所述训练集合进行训练,且对照图18总结的所述训练集合(图19a,n=1,416)及所述测试集合(图19b,n=1,416),以及依据肿瘤来源进行划分。图19c及19d提供针对依据肿瘤来源来划分的所述训练集合(图19c)及所述测试集合(图19d)中的肿瘤分数的信息。在更多的细节中,对于图19a及19b,当在训练集合(图19a)及测试集合(图19b)中通过wgbs(左手边条带,蓝色)、说明ch的wgs(中间条带,橘色),及说明ch的所述靶向测定(左手边条带,灰色)进行分析时,提供了针对所述训练集合及所述测试集合中的每个肿瘤类型(x轴)在98%特异性下的敏感度(y轴)。误差条代表95%的置信区间。在多个括号中指出每个癌症类型的多个样本的数量。来自事后检定(posthoc)的多发性骨髓瘤及白血病则单独表示。图19c及19d提供针对在所述训练集合(图19c)及所述测试集合(图19d)中的每个肿瘤类型(x轴)的多个参与者的一子集的cfdna肿瘤分数(y轴)的箱型图,所述多个参与者具有可进行定序的肿瘤正常组织及至少一突变cfdna读值(如括号中所指示)。描绘出中位数以及第一个及第三个四分位数。通过菱形(在98%特异性下通过wgbs侦测到)及空心圆(在98%特异性下通过wgbs未侦测到)表示各个参与者的cfdna肿瘤分数估计值。水平虚线指示出侦测的界限(>50%的侦测可能性)。图19d建立的是,本公开的多个方法可用于侦测在一受试者的无细胞核酸中的一肿瘤分数,甚至是在所述肿瘤分数f为0.100或更低时,在一些例子中,当所述受试者的所述肿瘤分数f为0.050或更低、0.050或更低、0.040或更低、0.030或更低,或甚至0.020或更低时。
图20a及20b说明通过将cfdnawgs与肿瘤wgs结果依照乳腺癌、结肠直肠癌、肺癌及其他癌症的总集合的阶段(图20a),及依照每个癌症类型(图20b)进行比较而计算出的cfdna肿瘤分数。具有cfdna中的至少一突变读值的多个样本被呈现出。通过三角形(训练集合)及圆形(测试集合)指示出各个参与者的多个肿瘤分数,其中符号颜色指出在98%特异性下的wgbs的侦测(侦测到:蓝色;未侦测到:橘色)。图20a包括没有乳癌、肺癌及结肠直肠癌的全部样本。图20b包括原始来源不明的两个神经内分泌、两个间皮瘤、两个胃肠道间质瘤、一个肛门及四个腺癌(未另作说明)。
示例13
甲基化状态向量的产生
图15为根据本公开的一实施例描述了一种将cfdna的一片段进行定序以获得一甲基化状态向量的过程1500的一流程图。
参考步骤1502,从所述生物样本获得所述多个cfdna片段(例如,如上文结合图2所描述)。参考步骤1520,对所述多个cfdna片段进行处理,以将多个未甲基化的包嘧啶转化成多个尿嘧啶。在一实施例中,对所述dna进行一亚硫酸氢盐处理法,其将cfdna的所述片段的所述多个未甲基化的胞嘧啶转化为多个尿嘧啶,而不会转化多个甲基化过的胞嘧啶。例如,在一些实施例中,将诸如ezdna甲基化tm-金色、ezdna甲基化tm-直接或ezdna甲基化tm-快速试剂盒(可从立莫研究公司(尔湾,加利福尼亚州)取得)的一商业试剂盒用于所述亚硫酸氢盐转化。在其他实施例中,使用一酶促反应来完成多个未甲基化的胞嘧啶变为尿嘧啶的转化。例如,所述转化可使用一种用于将多个未甲基化的胞嘧啶转化为多个尿嘧啶的市售试剂盒,例如apobec-seq(新英格兰生物实验室,伊普斯维奇,马萨诸塞州)。
从所述多个转化过的cfdna片段制备一定序文库(步骤1530)。可选择地,对于cfdna片段或多个基因组区域,1535富集了所述定序文库,其可使用多个杂交探针为癌症状态提供信息。所述多个杂交探针为多个短的寡核苷酸,所述多个短的寡核苷酸能够与多个特别指定的cfdna片段或目标区域杂交,并富集这些片段或区域,以进行随后的定序及分析。多个杂交探针可用于对研究者感兴趣的一组指定的cpg位点进行有针对性且高深度的分析。一旦制备完,就对所述定序文库或所述定序文库的一部分进行定序,以获得多个序列读值(1540)。所述多个序列读值可为一计算机可读的数字格式,以通过计算机软件进行处理及阐明。
