一种含七苯三胺-双荧光团结构的二胺单体及制备和应用,聚酰胺、聚酰亚胺及制备和应用的制作方法
本发明涉及电控荧光技术领域,尤其涉及一种含七苯三胺-双荧光团结构的二胺单体及制备和应用,聚酰胺、聚酰亚胺及制备和应用。
背景技术:
电控荧光是指材料的荧光对电化学氧化还原反应具有可逆的响应,其在光学显示、信息加密和传感等领域具有潜在的应用前景。电控荧光材料主要包括无机复合体系,可切换的荧光小分子和聚合物。相较而言,聚合物因其加工性强、结构易于修饰而具有更大的优势。聚酰胺和聚酰亚胺具有优异的物理和化学性能。然而,其刚性的骨架和分子链间强的相互作用,导致其溶解性较差。为了解决这个问题,可以在聚合物中引入脂环结构、醚键和非共面基团。尤其是在聚合物中引入星形的三苯胺(tpa)衍生物,不仅有效地提高了其溶解性,而且使聚合物具有了有趣的光电性能。
目前,相比于三苯胺结构,对苯二胺由于其单阳离子自由基更加稳定而受到更多的研究。然而,在对苯二胺结构中,形成单阳离子自由基的氧化电位仍然较高,且其双阳离子不稳定,使其在高电压下电活性较差,不利于电控荧光的稳定性;如hsiao,sheng-huei课题组(hsiaosh,chiuyt.synthesisandcharacterizationofnovelelectrochromicpoly(amide-imide)swithn,n'di(4-methoxyphenyl)-n,n'-diphenyl-p-phenylenediamineunits[j].rscadvances,2015,5(113):93591-93606.)制备一种含有对苯二胺的单体n,n'-双(4-氨基苯基)-n,n'–双(4-甲氧基苯基)-1,4-苯基二胺,用此二胺单体合成的聚酰胺第一重氧化电位高于0.6v,第二重氧化电位高于1v,且其双阳离子电化学循环稳定性较差。此外,对苯二胺结构的电压可调控范围较窄,而对于电控荧光材料来说高电压下可能使荧光淬灭更完全,荧光开关对比度更高。因此,通过延长对苯二胺的共轭长度来降低体系能量,有望进一步改善三苯胺衍生物的电化学稳定性。此外,三苯胺的荧光较弱,且聚酰胺、聚酰亚胺分子链间紧密的堆积致使材料荧光开/关对比度低、响应速度慢,这些限制了电控荧光器件的应用。
技术实现要素:
本发明的目的在于提供一种含七苯三胺-双荧光团结构的二胺单体及制备和应用,聚酰胺、聚酰亚胺及制备和应用,所述二胺单体制备的聚酰胺或聚酰亚胺可以在保持聚合物热稳定性的同时,提高其溶解性和成膜性,同时具有优异的电控荧光性能。
为了实现上述发明目的,本发明提供以下技术方案:
本发明提供了一种含七苯三胺-双荧光团结构的二胺单体,具有式i所示结构:
式i中,r为
r1为-ch3、-ch2ch3、-ch2ch2ch3、-ch(ch3)2、-(ch2)3ch3、-ch2ch(ch3)2、-c(ch3)3或-(ch2)4ch3。
优选的,所述含七苯三胺-双荧光团结构的二胺单体为:
本发明提供了上述技术方案所述含七苯三胺-双荧光团结构的二胺单体的制备方法,包括以下步骤:
将对烷氧基苯胺、第一卤代硝基苯、第一碱性催化剂和第一非质子极性溶剂混合,进行第一亲核取代反应,得到4,4'-二硝基-4”-烷氧基三苯胺;
将所述4,4'-二硝基-4”-烷氧基三苯胺、乙醇、第一催化剂和第一还原剂混合,进行第一还原反应,得到4,4'-二氨基-4”-烷氧基三苯胺;
将所述4,4'-二氨基-4”-烷氧基三苯胺、第二卤代硝基苯、第二碱性催化剂和第二非质子极性溶剂混合,进行第二亲核取代反应,得到4,4'-双[(4-硝基苯基)胺]4”-烷氧基三苯胺;
将所述4,4'-双[(4-硝基苯基)胺]4”-烷氧基三苯胺、rbr化合物、金属催化剂、助催化剂、相转移催化剂和反应溶剂混合,进行乌尔曼反应,得到二硝基化合物;
将所述二硝基化合物、有机溶剂、第二催化剂和第二还原剂混合,进行第二还原反应,得到含七苯三胺-双荧光团结构的二胺单体;
其中,rbr化合物中的r为
所述对烷氧基苯胺具有式ii所示结构:
r1为-ch3、-ch2ch3、-ch2ch2ch3、-ch(ch3)2、-(ch2)3ch3、-ch2ch(ch3)2、-c(ch3)3或-(ch2)4ch3。
优选的,所述第一卤代硝基苯包括对氟硝基苯、对氯硝基苯、对溴硝基苯或对碘硝基苯;所述第一碱性催化剂包括氟化铯、碳酸铯、碳酸钠、氢氧化钾或氢化钠;所述对烷氧基苯胺、第一卤代硝基苯和第一碱性催化剂的摩尔比为1:(2~2.5):(2~2.5);
所述第一非质子极性溶剂包括二甲基亚砜或n,n-二甲基甲酰胺;
所述第一亲核取代反应的温度为140~160℃,时间为12~24h。
优选的,所述第一催化剂包括钯/碳催化剂,所述4,4'-二硝基-4”-烷氧基三苯胺和第一催化剂的质量比为1:(0.2~0.5);
所述第一还原剂包括水合肼;所述第一还原剂与4,4'-二硝基-4”-烷氧基三苯胺的摩尔比为(5~25):1;
所述第一还原反应的温度为70~90℃,时间为10~15h。
优选的,所述4,4'-二氨基-4”-烷氧基三苯胺、第二卤代硝基苯和第二碱性催化剂的摩尔比为1:(2.3~2.5):(3~3.5);
所述第二卤代硝基苯包括对氟硝基苯、对氯硝基苯、对溴硝基苯或对碘硝基苯;所述第二碱性催化剂包括三乙胺或三丙胺;所述第二非质子极性溶剂包括二甲基亚砜或n,n-二甲基甲酰胺;
所述第二亲核取代反应的温度为110~130℃,时间为60~72h。
优选的,所述4,4'-双[(4-硝基苯基)胺]4”-烷氧基三苯胺、rbr化合物、金属催化剂、助催化剂和相转移催化剂的摩尔比为1:(2~4):(8~10):(8~10):(1~1.5);
所述金属催化剂包括铜粉、镍或碘化亚铜;
所述助催化剂包括碳酸钾、氢氧化钾、氢化钾、氢化钠、氢氧化钠、碳酸钠、碳酸铯或氟化铯;
所述相转移催化剂包括18-冠醚-6、15-冠醚-5、二苯并-18-冠醚-6、苯并-18-冠醚-6或苯并-15-冠醚-5;
所述乌尔曼反应的温度为150~170℃,时间为12~24h;
所述二硝基化合物与第二催化剂的质量比为1:(0.2~0.5),所述第二还原剂与二硝基化合物的摩尔比为(5~25):1。
本发明提供了上述技术方案所述含七苯三胺-双荧光团结构的二胺单体或上述技术方案所述制备方法制备的含七苯三胺-双荧光团结构的二胺单体在制备聚酰胺或聚酰亚胺中的应用。
本发明提供了一种含七苯三胺-双荧光团结构的聚酰胺,具有式iii所示结构:
其中,n=10~80且n为整数;所述含七苯三胺-双荧光团结构的聚酰胺的数均分子量为10300~83000;
ar包括
r为
r1为-ch3、-ch2ch3、-ch2ch2ch3、-ch(ch3)2、-(ch2)3ch3、-ch2ch(ch3)2、-c(ch3)3或-(ch2)4ch3。
本发明提供了上述技术方案所述含七苯三胺-双荧光团结构的聚酰胺的制备方法,包括以下步骤:
将含七苯三胺-双荧光团结构的二胺单体、二酸单体、亚磷酸三苯酯、吡啶、溶剂和助溶剂混合,进行缩聚反应,得到含七苯三胺-双荧光团结构的聚酰胺;
所述含七苯三胺-双荧光团结构的二胺单体为上述技术方案所述含七苯三胺-双荧光团结构的二胺单体或者上述技术方案所述制备方法制备得到的含七苯三胺-双荧光团结构的二胺单体;
所述二酸单体包括对苯二甲酸、1,4-环己烷二甲酸或4,4'-联苯二甲酸。
本发明提供了上述技术方案所述含七苯三胺-双荧光团结构的聚酰胺在电控荧光器件中的应用。
本发明提供了一种含七苯三胺-双荧光团结构的聚酰亚胺,具有式iv所示结构:
其中,m=10~80且n为整数,所述含七苯三胺-双荧光团结构的聚酰亚胺的数均分子量为10800~87000;
ar'包括
r为
r1为-ch3、-ch2ch3、-ch2ch2ch3、-ch(ch3)2、-(ch2)3ch3、-ch2ch(ch3)2、-c(ch3)3或-(ch2)4ch3。
本发明提供了上述技术方案所述含七苯三胺-双荧光团结构的聚酰亚胺的制备方法,包括以下步骤:
将含七苯三胺-双荧光团结构的二胺单体、二酐单体和反应溶剂混合,进行聚合反应,得到聚酰胺酸溶液;
将所述聚酰胺酸溶液、催化剂和脱水剂混合,进行热亚胺化,得到聚酰亚胺;