从所述多个序列读值,基于所述多个序列读值与一参考基因组的比对来确定对于每个cpg位点的一位置及甲基化状态(1550)。对于每个片段的一甲基化状态向量指定在所述参考基因组中的所述片段的一位置(例如,如通过在每个片段中的所述第一cpg位点的所述位置,或另一相似的度量所指定)、在所述片段中的多个cpg位点的数量,及在所述片段中的每个cpg位点的所述甲基化状态(1560)。
额外的实施例
使用肿瘤分数来评估一分类器的性能。本公开的另一方面提供一种评估一分类器的一性能的方法。所述方法包括:从一对应的受试者的一个别的生物样本获得包括有多个第一序列读值的电子形式的一个别的数据集,其用于多个受试者中的每个受试者,从而获得多个数据集。每个对应的受试者的所述个别的生物样本包括来自所述对应的受试者的多个无细胞核酸分子。将所述多个数据集中的每个个别的数据集应用至一分类器,从而获得对于所述个别的数据集的一对应的分类器的结果。所述分类器的结果指出所述多个受试者中的所述对应的受试者是否患有一第一癌症病况。再者,在应用上文描述的多个数据集之前,用所述多个数据集之外的数据训练所述分类器。
在一些实施例中,使用对应于所述受试者的所述数据集来估计在所述多个受试者的每个受试者的所述无细胞dna中的一估计出的肿瘤分数。接着,通过将对于每个个别的受试者的所述分类器的结果与所述个别的受试者的一临床观察进行比较,来根据跨越所述多个受试者的一估计出的肿瘤分数的函数计算出所述计算机的一性能,所述临床观察的推导独立于所述分类器对上所述个别的受试者的所述估计出的肿瘤分数。
以一整个群体的肿瘤定序为条件,而不仅仅是一单一的受试者。本公开提供一种分类一受试者的方法。所述方法包括:在一计算机系统中具有一个或多个处理器及一记忆体,所述记忆体存储通过所述一个或多个处理器执行的一个或多个程序,在所述计算机系统中获得来自所述受试者的一生物样本的电子形式的多个第一序列读值,其中所述生物样本包括多个无细胞核酸分子。所述多个无细胞核酸分子的所述多个第一序列读值用于辨识对一第一变体集合中的每个变体的支持。将所述多个第一序列读值中的一个别的序列读值视为支持所述第一变体集合中的一变体,这是在所述个别的序列读值含有所述变体的全部及一部分时。将所述多个第一序列读值中的一个别的序列读值视为不支持所述第一变体集合中的一变体,这是在所述个别的序列读值不含有所述变体的全部及一部分时。以此种方式,从所述多个第一序列读值中的支持或不支持所述第一变体集合中的每个变体的所述多个序列读值中,确定所述第一变体集合中的每个变体的一观察到的频率。将所述第一变体集合中的每个变体的所述观察到的频率与一第一参考集合中的一对应的参考频率进行比较。所述第一参考集合中的每个对应的参考频率为跨越一共同(相同)的第一类别的多个第一异常组织样本的所述对应变体的一频率。接着,将所述受试者分类。此分类步骤包括:当所述第一变体集合中的每个变体的所述观察到的频率满足一第一阈值时,将所述受试者视为患有与所述多个第一异常组织样本相关的一第一病况。所述第一阈值是通过所述第一参考集合中的每个参考频率来确定。
在一些实施例中,所述第一病况为来自一共同原发部位的癌症。在一些实施例中,所述第一病况为来自两个或更多个共同原发部位的癌症。所述第一病况为乳腺癌、肺癌、前列腺癌、结肠直肠癌、肾癌、子宫癌、胰腺癌、食道癌、淋巴瘤、头颈癌、卵巢癌、肝胆癌、黑色素瘤、子宫颈癌、多发性骨髓瘤、白血病、甲状腺癌、膀胱癌、胃癌,或其组合。
在一些实施例中,所述第一病况为乳腺癌、肺癌、前列腺癌、结肠直肠癌、肾癌、子宫癌、胰腺癌、食道癌、淋巴瘤、头颈癌、卵巢癌、肝胆癌、黑色素瘤、子宫颈癌、多发性骨髓瘤、白血病、甲状腺癌、膀胱癌或胃癌的一预定阶段。