所述含七苯三胺-双荧光团结构的二胺单体为上述技术方案所述含七苯三胺-双荧光团结构的二胺单体或者上述技术方案所述制备方法制备得到的含七苯三胺-双荧光团结构的二胺单体;
所述二酐单体包括均苯四甲酸二酐、氢化均苯四甲酸二酐或3,3',4,4'-联苯四甲酸二酐。
本发明提供了上述技术方案所述含七苯三胺-双荧光团结构的聚酰亚胺在电控荧光器件中的应用。
本发明提供了一种含七苯三胺-双荧光团结构的二胺单体,所述含七苯三胺-双荧光团结构的二胺单体含有双荧光团结构和七苯三胺结构,由该二胺单体制备的聚合物中会引入双荧光团结构(r所代表的萘、蒽、芘、芴或螺芴等荧光团结构)和七苯三胺结构,其中的双荧光团结构能够使聚合物具有高荧光强度,而且将具有助色团效应的三苯胺衍生物与荧光团相连可以进一步改善聚合物的荧光,确保聚合物具有高荧光强度和高荧光开/关对比度;其中的七苯三胺结构可以通过共振稳定单阳离子自由基和双阳离子,改善聚合物的电化学稳定性和电控荧光稳定性,且较宽的稳定电压范围可以有效调节聚合物的电控荧光行为;此外,高度共轭的七苯三胺结构能增强分子内的电荷传输能力,其扭曲的三维空间构型亦能减弱聚合物分子链间的堆积,双荧光团结构和七苯三胺结构的协同效应可以缩短电控荧光的响应时间。
本发明提供了含七苯三胺-双荧光团结构的聚酰胺或聚酰亚胺,所述聚酰胺或聚酰亚胺含有双荧光团结构和七苯三胺结构,可以在保持聚合物热稳定性的同时,提高其溶解性和成膜性,并赋予聚合物优异的电控荧光性能。根据实施例的记载,本发明提供的由含七苯三胺-双荧光团结构的二胺单体制备的聚酰胺或聚酰亚胺的电控荧光性能显著提高。
附图说明
图1为实施例1制备的4,4'-双[4-氨基苯基(6-甲氧基-2-萘基)胺]-4”-甲氧基三苯胺的氢核磁谱图;
图2为实施例1制备的4,4'-双[4-氨基苯基(6-甲氧基-2-萘基)胺]-4”-甲氧基三苯胺的红外谱图;
图3为实施例7制备的1,4-环己烷二甲酸型聚酰胺的红外谱图;
图4为实施例7制备的1,4-环己烷二甲酸型聚酰胺的dsc曲线图;
图5为实施例7制备的1,4-环己烷二甲酸型聚酰胺的tga图;
图6为实施例7制备的1,4-环己烷二甲酸型聚酰胺的循环伏安曲线图;
图7为实施例7制备的1,4-环己烷二甲酸型聚酰胺的第一重氧化还原态的电化学稳定性表征图;
图8为实施例7制备的1,4-环己烷二甲酸型聚酰胺的第二重氧化还原态的电化学稳定性表征图;
图9为实施例7制备的1,4-环己烷二甲酸型聚酰胺的电控荧光谱图;
图10为实施例7制备的1,4-环己烷二甲酸型聚酰胺的电控荧光响应时间谱图;
图11为实施例7制备的1,4-环己烷二甲酸型聚酰胺的电控荧光稳定性谱图。
具体实施方式
本发明提供了一种含七苯三胺-双荧光团结构的二胺单体,具有式i所示结构:
式i中,r为
r1为-ch3、-ch2ch3、-ch2ch2ch3、-ch(ch3)2、-(ch2)3ch3、-ch2ch(ch3)2、-c(ch3)3或-(ch2)4ch3。
在本发明中,所述含七苯三胺-双荧光团结构的二胺单体含有双荧光团结构和七苯三胺结构,由该二胺单体制备的聚合物中会引入双荧光团结构(r所代表的萘、蒽、芘、芴或螺芴等荧光团结构)和七苯三胺结构,其中的双荧光团结构能够使聚合物具有高荧光强度,而且将具有助色团效应的三苯胺衍生物与荧光团相连可以进一步改善聚合物的荧光,确保聚合物具有高荧光强度和高荧光开/关对比度;其中的七苯三胺结构可以通过共振稳定单阳离子自由基和双阳离子,改善聚合物的电化学稳定性和电控荧光稳定性,且较宽的稳定电压范围可以有效调节聚合物的电控荧光行为;此外,高度共轭的七苯三胺结构能增强分子内的电荷传输能力,其扭曲的三维空间构型亦能减弱聚合物分子链间的堆积,双荧光团结构和七苯三胺结构的协同效应可以缩短电控荧光的响应时间。
在本发明中,所述含七苯三胺-双荧光团结构的二胺单体优选为:
本发明提供了上述技术方案所述含七苯三胺-双荧光团结构的二胺单体的制备方法,包括以下步骤:
将对烷氧基苯胺、第一卤代硝基苯、第一碱性催化剂和第一非质子极性溶剂混合,进行第一亲核取代反应,得到4,4'-二硝基-4”-烷氧基三苯胺;
将所述4,4'-二硝基-4”-烷氧基三苯胺、乙醇、第一催化剂和第一还原剂混合,进行第一还原反应,得到4,4'-二氨基-4”-烷氧基三苯胺;
将所述4,4'-二氨基-4”-烷氧基三苯胺、第二卤代硝基苯、第二碱性催化剂和第二非质子极性溶剂混合,进行第二亲核取代反应,得到4,4'-双[(4-硝基苯基)胺]4”-烷氧基三苯胺;
将所述4,4'-双[(4-硝基苯基)胺]4”-烷氧基三苯胺、rbr化合物、金属催化剂、助催化剂、相转移催化剂和反应溶剂混合,进行乌尔曼反应,得到二硝基化合物;
将所述二硝基化合物、有机溶剂、第二催化剂和第二还原剂混合,进行第二还原反应,得到含七苯三胺-双荧光团结构的二胺单体;
其中,rbr化合物中的r为
所述对烷氧基苯胺具有式ii所示结构:
r1为-ch3、-ch2ch3、-ch2ch2ch3、-ch(ch3)2、-(ch2)3ch3、-ch2ch(ch3)2、-c(ch3)3或-(ch2)4ch3。
在本发明中,若无特殊说明,所需制备原料均为本领域技术人员熟知的市售商品。
本发明将对烷氧基苯胺、第一卤代硝基苯、第一碱性催化剂和第一非质子极性溶剂混合,进行第一亲核取代反应,得到4,4-二硝基-4-烷氧基三苯胺。在本发明中,所述对烷氧基苯胺具有式ii所示结构:
在本发明中,所述第一卤代硝基苯优选包括对氟硝基苯、对氯硝基苯、对溴硝基苯或对碘硝基苯;所述第一碱性催化剂优选包括氟化铯、碳酸铯、碳酸钠、氢氧化钾或氢化钠;所述对烷氧基苯胺、第一卤代硝基苯和第一碱性催化剂的摩尔比优选为1:(2~2.5):(2~2.5),更优选为1:(2.2~2.3):(2.2~2.4)。
在本发明中,所述第一非质子极性溶剂优选包括二甲基亚砜或n,n-二甲基甲酰胺;所述第一非质子极性溶剂的用量优选使得混合所得反应体系的总固含量为20~30%即可,所述总固含量更优选为25%。本发明对将对烷氧基苯胺、第一卤代硝基苯、第一碱性催化剂和第一非质子极性溶剂混合的过程没有特殊的限定,按照本领域熟知的过程能够将原料混合均匀即可。
在本发明中,所述第一亲核取代反应的温度优选为140~160℃,更优选为145~155℃,时间优选为12~24h,更优选为15~20h。本发明优选在搅拌以及氮气保护条件下进行所述第一亲核取代反应。本发明对所述搅拌的过程没有特殊的限定,能够保证反应顺利进行即可。
完成所述第一亲核取代反应后,本发明优选出料于不良溶剂(如甲醇、乙醇或水)中,将析出的沉淀物过滤,重结晶,得到橙黄色固体,烘箱干燥后得到4,4'-二硝基-4”-烷氧基三苯胺。本发明对所述出料、过滤、重结晶、干燥的过程没有特殊的限定,按照本领域熟知的过程进行即可。
得到4,4'-二硝基-4”-烷氧基三苯胺后,本发明将所述4,4'-二硝基-4”-烷氧基三苯胺、乙醇、第一催化剂和第一还原剂混合,进行第一还原反应,得到4,4'-二氨基-4”-烷氧基三苯胺。在本发明中,所述第一催化剂优选包括钯/碳催化剂,本发明所用钯/碳催化剂优选为市售质量分数为10%的钯/碳催化剂;所述4,4'-二硝基-4”-烷氧基三苯胺和第一催化剂的质量比优选为1:(0.2~0.5),更优选为1:(0.3~0.4)。在本发明中,所述第一还原剂优选包括水合肼;所述第一还原剂与4,4'-二硝基-4”-烷氧基三苯胺的摩尔比优选为(5~25):1,更优选为(10~20):1。在本发明中,所述水合肼优选以水合肼水溶液的形式使用,所述水合肼水溶液的质量分数优选为80%。
在本发明中,所述4,4'-二硝基-4”-烷氧基三苯胺、乙醇、第一催化剂和第一还原剂混合的过程优选为先将4,4'-二硝基-4”-烷氧基三苯胺溶于乙醇,然后加入第一催化剂,加热至回流(70~90℃),然后向所得溶液中滴加第一还原剂。在本发明中,乙醇的用量优选使得4,4'-二硝基-4”-烷氧基三苯胺和乙醇体系的固含量为5~10%即可。本发明对所述滴加的速率没有特殊的限定,按照本领域熟知的过程进行即可。
在本发明中,所述第一还原反应的温度为70~90℃,时间为10~15h;所述第一还原反应优选在搅拌回流条件下进行,所述回流的温度优选与第一还原反应的温度相同;本发明对所述搅拌的过程没有特殊的限定,能够保证反应顺利进行即可。
完成所述第一还原反应后,本发明优选将所得体系趁热过滤除去第一催化剂,然后在氮气气氛下冷却结晶,得到4,4'-二氨基-4”-烷氧基三苯胺。本发明对所述过滤和冷却结晶的过程没有特殊的限定,按照本领域熟知的过程进行即可。
在本发明中,以第一卤代硝基苯为对氟硝基苯,第一非质子极性溶剂为二甲基亚砜(dmso),第一催化剂为钯/碳催化剂以及第一还原剂为水合肼为例,所述第一亲核取代反应和第一还原反应的过程如下所示:
如上反应式所示,首先,在碱性条件下,对烷氧基苯胺氨基上的氢被对氟硝基苯取代得到4,4'-二硝基-4”-烷氧基三苯胺;然后以钯/碳为催化剂,水合肼为还原剂,将硝基还原为氨基,得到4,4'-二氨基-4”-烷氧基三苯胺。
得到4,4'-二氨基-4”-烷氧基三苯胺后,本发明将所述4,4'-二氨基-4”-烷氧基三苯胺、第二卤代硝基苯、第二碱性催化剂和第二非质子极性溶剂混合,进行第二亲核取代反应,得到4,4'-双[(4-硝基苯基)胺]-4”-烷氧基三苯胺。在本发明中,所述第二卤代硝基苯优选包括对氟硝基苯、对氯硝基苯、对溴硝基苯或对碘硝基苯;所述第二碱性催化剂优选包括三乙胺(tea)或三丙胺;所述第二非质子极性溶剂优选包括二甲基亚砜或n,n-二甲基甲酰胺;所述4,4'-二氨基-4”-烷氧基三苯胺、第二卤代硝基苯和第二碱性催化剂的摩尔比优选为1:(2.3~2.5):(3~3.5),更优选为1:2.4:(3.2~3.4)。
本发明利用三乙胺或三丙胺作为碱性催化剂,其可以与4,4'-二氨基-4”-烷氧基三苯胺中-nh2上的质子结合,增强氨基中氮原子的亲核性,加快与卤代硝基苯的反应速度。
本发明对所述4,4'-二氨基-4”-烷氧基三苯胺、第二卤代硝基苯、第二碱性催化剂和第二非质子极性溶剂混合的过程没有特殊的限定,按照本领域熟知的过程进行即可。在本发明中,所述第二非质子极性溶剂的用量优选使得混合所得反应体系的总固含量为20~30%即可,更优选为25%。
在本发明中,所述第二亲核取代反应的温度优选为110~130℃,更优选为115~125℃,时间优选为60~72h,更优选为65~70h。本发明优选在搅拌以及氮气保护条件下进行所述第二亲核取代反应。本发明对所述搅拌的过程没有特殊的限定,能够保证反应顺利进行即可。
完成所述第二亲核取代反应后,本发明将所得体系冷却至室温后出料于甲醇(或乙醇)和水(甲醇(或乙醇)与水的体积比优选为:1:(1~3))的混合液中,将析出的沉淀物过滤,重结晶后得到4,4'-双[(4-硝基苯基)胺]4”-烷氧基三苯胺。本发明对所述出料、过滤和重结晶的过程没有特殊的限定,按照本领域熟知的过程进行即可。
在本发明中,以第二卤代硝基苯为对氟硝基苯、第二碱性催化剂为三乙胺(tea)以及第二非质子极性溶剂为二甲基亚砜(dmso)为例,所述第二亲核取代反应的过程如下所示:
如上述反应式所示,在碱性条件下,4,4'-二氨基-4”-烷氧基三苯胺中两个氨基上的质子分别被对氟硝基苯取代,生成取代产物4,4'-双[(4-硝基苯基)胺]4”-烷氧基三苯胺。
得到4,4'-双[(4-硝基苯基)胺]4”-烷氧基三苯胺后,本发明将所述4,4'-双[(4-硝基苯基)胺]4”-烷氧基三苯胺、rbr化合物、金属催化剂、助催化剂、相转移催化剂和反应溶剂混合,进行乌尔曼反应,得到二硝基化合物。在本发明中,所述金属催化剂优选包括铜粉、镍或碘化亚铜;所述助催化剂优选包括碳酸钾、氢氧化钾、氢化钾、氢化钠、氢氧化钠、碳酸钠、碳酸铯或氟化铯;所述相转移催化剂优选包括18-冠醚-6、15-冠醚-5、二苯并-18-冠醚-6、苯并-18-冠醚-6或苯并-15-冠醚-5。
在本发明中,所述rbr化合物中的r为
在本发明中,所述4,4'-双[(4-硝基苯基)胺]4”-烷氧基三苯胺、rbr化合物、金属催化剂、助催化剂和相转移催化剂的摩尔比优选为1:(2~4):(8~10):(8~10):(1~1.5),更优选为1:(2.5~3.5):(8.5~9.5):(8.5~9.5):(1.2~1.3)。
在本发明中,所述反应溶剂优选包括邻二氯苯、二氯甲苯、1,2,4-三氯苯或邻二甲氧基苯;所述反应溶剂的用量优选使得混合所得反应体系的总固含量为15~45%即可,优选为20~35%,更优选为25~30%。本发明对所述4,4'-双[(4-硝基苯基)胺]4”-烷氧基三苯胺、rbr化合物、金属催化剂、助催化剂、相转移催化剂和反应溶剂混合的过程没有特殊的限定,按照本领域熟知的过程进行即可。
在本发明中,所述乌尔曼反应的温度优选为150~170℃,更优选为155~165℃,时间优选为12~24h,更优选为15~20h;所述乌尔曼反应优选在搅拌以及氮气保护条件下进行,本发明对所述搅拌的过程没有特殊的限定,能够保证反应顺利进行即可。
完成所述乌尔曼反应后,本发明将所得体系趁热抽滤,将所得滤液缓慢倾倒于不良溶剂中(如甲醇、乙醇或水)析出固体,将所得粗产物用柱层析进行提纯,得到含“4,4'-双(荧光团胺)-4”-烷氧基三苯胺”结构的二硝基化合物。本发明对所述抽滤、析出和提纯的过程没有特殊的限定,按照本领域熟知的过程进行即可。
得到二硝基化合物后,本发明将所述二硝基化合物、有机溶剂、第二催化剂和第二还原剂混合,进行第二还原反应,得到含七苯三胺-双荧光团结构的二胺单体。在本发明中,所述有机溶剂优选包括二氧六环或四氢呋喃;所述第二催化剂优选为pd/c,所述第二还原剂优选为水合肼。在本发明中,所述第二还原反应所用第二催化剂和第二还原剂的规格要求优选与第一还原反应所用第一催化剂和第一还原剂的规格要求相同。
在本发明中,所述二硝基化合物与第二催化剂的质量比优选为1:(0.2~0.5),更优选为1:(0.3~0.4),所述第二还原剂与二硝基化合物的摩尔比优选为(5~25):1,更优选为(10~20):1。
在本发明中,所述二硝基化合物、有机溶剂、第二催化剂和第二还原剂混合的过程优选为先将所述二硝基化合物溶于有机溶剂中,然后向所得溶液中加入第二催化剂,加热至回流,然后向所得溶液中滴加第二还原剂。本发明对所述滴加的速率没有特殊的限定,按照本领域熟知的过程进行即可。
在本发明中,当所述有机溶剂为二氧六环时,所述回流的温度为90~110℃;当所述有机溶剂为四氢呋喃时,所述回流的温度为60~80℃;所述有机溶剂的用量优选使得二硝基化合物与有机溶剂混合所得体系总固含量为5~10%即可。
在本发明中,所述第二还原反应的温度优选与所述有机溶剂的回流温度相同;所述第二还原反应的时间优选为1~72h,更优选为10~60h,所述第二还原反应优选在搅拌条件下进行,本发明对所述搅拌的过程没有特殊的限定,能够保证反应顺利进行即可。
完成所述第二还原反应后,本发明优选将所得体系趁热过滤除去催化剂,减压浓缩滤液至原体积的1/2~1/5,在氮气气氛下冷却结晶,得到含“4,4'-双(荧光团胺)-4”-烷氧基三苯胺”结构的二胺单体。本发明对所述过滤、减压浓缩和冷却结晶的过程没有特殊的限定,按照本领域熟知的过程进行即可。
在本发明中,以金属催化剂为铜粉,助催化剂为碳酸钾,相转移催化剂为18-冠醚-6,反应溶剂为邻二氯苯,第二催化剂为pd/c,第二还原剂为水合肼为例,所述乌尔曼反应和第二还原反应的过程如下所示:
如上述反应式所示,4,4'-双[(4-硝基苯基)胺]4”-烷氧基三苯胺先与溴代荧光团发生乌尔曼反应,导致二级胺上的氢原子被荧光团取代,得到4,4'-双(荧光团胺)-4”-烷氧基三苯胺”结构的二硝基化合物,硝基在钯碳催化下,被水合肼还原,得到4,4'-双(荧光团胺)-4”-烷氧基三苯胺”结构的二胺单体。
本发明提供了上述技术方案所述含七苯三胺-双荧光团结构的二胺单体或上述技术方案所述制备方法制备的含七苯三胺-双荧光团结构的二胺单体在制备聚酰胺或聚酰亚胺中的应用。本发明对将所述含七苯三胺-双荧光团结构的二胺单体用于制备聚酰胺或聚酰亚胺的方法没有特殊的限定,按照本领域熟知的过程进行即可。
本发明提供了一种含七苯三胺-双荧光团结构的聚酰胺,具有式iii所示结构:
其中,n=10~80且n为整数;所述含七苯三胺-双荧光团结构的聚酰胺的数均分子量为10300~83000;
ar包括