在一些实施例中,所述第一病况为一癌症的一预定亚型(例如,乳腺癌、肺癌、前列腺癌、结肠直肠癌、肾癌、子宫癌、胰腺癌、食道癌、淋巴瘤、头颈癌、卵巢癌、肝胆癌、黑色素瘤、子宫颈癌、多发性骨髓瘤、白血病、甲状腺癌、膀胱癌或胃癌)。
在一些实施例中,所述第一变体集合中的一变体为与一预定的基因组位置相关的一单核苷酸变体、与一预定的基因组位置相关的一插入突变、与一预定的基因组位置相关的一缺失突变、一体细胞拷贝数的改变、与一预定的基因组位点相关的一核酸重组,或与一预定的基因组位置相关的一异常的甲基化模式。
在一些实施例中,所述多个第一异常组织样本为多个肿瘤样本。在一些实施例中,所述第一变体集合是由一单一变体组成,所述单一变体为位在所述受试者的所述基因组中的一单一位点处的一单一的遗传变体。在一些实施例中,所述第一变体集合是由一第一变体及一第二变体组成,所述第一变体为位在所述受试者的所述基因组中的一第一位点处的一第一遗传变异,且所述第二变体为位在所述受试者的所述基因组中的一第二位点处的一第二遗传变异。
在一些实施例中,所述第一变体集合是由一第一变体、一第二变体及一第三变体组成,所述第一变体为位在所述受试者的所述基因组中的一第一位点处的一第一遗传变异,所述第二变体为位在所述受试者的所述基因组中的一第二位点处的一第二遗传变异,且所述第三变体为位在所述受试者的所述基因组中的一第三位点处的一第三遗传变异。
在一些实施例中,所述第一变体集合是由介于2至20个之间的变体组成,其中所述第一变体集合中的每个变体为所述受试者的所述基因组中的(可选择地位于一不同位点处的)一不同的遗传变异。在一些实施例中,所述第一变体集合是由介于2至200个之间的变体组成,其中所述第一变体集合中的每个变体为所述受试者的所述基因组中的(可选择地位于一不同位点处的)一不同的遗传变异。
在一些实施例中,所述变体集合包括200个变体、包括300个变体、包括400个变体、包括500个变体、包括750个变体、包括1000个变体、包括2000个变体,或包括5000个变体,其中所述第一变体集合中的每个变体为所述受试者的所述基因组中的(可选择地位于一不同位点处的)一不同的遗传变异。
在一些实施例中,所述比较的步骤包括:从所述第一变体集合中的每个变体的所述观察到的频率计算出在所述人类受试者的所述cfdna中的一单一的估计出的ctdna。在多个这样的实施例中,所述第一阈值为从所述第一参考集合中的每个参考频率的数值所确定的所述人类受试者的所述cfdna中的一单一的预期的ctdna分数。在一些这样的实施例中,在所述cfdna中的所述单一的预期的ctdna分数介于0.5x10-4至1.5x10-4之间,并且所述第一病况为黑色素瘤。在一些这样的实施例中,在所述cfdna中的所述单一的预期的ctdna分数介于0.5x10-3至1x10-2之间,并且所述第一病况为肾癌、子宫癌、甲状腺癌、前列腺癌、乳腺癌、膀胱癌、胃癌、子宫颈癌或其组合。在一些这样的实施例中,在所述cfdna中的所述单一的预期的ctdna分数介于1x10-2至0.8之间,并且所述第一病况为肺癌、食道癌、头颈癌、结肠直肠癌、肛门直肠癌、卵巢癌、肝胆癌、胰腺癌、淋巴瘤或其组合。
在一些实施例中,所述使用的步骤包括:将所述多个第一序列读值中的一个别的序列读值与一参考基因组中的一区域进行比对,以确定所述个别的序列读值是否含有一变体的全部或一部分。在一些实施例中,所述使用的步骤包括:将所述多个第一序列读值中的一个别的序列读值与多个变体的一查找表进行比对,以确定所述个别的序列读值是否含有一变体的全部或一部分。在一些实施例中,所述使用的步骤包括:将所述多个第一序列读值中的一序列读值与一查找表中的每个条目进行比对,其中所述查找表中的每个条目代表一基因组的一不同部分。
在一些实施例中,所述比较的步骤包括:从所述第一变体集合中的每个变体的所述观察到的频率计算出在所述人类受试者的所述无细胞dna(cfdna)中的一单一的估计出的循环肿瘤dna(ctdna)分数。在多个这样的实施例中,当所述单一的估计出的循环肿瘤dna(ctdna)分数超过1x10-3,且所述第一病况为第ii期、第iii期或第iv期乳腺癌时,所述第一变体集合中的每个变体的所述观察的频率满足所述第一阈值。
在一些实施例中,所述方法进一步包括:使用所述多个第一序列读值来辨识对一第二变体集合中的每个变体的支持,其中将所述多个第一序列读值中的一个别的序列读值视为支持所述第二变体中的一变体,这是在所述个别的序列读值含有所述第二变体的全部或一部分时,并且将所述多个第一序列读值中的一个别的序列读值视为不支持所述第二变体中的一变体,这是在所述个别的序列读值不含有所述个别的第二变体时。以此种方式,从所述多个第一序列读值中的支持或不支持所述第二变体中的一变体的所述多个序列读值中,确定所述第二变体中的每个变体的一观察到的频率。将所述第二变体中的每个变体的所述观察到的频率与一第二参考集合中的一对应的第二参考频率进行比较。在多个这样的实施例中,所述第二参考集合中的每个对应的第二参考频率为跨越一共同(相同)的第二类别的多个第二异常组织样本的所述对应变体的一频率。进一步地,在多个这样的实施例中,所述分类所述人类受试者的步骤进一步包括:当所述第二变体集合中的每个变体的所述观察到的频率满足一第二阈值时,将所述人类受试者视为患有与所述多个第二异常组织样本相关的一第二病况,其中所述第二阈值是通过所述第二参考集合中的每个参考频率来确定。
在一些实施例中,所述受试者为一人类受试者。在一些实施例中,所述生物样本包括所述受试者的血液、全血、血浆、血清、尿液、脑脊髓液、粪便、唾液、汗水、眼泪、胸水、心包液或腹膜液。
本公开的另一方面提供一种计算机系统,所述计算机系统包括一个或多个处理器及一记忆体,所述记忆体存储通过所述一个或多个处理器执行的一个或多个程序。所述一个或多个程序包括用于通过一方法来分类一受试者的多个指令。所述方法包括:(a)从所述受试者的一生物样本获得电子形式的多个第一序列读值,其中所述生物样本包括多个无细胞核酸分子。所述方法进一步包括:(b)使用所述多个无细胞核酸分子的所述多个第一序列读值来辨识对一第一变体集合中的每个变体的支持。在此,将所述多个第一序列读值中的一个别的序列读值视为支持所述第一变体集合中的一变体,这是在所述个别的序列读值含有所述变体的全部及一部分时,并且将所述多个第一序列读值中的一个别的序列读值视为不支持所述第一变体集合中的一变体,这是在所述个别的序列读值不含有所述变体时。以此种方式,从所述多个第一序列读值中的支持或不支持所述第一变体集合中的每个变体的所述多个序列读值中,确定所述第一变体集合中的每个变体的一观察到的频率。将所述第一变体集合中的每个变体的所述观察到的频率与一第一参考集合中的一对应的参考频率进行比较。在多个这样的实施例中,所述第一参考集合中的每个对应的参考频率为跨越一共同(相同)的第一类别的多个第一异常组织样本的所述对应变体的一频率。接着,将所述受试者分类。此分类步骤包括:当所述第一变体集合中的每个变体的所述观察到的频率满足一第一阈值时,将所述受试者视为患有与所述多个第一异常组织样本相关的一第一病况,其中所述第一阈值是通过所述第一参考集合中的每个参考频率来确定。