r为
r1为-ch3、-ch2ch3、-ch2ch2ch3、-ch(ch3)2、-(ch2)3ch3、-ch2ch(ch3)2、-c(ch3)3或-(ch2)4ch3。
在本发明中,所述n优选为20~60,更优选为30~50,进一步优选为35~45。
本发明所述聚酰胺含有双荧光团结构和七苯三胺结构,其中的双荧光团结构能够提高聚酰胺的荧光强度,而且将具有助色团效应的三苯胺衍生物与荧光团相连可以进一步改善聚酰胺的荧光,所述聚酰胺具有高荧光强度和高荧光开/关对比度;其中的七苯三胺结构可以通过共振稳定单阳离子自由基和双阳离子,改善聚酰胺的电化学稳定性和电控荧光稳定性,且较宽的稳定电压范围可以有效调节聚酰胺的电控荧光行为;此外,高度共轭的七苯三胺结构能增强分子内的电荷传输能力,其扭曲的三维空间构型亦能减弱聚合物分子链间的堆积,双荧光团结构和七苯三胺结构的协同效应可以缩短电控荧光的响应时间。
本发明提供了上述技术方案所述含七苯三胺-双荧光团结构的聚酰胺的制备方法,包括以下步骤:
将含七苯三胺-双荧光团结构的二胺单体、二酸单体、亚磷酸三苯酯、吡啶、溶剂和助溶剂混合,进行缩聚反应,得到含七苯三胺-双荧光团结构的聚酰胺;
所述含七苯三胺-双荧光团结构的二胺单体为上述技术方案所述含七苯三胺-双荧光团结构的二胺单体或者上述技术方案所述制备方法制备得到的含七苯三胺-双荧光团结构的二胺单体;
所述二酸单体包括对苯二甲酸、1,4-环己烷二甲酸或4,4'-联苯二甲酸。
在本发明中,所述含七苯三胺-双荧光团结构的二胺单体和二酸单体的摩尔比优选为1:(0.8~1.2),更优选为1:(0.9~1.1)。在本发明中,所述溶剂优选为n-甲基吡咯烷酮;所述溶剂的用量优选使得含七苯三胺-双荧光团结构的二胺单体、二酸单体和溶剂混合所得反应体系的总固含量为15~35%即可。
在本发明中,所述助溶剂优选为氯化钙;所述助溶剂与所述二胺单体和二酸单体质量之和的质量比优选为(0.1~0.5):1,更优选为(0.2~0.3):1。
在本发明中,所述亚磷酸三苯酯与含七苯三胺-双荧光团结构的二胺单体的摩尔比优选为(2~4):1,更优选为3:1;所述吡啶与含七苯三胺-双荧光团结构的二胺单体的摩尔比优选为(4~8):1,更优选为(5~6):1;所述亚磷酸三苯酯和吡啶作为缩合剂。
在本发明中,所述含七苯三胺-双荧光团结构的二胺单体、二酸单体、亚磷酸三苯酯、吡啶、溶剂和助溶剂混合的过程优选为先将所述含七苯三胺-双荧光团结构的二胺单体、二酸单体和溶剂混合,然后向所得混合液中加入亚磷酸三苯酯和吡啶,最后向所得混合物料中加入助溶剂。
在本发明中,所述缩聚反应优选在氮气保护下进行,所述缩聚反应的温度优选为100~130℃,时间优选为2~5h;完成所述缩聚反应后,本发明优选将所得产物体系出料于乙醇,过滤后将所得产物用乙醇和水加热至回流洗涤,干燥,得到含七苯三胺-双荧光团结构的聚酰胺。本发明对所述出料、过滤、加热回流、洗涤和干燥的过程没有特殊的限定,按照本领域熟知的过程进行即可。
在本发明中,所述含七苯三胺-双荧光团结构的聚酰胺的制备过程如下所示:
本发明提供了上述技术方案所述含七苯三胺-双荧光团结构的聚酰胺在电控荧光器件中的应用。本发明对所述应用的方法没有特殊的限定,按照本领域熟知的过程将所述含七苯三胺-双荧光团结构的聚酰胺用于电控荧光器件即可。
本发明提供了一种含七苯三胺-双荧光团结构的聚酰亚胺,具有式iv所示结构:
其中,m=10~80且m为整数,所述含七苯三胺-双荧光团结构的聚酰亚胺的数均分子量为10800~87000;
ar'包括
r为
r1为-ch3、-ch2ch3、-ch2ch2ch3、-ch(ch3)2、-(ch2)3ch3、-ch2ch(ch3)2、-c(ch3)3或-(ch2)4ch3。
在本发明中,所述m优选为20~60,更优选为30~50,进一步优选为35~45。
本发明所述聚酰亚胺含有双荧光团结构和七苯三胺结构,其中的双荧光团结构能够提高聚酰亚胺的荧光强度,而且将具有助色团效应的三苯胺衍生物与荧光团相连可以进一步改善聚酰亚胺的荧光,所述聚酰亚胺具有高荧光强度和高荧光开/关对比度;其中的七苯三胺结构可以通过共振稳定单阳离子自由基和双阳离子,改善聚酰亚胺的电化学稳定性和电控荧光稳定性,且较宽的稳定电压范围可以有效调节聚酰亚胺的电控荧光行为;此外,高度共轭的七苯三胺结构能增强分子内的电荷传输能力,其扭曲的三维空间构型亦能减弱聚合物分子链间的堆积,双荧光团结构和七苯三胺结构的协同效应可以缩短电控荧光的响应时间。
本发明提供了上述技术方案所述含七苯三胺-双荧光团结构的聚酰亚胺的制备方法,包括以下步骤:
将含七苯三胺-双荧光团结构的二胺单体、二酐单体和反应溶剂混合,进行聚合反应,得到聚酰胺酸溶液;
将所述聚酰胺酸溶液、催化剂和脱水剂混合,进行热亚胺化,得到聚酰亚胺;
所述含七苯三胺-双荧光团结构的二胺单体为上述技术方案所述含七苯三胺-双荧光团结构的二胺单体或者上述技术方案所述制备方法制备得到的含七苯三胺-双荧光团结构的二胺单体;
所述二酐单体包括均苯四甲酸二酐、氢化均苯四甲酸二酐或3,3',4,4'-联苯四甲酸二酐。
本发明将含七苯三胺-双荧光团结构的二胺单体、二酐单体和反应溶剂混合,进行聚合反应,得到聚酰胺酸溶液。在本发明中,所述二酐单体包括均苯四甲酸二酐(即1,2,4,5-均苯四甲酸二酐)、氢化均苯四甲酸二酐或3,3',4,4'-联苯四甲酸二酐;所述二酐单体与含七苯三胺-双荧光团结构的二胺单体的摩尔比优选为1:(0.8~1.2),更优选为1:(0.9~1.0)。
在本发明中,所述反应溶剂优选为n,n-二甲基乙酰胺;所述反应溶剂的用量优选使得混合所得反应体系的固含量为5~30%即可,更优选为10~20%。
在本发明中,所述混合的过程优选为先将二胺单体加入到反应溶剂中,全部溶解后,分多次加入二酐单体。本发明对多次加入的过程没有特殊的限定,按照本领域熟知的操作进行即可。
在本发明中,所述聚合反应优选在氮气条件下进行,所述聚合反应的温度优选为室温,所述聚合反应的时间优选为4~18h,更优选为5~15h,进一步优选为8~12h。
完成所述聚合反应后,本发明优选不进行任何处理,直接将所得聚酰胺酸溶液、催化剂和脱水剂混合,进行热亚胺化,得到聚酰亚胺。在本发明中,所述催化剂优选为吡啶,所述脱水剂优选为乙酸酐,所述脱水剂与含七苯三胺-双荧光团结构的二胺单体的摩尔比优选为(2~5):1,更优选为(3~4):1;所述催化剂与含七苯三胺-双荧光团结构的二胺单体的摩尔比优选为(1~5):1,更优选为(2~3):1。
本发明对所述聚酰胺酸溶液、催化剂和脱水剂混合的过程没有特殊的限定,能够将原料混合均匀即可。在本发明中,所述热亚胺化的温度优选为100~120℃,更优选为105~110℃,时间优选为3~8h,更优选为5~6h。
完成所述热亚胺化后,本发明优选将所得产物体系出料于乙醇,然后将产物用乙醇加热至回流洗涤,干燥,得到含七苯三胺-双荧光团结构的聚酰亚胺。本发明对所述出料、加热回流、洗涤和干燥的过程没有特殊的限定,按照本领域熟知的过程进行即可。
在本发明中,所述聚酰亚胺的制备过程如下所示:
本发明提供了上述技术方案所述含七苯三胺-双荧光团结构的聚酰亚胺在电控荧光器件中的应用。本发明对所述应用的方法没有特殊的限定,按照本领域熟知的过程将所述含七苯三胺-双荧光团结构的聚酰亚胺用于电控荧光器件即可。
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
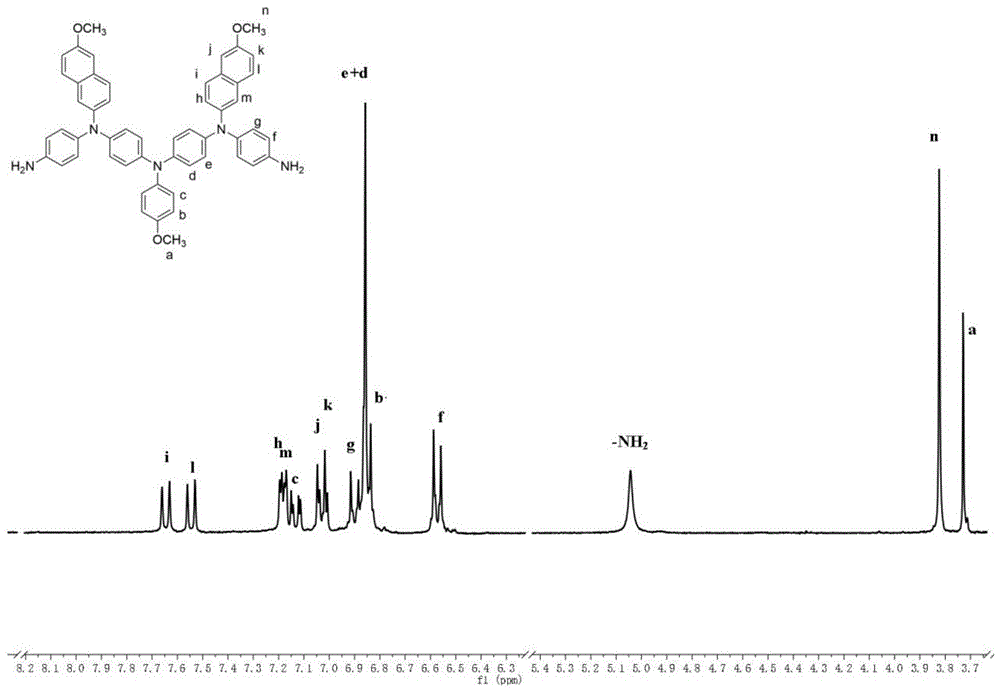
实施例1
4,4'-双[4-氨基苯基(6-甲氧基-2-萘基)胺]-4”-甲氧基三苯胺的制备,其结构式如下所示:
向装有机械搅拌装置和冷凝管的500ml三颈烧瓶中,加入15.00g(121.8mmol)的对甲氧基苯胺,43.00g(304.51mmol)的对氟硝基苯,46.25g(304.5mmol)氟化铯,加入158.2ml的二甲基亚砜,体系固含量为25%,在搅拌,氮气保护下,于150℃下反应12h;冷却至室温后出料于乙醇中并充分搅拌,过滤得到的粗产物用n,n-二甲基乙酰胺和乙醇重结晶,干燥后得到39.60g橙色的4,4′-二硝基-4″-甲氧基三苯胺,产率为89%;
向装有磁力搅拌子和冷凝管的500ml三颈烧瓶中,加入20.00g(54.78mmol)4,4′-二硝基-4″-甲氧基三苯胺和225ml乙醇溶剂,使得体系固含量为10%,然后加入6.00g质量分数为10%的pd/c,加热至75℃回流后,向溶液中缓慢滴加68.55g(水合肼的摩尔量为1095.6mmol)质量分数为80%的水合肼水溶液,继续回流搅拌10h,趁热过滤除去pd/c,滤液通氮气,冷却过程中逐渐析出白色晶体4,4′-二氨基-4″-甲氧基三苯胺14.11g,产率为84.3%。
向装有磁力搅拌子和冷凝管的250ml三颈烧瓶中,加入13.00g(42.57mmol)4,4′-二氨基-4″-甲氧基三苯胺,14.42g(102.17mmol)对氟硝基苯,14.00g(138.35mmol)三乙胺,加入67ml二甲基亚砜为溶剂,体系固含量为27%,在氮气保护下,于120℃反应72小时,将反应液冷却至室温后倒入搅拌条件下的甲醇/水溶液(甲醇与水的体积比为1:2)中,析出棕色粉末,粗产物用甲苯重结晶,得到9.09g4,4′-双[(4-硝基苯基)胺]-4″-甲氧基三苯胺,产率为39%。
向装有磁力搅拌子和冷凝管的250ml三颈烧瓶中,加入4.50g(8.22mmol)4,4′-双[(4-硝基苯基)胺]4″-甲氧基三苯胺,4.88g(17.87mmol)2-溴-6-甲氧基萘,4.18g(65.77mmol)铜粉,9.09g(65.77mmol)碳酸钾,2.16g(8.22mmol)18-冠醚-6,加入16.8ml邻二氯苯,体系固含量为30%,在氮气保护下,于160℃下反应24h,反应结束后趁热过滤,向滤液中加入乙醇析出固体,所得粗产物用柱层析提纯(展开剂:二氯甲烷:石油醚=8:1),得到2.91g4,4'-双[4-硝基苯基(6-甲氧基-2-萘基)胺]-4”-甲氧基三苯胺红色粉末,产率为41.6%。
向装有磁力搅拌子和冷凝管的250ml三颈烧瓶中,加入2.30g(2.71mmol)4,4'-双[4-硝基苯基(6-甲氧基-2-萘基)胺]-4”-甲氧基三苯胺,加入20ml二氧六环作为溶剂,使得体系固含量为10%,然后加入0.69g质量分数为10%的pd/c,加热至100℃回流后,向溶液中缓慢滴加3.40g(水合肼的摩尔量为54.33mmol)质量分数为80%的水合肼水溶液,滴加完成后,继续回流搅拌20h,趁热过滤除去pd/c,减压浓缩滤液至原体积的1/3,在氮气气氛下冷却结晶,得到浅绿色4,4'-双[4-氨基苯基(6-甲氧基-2-萘基)胺]-4”-甲氧基三苯胺粉末1.79g,产率为84%。
图1为4,4'-双[4-氨基苯基(6-甲氧基-2-萘基)胺]-4”-甲氧基三苯胺的氢核磁谱图,由图可知,h原子的化学位移归属明确,可一一对应,为所得4,4'-双[4-氨基苯基(6-甲氧基-2-萘基)胺]-4”-甲氧基三苯胺的结构,核磁数据为:1hnmr(300mhz,dmso-d6)δ7.65(d,j=8.8hz,1h),7.55(d,j=9.0hz,1h),7.19(d,j=2.5hz,1h),7.17(d,j=2.3hz,1h),7.13(d,j=8.7,2.3hz,1h),7.04(d,j=2.7hz,1h),7.01(d,j=2.9hz,1h),6.90(d,j=8.9hz,2h),6.86(d,j=2.3hz,4h),6.84(d,j=2.1hz,1h),6.61(d,2h),5.04(s,2h),3.82(d,j=0.6hz,3h),3.73(d,j=0.6hz,1.5h).
图2实施例1制备的4,4'-双[4-氨基苯基(6-甲氧基-2-萘基)胺]-4”-甲氧基三苯胺的红外谱图,该图说明了制备的二胺单体的结构,其中3448cm-1和3369cm-1为氨基的振动吸收峰。
实施例2
4,4'-双[4-氨基苯基(2-萘基)胺]-4”-乙氧基三苯胺的制备,其结构式如下所示:
向装有机械搅拌装置和冷凝管的500ml三颈烧瓶中,加入20.00g(145.8mmol)的对乙氧基苯胺,57.42g(364.5mmol)的对氯硝基苯,118.76g(364.5mmol)碳酸铯,加入195ml的二甲基亚砜,体系固含量为25%。在搅拌,氮气保护下,于150℃下反应12h;冷却至室温后出料于甲醇中并充分搅拌,过滤所得粗产物用n,n-二甲基乙酰胺和乙醇重结晶,干燥后得到47.01g橙色的4,4′-二硝基-4″-乙氧基三苯胺,产率为85%;
向装有磁力搅拌子和冷凝管的500ml三颈烧瓶中,加入30.00g(79.08mmol)4,4′-二硝基-4″-乙氧基三苯胺和340ml乙醇溶剂,使得体系固含量为10%,然后加入9.00g质量分数为10%的pd/c,加热至75℃回流后,向溶液中缓慢滴加99.00g(水合肼的摩尔量为1582.1mmol)质量分数为80%的水合肼水溶液,继续回流搅拌10h,趁热过滤除去pd/c,滤液通氮气,冷却过程中逐渐析出白色晶体4,4′-二氨基-4″-乙氧基三苯胺21g,产率为83.2%。
向装有磁力搅拌子和冷凝管的250ml三颈烧瓶中,加入15.00g(46.96mmol)4,4′-二氨基-4″-乙氧基三苯胺,15.90g(112.71mmol)对氟硝基苯,21.80g(152.15mmol)三丙胺,加入88mln,n-二甲基甲酰胺为溶剂,固含量为27%。在氮气保护下,于120℃反应72小时,将反应液冷却至室温后倒入搅拌条件下的乙醇/水溶液(甲醇与水的体积比为1:2)中,析出棕色粉末,粗产物用甲苯重结晶,得到11.08g4,4′-双[(4-硝基苯基)胺]-4″-乙氧基三苯胺,产率为42%。
向装有磁力搅拌子和冷凝管的250ml三颈烧瓶中,加入7.00g(12.46mmol)4,4′-双[(4-硝基苯基)胺]-4″-乙氧基三苯胺,6.45g(31.16mmol)2-溴萘,5.85g(99.68mmol)镍,3.99g(99.68mmol)氢化钾,2.74g(12.46mmol)15-冠醚-5,加入24ml二氯甲苯,体系固含量为30%,在氮气保护下,于160℃下反应24h,反应结束后趁热过滤,向滤液中加入甲醇析出固体,所得粗产物用柱层析提纯(展开剂:二氯甲烷:石油醚=6:1),得到5.9g4,4'-双[4-硝基苯基(2-萘基)胺]-4”-乙氧基三苯胺红色粉末,产率为58%。
向装有磁力搅拌子和冷凝管的250ml三颈烧瓶中,加入3.0g(3.69mmol)4,4'-双[4-硝基苯基(2-萘基)胺]-4”-乙氧基三苯胺,加入26ml四氢呋喃作为溶剂,使得体系固含量为10%,然后加入0.9g质量分数为10%的pd/c,加热至65℃回流后,向溶液中缓慢滴加4.61g(水合肼的摩尔量为73.71mmol)质量分数为80%的水合肼水溶液,滴加完成后,继续回流搅拌20h,趁热过滤除去pd/c,减压浓缩滤液至原体积的1/3,在氮气气氛下冷却结晶,得到浅绿色的4,4'-双[4-氨基苯基(2-萘基)胺]-4”-乙氧基三苯胺粉末2.5g,产率为89.7%。
实施例3
4,4'-双[4-氨基苯基(2-9,9′-螺二芴基)胺]-4”-异丙氧基三苯胺的制备,其结构式如下:
向装有机械搅拌装置和冷凝管的500ml三颈烧瓶中,加入15.00g(99.2mmol)的对异丙氧基苯胺,30.79g(218.2mmol)的对氟硝基苯,12.24g(218.2mmol)氢氧化钾,加入125ml的二甲基亚砜,体系固含量为25%。在搅拌,氮气保护下,于150℃下反应12h;冷却至室温后出料于乙醇中并充分搅拌,过滤所得粗产物用n,n-二甲基乙酰胺和乙醇重结晶,干燥后得到30.5g橙色的4,4′-二硝基-4″-异丙氧基三苯胺,产率为78.2%;
向装有磁力搅拌子和冷凝管的500ml三颈烧瓶中,加入25.00g(63.55mmol)4,4′-二硝基-4″-异丙氧基三苯胺和280ml乙醇作为溶剂,使得体系固含量为10%,然后加入7.50g质量分数为10%的pd/c,加热至75℃回流后,向溶液中缓慢滴加79.52g(水合肼的摩尔量为1270.9mmol)质量分数为80%的水合肼水溶液,继续回流搅拌10h,趁热过滤除去pd/c,滤液通氮气,冷却过程中逐渐析出白色晶体4,4′-二氨基-4″-异丙氧基三苯胺18.4g,产率为86.8%。