本公开的另一方面提供一种存储一个或多个程序的非暂时性计算机可读存储介质,以便对一受试者进行分类。所述一个或多个程序配置成通过一计算机来执行。所述一个或多个程序包括多个指令用于:从所述受试者的一生物样本获得电子形式的多个第一序列读值,其中所述生物样本包括多个无细胞核酸分子。所述一个或多个程序进一步包括多个指令用于:使用所述多个无细胞核酸分子的所述多个第一序列读值来辨识对一第一变体集合中的每个变体的支持。在多个这样的实施例中,将所述多个第一序列读值中的一个别的序列读值视为支持所述第一变体集合中的一变体,这是在所述个别的序列读值含有所述变体的全部及一部分时,并且将所述多个第一序列读值中的一个别的序列读值视为不支持所述第一变体集合中的一变体,这是在所述个别的序列读值不含有所述变体时。以此种方式,从所述多个第一序列读值中的支持或不支持所述第一变体集合中的每个变体的所述多个序列读值中,确定所述第一变体集合中的每个变体的一观察到的频率。将所述第一变体集合中的每个变体的所述观察到的频率与一第一参考集合中的一对应的参考频率进行比较。在多个这样的实施例中,所述第一参考集合中的每个对应的参考频率为跨越一共同(相同)的第一类别的多个第一异常组织样本的所述对应变体的一频率。所述一个或多个程序进一步包括多个指令用于:分类所述受试者。所述分类步骤包括:当所述第一变体集合中的每个变体的所述观察到的频率满足一第一阈值时,将所述受试者视为患有与所述多个第一异常组织样本相关的一第一病况。在此,所述第一阈值是通过所述第一参考集合中的每个参考频率来确定。
从来自一个体的读值信息估计cfdna肿瘤分数,而不需要直接分析所述异常组织。
在多个可替代的实施例中,不需要使用来自一异常组织的多个序列读值126来估计cfdna肿瘤分数。在一些这样的实施例中,使用来自所述生物样本的多个序列读值140来辨识源自于肿瘤的多个特征(例如,多个小变体),所述生物样本含有所述无细胞核酸。接着,以这些变体中的其中一个的所述观察到的频率为条件,估计所述潜在的肿瘤分数。
在一些这样的实施例中,为了确保一特定的突变为所述受试者的所述cfdna中的单一的估计出的ctdna分数的一合适的替代物,所选择的变体为一个具有所述最高频率以外的变体,这是在此变体具有非源自于所述异常组织的一高概率的假定基础上。为了进行说明,考虑到一例子,在所述例子中,将一生物样本的所述无细胞核酸进行定序,并且找到具有一第一频率132-1的一第一变体130-1及具有一第二频率132-2的一第二变体130-2,其中所述第一参考频率132-1高于所述第二参考频率132-2。在此例子中,仅有所述第二变体132-2被假定为与所述特定受试者的未测量过的异常组织相关的病况的一合适的替代物。
在一些这样的实施例中,为了确保一特定的突变为所述受试者的所述cfdna中的单一的估计出的ctdna分数的一合适的替代物,将已知与研究中的病况不相关的多个变体(例如,经常与白细胞相关的多个变体)排除在考虑之外。
在一些实施例中,在所述个别的变体144具有在含有所述无细胞核酸的所述生物样本(例如,血液样本)中观察到的所有变体的第二高的频率的基础上,将一个别的变体144用于估计肿瘤分数。例如,假如此变体的所述频率(支持所述变体的覆盖所述基因组中的所述变体的所述位置的多个观察到的序列读值的数量除以覆盖所述基因组中的所述变体的所述位置的多个观察到的序列读值的总数量)为百分之十,则在所述受试者的所述cfdna中的所述单一的估计出的ctdna分数为百分之十。
在一些实施例中,将所述第一变体集合142的一个别的变体144用于估计肿瘤分数,这是在所述个别的变体具有在含有所述无细胞核酸的所述生物样本(例如,血液样本)中观察到的所有变体的第三高的频率的基础上。例如,假如此变体的所述频率(支持所述变体的覆盖所述基因组中的所述变体的所述位置的多个观察到的序列读值的数量除以覆盖所述基因组中的所述变体的所述位置的多个观察到的序列读值的总数量)为百分之十,则在所述受试者的所述cfdna中的所述单一的估计出的ctdna分数为百分之十。
在一些这样的实施例中,未使用所述异常组织样本,而是仅使用含有所述无细胞核酸的所述生物样本的所述实施例可用于计算低于约百分之一的多个单一的估计出的肿瘤分数。