向装有磁力搅拌子和冷凝管的250ml三颈烧瓶中,加入15.00g(44.99mmol)4,4′-二氨基-4″-异丙氧基三苯胺,17.72g(112.47mmol)对氯硝基苯,20.88g(145.77mmol)三丙胺,加入76mln,n-二甲基甲酰胺为溶剂,体系固含量为27%。在氮气保护下,于120℃反应72小时,将反应液冷却至室温后倒入搅拌条件下的乙醇/水溶液(甲醇与水的体积比为1:2)中,析出棕色粉末,粗产物用甲苯重结晶,得到10.5g4,4′-双[(4-硝基苯基)胺]-4″-异丙氧基三苯胺,产率为40.5%。
向装有磁力搅拌子和冷凝管的250ml三颈烧瓶中,加入4.66g(8.1mmol)4,4′-双[(4-硝基苯基)胺]4″-异丙氧基三苯胺,8.00g(20.24mmol)2-溴-9,9'-螺二芴,4.12g(64.80mmol)铜粉,3.64g(64.80mmol)氢氧化钾,2.92g(8.1mmol)二苯并-18-冠醚-6,加入23ml邻二氯苯,体系固含量为30%,在氮气保护下,于160℃下反应24h,反应结束后趁热过滤,向滤液中加入乙醇析出固体,所得粗产物用柱层析提纯(展开剂:二氯甲烷:石油醚=5:1),得到6.14g4,4'-双[4-硝基苯基(2-9,9′-螺二芴基)胺]-4”-异丙氧基三苯胺红色粉末,产率为62.97%。
向装有磁力搅拌子和冷凝管的250ml三颈烧瓶中,加入3.0g(2.49mmol)4,4'-双[4-硝基苯基(2-9,9′-螺二芴基)胺]-4”-异丙氧基三苯胺,加入26ml四氢呋喃作为溶剂,使得体系固含量为10%,然后加入0.9g质量分数为10%的pd/c,加热至65℃回流后,向溶液中缓慢滴加3.11g(水合肼的摩尔量为49.75mmol)质量分数为80%的水合肼水溶液,滴加完成后,继续回流搅拌20h,趁热过滤除去pd/c,减压浓缩滤液至原体积的1/3,在氮气气氛下冷却结晶,即得到本发明所述的4,4'-双[4-氨基苯基(2-9,9′-螺二芴基)胺]-4”-异丙氧基三苯胺2.48g,产率为87.02%。
实施例4
4,4'-双[4-氨基苯基(9,9-二甲基-2-芴基)胺]-4”-丁氧基三苯胺的制备,其结构式如下:
向装有机械搅拌装置和冷凝管的500ml三颈烧瓶中,加入18.00g(108.93mmol)的对丁氧基苯胺,55.01g(272.33mmol)的对溴硝基苯,28.86g(272.33mmol)碳酸钠,加入154ml的n,n-二甲基甲酰胺,体系固含量为25%。在搅拌,氮气保护下,于150℃下反应12h;冷却至室温后出料于水醇中并充分搅拌,过滤得到的粗产物用n,n-二甲基乙酰胺和乙醇重结晶,干燥后得到32.11g橙色的4,4′-二硝基-4″-丁氧基三苯胺,产率为72.3%;
向装有磁力搅拌子和冷凝管的500ml三颈烧瓶中,加入25.00g(61.36mmol)4,4′-二硝基-4″-丁氧基三苯胺,加入280ml乙醇作为溶剂,使得体系固含量为10%,然后加入7.50g质量分数为10%的pd/c,加热至75℃回流后,向溶液中缓慢滴加76.79g(水合肼的摩尔量为1227.13mmol)质量分数为80%的水合肼水溶液,继续回流搅拌10h,趁热过滤除去pd/c,滤液通氮气,冷却过程中逐渐析出白色晶体4,4′-二氨基-4″-丁氧基三苯胺18.5g,产率为88.1%。
向装有磁力搅拌子和冷凝管的250ml三颈烧瓶中,加入15.00g(43.17mmol)4,4′-二氨基-4″-丁氧基三苯胺,16.32g(103.61mmol)对氯硝基苯,14.15g(139.87mmol)三乙胺,加入77ml二甲基亚砜为溶剂,体系固含量为27%。在氮气保护下,于120℃反应72小时,将反应液冷却至室温后倒入搅拌条件下的甲醇/水溶液(甲醇与水的体积比为1:2)中,析出棕色粉末,粗产物用甲苯重结晶,得到11.00g4,4′-双[(4-硝基苯基)胺]-4″-丁氧基三苯胺,产率为43.2%。
向装有磁力搅拌子和冷凝管的250ml三颈烧瓶中,加入7.00g(11.87mmol)4,4′-双[(4-硝基苯基)胺]4″-丁氧基三苯胺,8.11g(29.68mmol)2-溴-9,9-二甲基芴,18.09g(94.96mmol)碘化亚铜,2.28g(94.96mmol)氢化钠,3.71g(11.87mmol)苯并-18-冠醚-6,加入27ml1,2,4-三氯苯,体系固含量为30%,在氮气保护下,于160℃下反应24h,反应结束后趁热过滤,向滤液中加入水中析出固体,所得粗产物用柱层析提纯(展开剂:二氯甲烷:石油醚=5:1),得到7.5g4,4'-双[4-硝基苯基(9,9-二甲基-2-芴基)胺]-4”-丁氧基三苯胺红色粉末,产率为64.8%。
向装有磁力搅拌子和冷凝管的250ml三颈烧瓶中,加入4.00g(4.11mmol)4,4'-双[4-硝基苯基(9,9-二甲基-2-芴基)胺]-4”-丁氧基三苯胺,加入35ml二氧六环作为溶剂,使得体系固含量为10%,然后加入1.20g质量分数为10%的pd/c,加热至100℃回流后,向溶液中缓慢滴加5.14g(水合肼的摩尔量为82.10mmol)质量分数为80%的水合肼水溶液,滴加完成后,继续回流搅拌20h,趁热过滤除去pd/c,减压浓缩滤液至原体积的1/3,在氮气气氛下冷却结晶,即得到本发明所述的4,4'-双[4-氨基苯基(9,9-二甲基-2-芴基)胺]-4”-丁氧基三苯胺2.9g,产率为77.3%。
实施例5
4,4'-双[4-氨基苯基(2-芘基)胺]-4”-异丁氧基三苯胺的制备,其结构式如下:
向装有机械搅拌装置和冷凝管的500ml三颈烧瓶中,加入15.00g(90.78mmol)的对异丁氧基苯胺,49.73g(199.71mmol)的对碘硝基苯,4.19g(199.71mmol)氢化钠,加入118ml的n,n-二甲基甲酰胺,体系固含量为25%。在搅拌,氮气保护下,于150℃下反应12h;冷却至室温后出料于甲醇中并充分搅拌,过滤所得粗产物用n,n-二甲基乙酰胺和乙醇重结晶,干燥后得到28.48g橙色的4,4′-二硝基-4″-异丁氧基三苯胺,产率为77.01%;
向装有磁力搅拌子和冷凝管的500ml三颈烧瓶中,加入25.00g(61.36mmol)4,4′-二硝基-4″-异丁氧基三苯胺,加入280ml乙醇作为溶剂,使得体系固含量为10%,然后加入7.50g质量分数为10%的pd/c,加热至75℃回流后,向溶液中缓慢滴加76.79g(水合肼的摩尔量为1227.13mmol)质量分数为80%的水合肼水溶液,继续回流搅拌10h,趁热过滤除去pd/c,滤液通氮气,冷却过程中逐渐析出白色晶体4,4′-二氨基-4″-异丁氧基三苯胺17.6g,产率为82.5%。
向装有磁力搅拌子和冷凝管的250ml三颈烧瓶中,加入15.00g(43.17mmol)4,4′-二氨基-4″-异丁氧基三苯胺,14.62g(103.61mmol)对氟硝基苯,14.15g(139.87mmol)三乙胺,加入73ml二甲基亚砜为溶剂,体系固含量为27%,在氮气保护下,于120℃反应72小时,将反应液冷却至室温后倒入搅拌条件下的甲醇/水溶液(甲醇与水的体积比为1:2)中,析出棕色粉末,粗产物用甲苯重结晶,得到9.77g4,4′-双[(4-硝基苯基)胺]-4″-异丁氧基三苯胺,产率为38.4%。
向装有磁力搅拌子和冷凝管的250ml三颈烧瓶中,加入4.00g(6.78mmol)4,4′-双[(4-硝基苯基)胺]4″-异丁氧基三苯胺,4.77g(16.96mmol)2-溴芘,3.18g(54.24mmol)镍,5.75g(54.24mmol)碳酸钠,1.82g(6.78mmol)苯并-15-冠醚-5,加入15.7ml二氯甲苯,体系固含量为30%,在氮气保护下,于160℃下反应24h,反应结束后趁热过滤,向滤液中加入甲醇析出固体,所得粗产物用柱层析提纯(展开剂:二氯甲烷:石油醚=5:1),得到4.36g4,4'-双[4-硝基苯基(2-芘基)胺]-4”-异丁氧基三苯胺红色粉末,产率为64.8%。
向装有磁力搅拌子和冷凝管的250ml三颈烧瓶中,加入3.00g(3.03mmol)4,4'-双[4-硝基苯基(2-芘基)胺]-4”-异丁氧基三苯胺,加入26ml二氧六环作为溶剂,使得体系固含量为10%,然后加入0.9g质量分数为10%的pd/c,加热至100℃回流后,向溶液中缓慢滴加3.79g(水合肼的摩尔量为60.53mmol)质量分数为80%的水合肼水溶液,滴加完成后,继续回流搅拌12h,趁热过滤除去pd/c,减压浓缩滤液至原体积的1/3,在氮气气氛下冷却结晶,得到4,4'-双[4-氨基苯基(2-芘基)胺]-4”-异丁氧基三苯胺2.38g,产率为84.6%。
实施例6
4,4'-双[4-氨基苯基(2-蒽基)胺]-4”-戊氧基三苯胺的制备,其结构式如下:
向装有机械搅拌装置和冷凝管的500ml三颈烧瓶中,加入16.00g(89.25mmol)的对戊氧基苯胺,49.73g(223.15mmol)的对氟硝基苯,33.89g(223.15mmol)氟化铯,加入129.5ml的二甲基亚砜,体系固含量为25%,在搅拌、氮气保护下,于150℃下反应12h;冷却至室温后出料于甲醇中并充分搅拌,过滤所得粗产物用n,n-二甲基乙酰胺和乙醇重结晶,干燥后得到30.51g橙色的4,4′-二硝基-4″-戊氧基三苯胺,产率为81.12%;
向装有磁力搅拌子和冷凝管的500ml三颈烧瓶中,加入25.00g(59.32mmol)4,4′-二硝基-4″-戊氧基三苯胺,加入280ml乙醇作为溶剂,使得体系固含量为10%,然后加入7.50g质量分数为10%的pd/c,加热至75℃回流后,向溶液中缓慢滴加74.24g(水合肼的摩尔量为1186.37mmol)质量分数为80%的水合肼溶液,继续回流搅拌10h,趁热过滤除去pd/c,滤液通氮气,冷却过程中逐渐析出白色晶体4,4′-二氨基-4″-戊氧基三苯胺18.05g,产率为84.2%。
向装有磁力搅拌子和冷凝管的250ml三颈烧瓶中,加入15.00g(41.49mmol)4,4′-二氨基-4″-戊氧基三苯胺,14.05g(99.58mmol)对氟硝基苯,13.64g(134.84mmol)三乙胺,加入71.4ml二甲基亚砜为溶剂,体系固含量为27%,在氮气保护下,于120℃反应60小时,将反应液冷却至室温后倒入搅拌条件下的甲醇/水溶液(甲醇与水的体积比为1:2)中,析出棕色粉末,粗产物用甲苯重结晶,得到9.74g4,4′-双[(4-硝基苯基)胺]-4″-戊氧基三苯胺,产率为38.9%。
向装有磁力搅拌子和冷凝管的250ml三颈烧瓶中,加入5.00g(8.28mmol)4,4′-双[(4-硝基苯基)胺]4″-戊氧基三苯胺,5.33g(20.71mmol)2-溴蒽,4.21g(66.24mmol)铜粉,21.58g(66.24mmol)碳酸铯,2.19g(8.28mmol)18-冠醚-6,加入19ml邻二氯苯,体系固含量为30%,在氮气保护下,于160℃下反应24h,反应结束后趁热过滤,向滤液中加入甲醇析出固体,所得粗产物用柱层析提纯(展开剂:二氯甲烷:石油醚=5:1),得到4.57g4,4'-双[4-硝基苯基(2-蒽基)胺]-4”-戊氧基三苯胺红色粉末,产率为57.7%。
向装有磁力搅拌子和冷凝管的250ml三颈烧瓶中,加入3.00g(3.14mmol)4,4'-双[4-硝基苯基(2-蒽基)胺]-4”-戊氧基三苯胺,加入26ml二氧六环作为溶剂,使得体系固含量为10%,然后加入0.9g质量分数为10%的pd/c,加热至100℃回流后,向溶液中缓慢滴加3.93g(水合肼的摩尔量为62.72mmol)质量分数为80%的水合肼溶液,滴加完成后,继续回流搅拌12h,趁热过滤除去pd/c,减压浓缩滤液至原体积的1/3,在氮气气氛下冷却结晶,即得到本发明所述的4,4'-双[4-氨基苯基(2-蒽基)胺]-4”-戊氧基三苯胺2.45g,产率为87.5%。
实施例7
4,4'-双[4-氨基苯基(6-甲氧基-2-萘基)胺]-4”-甲氧基三苯胺与1,4-环己烷二甲酸聚合制备1,4-环己烷二甲酸型聚酰胺,其结构如下:
将0.8000g(1mmol)4,4'-双[4-氨基苯基(6-甲氧基-2-萘基)胺]-4”-甲氧基三苯胺(由实施例1制备得到)、0.1722g(1mmol)1,4-环己烷二甲酸,0.1500g(1.35mmol)cacl2、1.0ml(3.82mmol)亚磷酸三苯酯、0.5ml(6.21mmol)吡啶和2.86mln-甲基吡咯烷酮混合,体系固含量为25%。在氮气气氛下,110℃反应4h;反应完成后,冷却至室温,出料至100ml乙醇中,然后过滤,得到浅绿色纤维状物;将所得浅绿纤维状物质依次用乙醇、水、乙醇回流洗料30min,然后在真空烘箱于90℃烘干,得到1,4-环己烷二甲酸型聚酰胺0.8006g,标记为7a。
性能测试
1)对实施例7制备的1,4-环己烷二甲酸型聚酰胺进行红外表征,结果见图3,图3为实施例7制备的1,4-环己烷二甲酸型聚酰胺7a的红外谱图,3317cm-1为n-h伸缩振动吸收峰,1679cm-1为c=o伸缩振动吸收峰,证明了1,4-环己烷二甲酸型聚酰胺被成功制备;
2)利用积分球对实施例7制备的1,4-环己烷二甲酸型聚酰胺进行荧光量子产率测定,结果见表1;由表1可知,1,4-环己烷二甲酸型聚酰胺在n-甲基吡咯烷酮稀溶液及薄膜态的荧光量子产率分别高达17.31%和9.07%,证明了双荧光团和具有助色团效应的三苯胺衍生物的引入使聚酰胺具有高荧光强度;
表1实施例7制备的1,4-环己烷二甲酸型聚酰胺的荧光量子产率
3)对实施例7制备的1,4-环己烷二甲酸型聚酰胺进行热性能测试,结果见图4,图4为实施例7制备的1,4-环己烷二甲酸型聚酰胺的dsc曲线图,由图可知,其玻璃化转变温度为237℃,说明其具有很好的热性能;
4)对实施例7制备的1,4-环己烷二甲酸型聚酰胺进行热重性能测试,图5为实施例7制备的1,4-环己烷二甲酸型聚酰胺的tga图;由图可知,在氮气气氛下,5%失重温度为378℃,10%失重温度为432℃,说明其具有良好的热稳定性;
5)对实施例7制备的1,4-环己烷二甲酸型聚酰胺进行循环伏安性能的测试,方法如下:将1,4-环己烷二甲酸型聚酰胺溶解于n-甲基吡咯烷酮中,使得溶液的浓度为3mg/ml,将其滴涂于ito玻璃板上,烘干后作为工作电极,铂丝作为对电极,ag/agcl作为参比电极,以0.1m四丁基高氯酸铵(tbap)的乙腈溶液作为电解质。基于该三电极体系,通过电化学工作站测试聚合物的循环伏安性能。在计算机上打开chi660e电化学工作站软件,进入设置-实验技术-选择cyclicvoltammetry;接着进行实验参数(起始电位,最高电位,扫描速度,扫描次数等)设置;连接三电极,即可测试循环伏安图并进行数据保存。图6为实施例7制备的1,4-环己烷二甲酸型聚酰胺的循环伏安曲线图;由图可知,所述聚酰胺有三个准可逆的氧化还原峰:0.58/0.32v,0.84/0.66v和1.39/1.12v,对应三个氮依次得失电子,起始氧化电位为0.3v,较低的氧化电位是由于高度共轭的七苯三胺结构可以提高分子的homo能级,降低起始氧化电位;
7)对实施例7制备的1,4-环己烷二甲酸型聚酰胺进行电化学稳定性测试,方法与循环伏安性能的测试方法类似,不同之处在于循环伏安性能的测试将实验中的扫描圈数设置为1圈,电化学稳定性测试实验参数中的扫描圈数根据预估所合成聚合物的性质设定,如本实验中第一重氧化还原稳定性测试设定扫描500圈,第二重氧化还原稳定性测试设定扫描300圈。第一重氧化还原稳定性测试如图7所示,经过500圈循环后曲线峰电流下降不明显,表明单阳离子自由基优异的电化学稳定性;第二重氧化还原稳定性测试如图8所示,经过300圈循环曲线有略微向内收缩的现象,表明双阳离子自由基稳定,证明了高度共轭的七苯三胺结构可以通过共振稳定单阳离子自由基和双阳离子。