在一些实施例中,将依照频率排序第二高的所述变体使用作为真实肿瘤分数(所述受试者的所述cfdna中的单一的估计出的ctdna)的代表物(proxy)。例如,假如此变体的所述频率(支持所述变体的覆盖所述基因组中的所述变体的所述位置的多个观察到的序列读值的数量除以覆盖所述基因组中的所述变体的所述位置的多个观察到的序列读值的总数量)为百分之十,则在所述受试者的所述cfdna中的所述单一的估计出的ctdna分数为百分之十。
在一些实施例中,将依照频率排序第三高的所述变体使用作为真实肿瘤分数(所述受试者的所述cfdna中的单一的估计出的ctdna)的代表物。例如,假如此变体的所述频率(支持所述变体的覆盖所述基因组中的所述变体的所述位置的多个观察到的序列读值的数量除以覆盖所述基因组中的所述变体的所述位置的多个观察到的序列读值的总数量)为百分之十,则在所述受试者的所述cfdna中的所述单一的估计出的ctdna分数为百分之十。
在一些实施例中,将来自含有无细胞核酸的所述生物样本的所述受试者的所述cfdna中的所述单一的估计出的ctdna用作为一参考基础,所述参考基础用于在之后的多个时间点从相同受试者取得的多个生物样本,以便确定一段时间内的所述受试者的所述肿瘤分数的变化。
结论
复数个示例可作为单一的实例而被提供用于本文所描述的多个组件、操作或结构。最后,在各种组件、操作及数据存储之间的边界在某种程度上为任意的,并且在特定的说明性配置的上下文中说明了多个特定的操作。功能的其他分配可被设想,并且可落入所述(多个)实施方式的范围内。通常,在示例配置中作为多个单独组件所呈现出的结构及功能可被实施为一组合结构或组件。同样地,作为一单一的组件所呈现出的结构及功能可被实施为多个单独的组件。这些与其他变型、修改、附加及改进皆落入所述(多个)实施方式的范围内。
还应当理解,虽然在本文中可使用多个术语第一、第二等来描述各种元件,但是这些元件不应受到这些术语的限制。这些术语仅用于区分一个元件及另一个元件。例如,在不脱离本公开的范围的情况下,一第一受试者可被称为一第二受试者,并且同样地,一第二受试者可被称为一第一受试者。所述第一受试者及所述第二受试者皆为受试者,但是他们不是相同的受试者。
本公开中使用的术语仅出于描述多个特定实施例的目的,并且不旨在限制本发明。如在本发明的描述及所附权利要求中所使用,单数形式“一”(“a”,“an”)及“所述”也旨在包括复数形式,除非上下文另外明确指出。还应当理解,本文所使用的术语“及/或”指的是且涵盖一个或多个相关联的所列项目的任何及所有可能的组合。应当进一步理解的是,在此说明书中使用时,术语“包含”(comprises”及/或“comprising”)具体说明所陈述的特征、整数、步骤、操作、元件及/或组件的存在,但是不排除一个或多个其他特征、整数、步骤、操作、元件、组件及/或其群组的存在或增加。
如本文所使用,取决于上下文,术语“假如”可被解释成表示“何时”,或“一旦”,或“响应于确定”,或“响应于侦测”。同样地,取决于上下文,片语“假如确定”或“假如侦测到[一陈述的状况或事件]”可被解释成表示“一旦确定”,或“响应于确定”,或“一旦侦测到(所述陈述的状况或事件(”,或“响应于侦测到(所述陈述的状况或事件)”。
前面的描述包括体现多个说明性实施方式的多个示例系统、方法、技术、指令序列及计算机器程序产品。为了解释的目的,阐述了许多具体的细节,以便提供对发明主题的各种实施方式的理解。然而,对于本领域技术人员显而易见的是,可在没有这些具体细节的情况下,实践本发明主题的多个实施方式。通常,没有详细显示出多个众所周知的指令示例、实验步骤、结构及技术。
为了说明的目的,已经参考多个具体实施方式来描述前面的描述。然而,以上的说明性讨论并非旨在穷举,或将所述多个实施方式限制为多个所公开的精确形式。鉴于以上的多个教示,许多修改及变形是可能的。所述多个实施方式被选择及描述,以便最佳地解释多个原理及其实践应用,从而使本领域的其他技术人员能够最佳地利用所述多个实施方式及具有各种修改的各种实施方式,以适合于所设想的特定用途。
- 还没有人留言评论。精彩留言会获得点赞!