8)对实施例7制备的1,4-环己烷二甲酸型聚酰胺进行电控荧光性能测试,测试方法如下:基于上述5)中的三电极体系(涂有聚合物薄膜的ito作为工作电极,铂丝作为对电极,ag/agcl作为参比电极),将电化学工作站和荧光光谱仪联用,将这三种电极放在装有电解质溶液(如0.1mtbap/ch3cn)的荧光池中。在电化学工作站amperometrici-tcurve模式下设置电压及持运行时间,程序结束后,用荧光光谱仪监测聚合物薄膜的荧光发射光谱。例如,先施加0v电压,用荧光光谱仪监测发射荧光的强度,以0.1v为间隔逐渐增大电压,荧光强度会发生明显的变化。当施加电压至荧光强度不发生明显变化,该测试结束,将电控荧光数据保存后作图。所述1,4-环己烷二甲酸型聚酰胺的电控荧光谱图如图9所示,当外加电压从0v逐渐升高时,479nm处的荧光强度逐渐降低,薄膜的绿色荧光淬灭;反向施加电压时,薄膜的绿色荧光恢复,说明该聚酰胺具有可逆的电控荧光行为;其中,当电压从0v升到0.6v时,单阳离子自由基产生,最大发射峰处荧光开关对比度高达225,当电压从0v升到0.9v时,生成双阳离子,最大发射峰处荧光开关对比度为369,聚合物在高电压下荧光开关对比度更高,这是因为高电压下材料的荧光淬灭更加完全。
9)对实施例7制备的1,4-环己烷二甲酸型聚酰胺进行电控荧光响应测试,所得电控荧光响应时间谱图如图10所示,持续时间20s,方波电压为0~0.9v时,荧光开/关时间为2.9s/1.4s,这种快速的响应速度一方面是由于体积较大的七苯三胺-双荧光团结构能有效减弱聚合物分子链的堆积作用,加快离子的传输;另一方面是因为共轭的七苯三胺结构可以加快分子内的电荷传输。
10)对实施例7制备的1,4-环己烷二甲酸型聚酰胺进行电控荧光转换性能测试,方法如下:基于上述三电极体系,将电化学工作站和荧光光谱仪联用。通过电化学工作站chronoamperometry模式设置参数(起始电压,最高电压,脉冲持续时间等)施加方波电压,同时用荧光光谱仪记录荧光强度随时间的变化。该测试可得到电控荧光开关时间和电控荧光稳定性数据;所得1,4-环己烷二甲酸型聚酰胺在第二重氧化还原态电控荧光稳定性谱图如图11所示,持续时间为20s,方波电压为0~0.9v时,经过300次转换后,荧光开关对比度几乎没有变化,说明三苯胺衍生物七苯三胺的引入使聚合物即使在高电压下仍具有优异的电控荧光循环稳定性。
实施例8
4,4'-双[4-氨基苯基(6-甲氧基-2-萘基)胺]-4”-甲氧基三苯胺与对苯二甲酸聚合制备对苯二甲酸型聚酰胺,其结构如下:
向装有磁力搅拌子和冷凝管的100ml三颈烧瓶中,加入实施例1制得的0.8000g(1mmol)4,4'-双[4-氨基苯基(6-甲氧基-2-萘基)胺]-4”-甲氧基三苯胺,0.1661g(1mmol)对苯二甲酸,0.1500g(1.35mmol)cacl2,加入1.0ml(3.82mmol)亚磷酸三苯酯,0.5ml(6.21mmol)吡啶,2.84mln-甲基吡咯烷酮,体系固含量为25%,在氮气气氛下,110℃反应3.5h。反应完毕后,冷却至室温,出料至乙醇中得到浅绿色纤维状产物,依次用乙醇、水、乙醇回流洗料30min,真空烘箱90℃烘干,得到对苯二甲酸型聚酰胺,质量为0.7922g,标记为8a。
实施例9
4,4'-双[4-氨基苯基(6-甲氧基-2-萘基)胺]-4”-甲氧基三苯胺与4,4'-联苯二甲酸聚合制备4,4'-联苯二甲酸型聚酰胺,其结构如下:
向装有磁力搅拌子和冷凝管的100ml三颈烧瓶中,加入实施例1制得的0.8000g(1mmol)4,4'-双[4-氨基苯基(6-甲氧基-2-萘基)胺]-4”-甲氧基三苯胺,0.2422g(1mmol)4,4'-联苯二甲酸,0.1500g(1.35mmol)cacl2,加入1.0ml(3.82mmol)亚磷酸三苯酯,0.5ml(6.21mmol)吡啶,3.07mln-甲基吡咯烷酮,体系固含量为25%,在氮气气氛下,110℃反应3.5h。反应完毕后,冷却至室温,出料至乙醇中得到浅绿色纤维状产物,依次用乙醇、水、乙醇回流洗料30min,真空烘箱90℃烘干,得到4,4'-联苯二甲酸型聚酰胺,质量为0.8598g,标记为9a。
实施例10
4,4'-双[4-氨基苯基(6-甲氧基-2-萘基)胺]-4”-甲氧基三苯胺与均苯四甲酸二酐聚合制备均苯四甲酸二酐型聚酰亚胺,其结构如下:
向装有磁力搅拌子和冷凝管的100ml三颈烧瓶中,加入实施例1制得的0.8000g(1mmol)4,4'-双[4-氨基苯基(6-甲氧基-2-萘基)胺]-4”-甲氧基三苯胺,3.26ml的n,n-二甲基乙酰胺作为溶剂,分多次加入0.2181g(1mmol)的1,2,4,5-均苯四甲酸二酐,体系固含量为25%。室温下反应8h后,加入0.28ml(2.96mmol)乙酸酐和0.4ml(5.00mmol)吡啶。氮气气氛下,110℃反应5h,出料于乙醇,得到的聚合物用乙醇加热至回流洗涤,真空烘箱90℃烘干,得到均苯四甲酸二酐型的聚酰亚胺0.8401g。标记为10a。
实施例11
4,4'-双[4-氨基苯基(6-甲氧基-2-萘基)胺]-4”-甲氧基三苯胺与氢化均苯四甲酸二酐聚合制备氢化均苯四甲酸二酐型聚酰亚胺,其结构如下:
向装有磁力搅拌子和冷凝管的100ml三颈烧瓶中,加入实施例1制得的0.8000g(1mmol)4,4'-双[4-氨基苯基(6-甲氧基-2-萘基)胺]-4”-甲氧基三苯胺,3.3ml的n,n-二甲基乙酰胺作为溶剂,分多次加入0.2242g(1mmol)的氢化均苯四甲酸二酐,固含量为25%。室温下反应8h后,加入0.28ml(2.96mmol)乙酸酐和0.4ml(5.00mmol)吡啶。氮气气氛下,110℃反应5h,出料于乙醇,得到的聚合物用乙醇加热至回流洗涤,真空烘箱90℃烘干,得到氢化均苯四甲酸二酐型聚酰亚胺0.8340g。标记为11a。
实施例12
4,4'-双[4-氨基苯基(6-甲氧基-2-萘基)胺]-4”-甲氧基三苯胺与3,3',4,4'-联苯四甲酸二酐聚合制备3,3',4,4'-联苯四甲酸二酐型聚酰亚胺,其结构如下:
向装有磁力搅拌子和冷凝管的100ml三颈烧瓶中,加入实施例1制得的0.8000g(1mmol)4,4'-双[4-氨基苯基(6-甲氧基-2-萘基)胺]-4”-甲氧基三苯胺,3.53ml的n,n-二甲基乙酰胺作为溶剂,分多次加入0.2942g(1mmol)的3,3',4,4'--联苯四甲酸二酐,固含量为25%。室温下反应8h后,加入0.28ml(2.96mmol)乙酸酐和0.4ml(5.00mmol)吡啶;氮气气氛下,110℃反应5h,出料于乙醇,得到的聚合物用乙醇加热至回流洗涤,真空烘箱90℃烘干,得到3,3',4,4'-联苯四甲酸二酐型聚酰亚胺0.9119g,记为12a。
性能测试
1)按照实施例7所述性能测试方法,对实施例8~12制备的聚酰胺或聚酰亚胺进行荧光相关性能测试,结果发现,实施例8~12制备的聚酰胺或聚酰亚胺具有与实施例7制备的7a相似的荧光性能。
2)对实施例7~12制备得到的聚酰胺或聚酰亚胺在n-甲基吡咯烷酮(nmp)、二甲基乙酰胺(dmac)、二甲基甲酰胺(dmf)、二甲基亚砜(dmso)、四氢呋喃(thf)和氯仿(chcl3)中的溶解性进行测试,其测试结果如表2所示:
表2实施例7~12制备的聚酰胺或聚酰亚胺在6种溶剂中的溶解性
注:用于测定溶解性的溶液浓度为10mg/ml;++:室温下可溶;+:加热可溶;+-:加热下部分可溶;-:加热不溶。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种含正十二烷基鼠李糖苷与硫代苯甲酰胺的农药组合物及应用的制造方法与工艺
- 一种啶氧菌酯的制备方法与流程
- 一种含有肟菌酯和榄香素的高效杀菌组合物的制造方法与工艺
- 酪氨酸激酶抑制剂的制造方法与工艺
- 杀虫、杀螨、杀线虫、杀软体动物、杀菌或杀细菌组合物及病害虫的防除方法与制造工艺
- 2-氯-3-(甲基硫烷基)-N-(1-甲基-1H-四唑-5-基)-4-(三氟甲基)苯甲酰胺或其盐在耐受HPPD抑制剂除草剂的转基因作物植物的区域中防治不想要的植物的用途的制造方法与工艺
- 手性8‑羟基升补身烷倍半萜酰胺类化合物及作为农用杀菌剂的用途的制造方法与工艺
- N‑甲基‑4‑羟基苯甲酰胺的制备方法与制造工艺
- 一种邻乙氧基苯甲酰胺药物中间体邻乙氧基苯甲酸乙酯的合成方法与制造工艺
- 一种含噻虫胺﹒毒死蜱﹒氯虫苯甲酰胺的杀虫组合物及制备方法与制造工艺