一组肺癌DNA甲基化分子标志物及其在制备用于肺癌早期诊断试剂盒中的应用
本发明涉及生物检测技术领域,具体涉及一组肺癌的dna甲基化分子标志物及其在制备用于肺癌诊断试剂盒中的应用。
背景技术:
肺癌是威胁人群健康和生命的最大的恶性肿瘤之一,2020年全球约有180万人死于肺癌,死亡率远超其他类型癌症,位居第一;也是我国发病率和死亡率最高的肿瘤。肺癌主要分成两类:非小细胞肺癌(nsclc)和小细胞肺癌。nsclc是最主要类型,占总肺癌80%以上。nsclc主要包括两个亚型即腺癌和鳞状细胞癌。腺癌占所有肺癌类型的40%,约占nsclc的55%左右。非小细胞肺癌增殖、侵袭速度较慢,因此更不容易被发现和诊断。大部分非小细胞肺癌患者被确诊时,都已经发展为肿瘤的中晚期,错过绝佳的治疗时间。在我国,肺癌的早期诊断率只有15%,但是这些肺癌病人5年生存率可达到50%~60%,显著高于平均水平15%。因此,在肺癌发生早期进行鉴别诊断,并开展针对性治疗是解决肺癌这一重大难题的重要途径。
生物标志物是指能将机体的生理和疾病状态区分开来的生物分子,筛选到可用于疾病早期发现、早期诊断的生物标志物可大大提高患者的临床治疗效果。近年来,随着研究的深入,研究者发现dna序列变异之外的调控机制,例如dna甲基化调控机制,在肿瘤发生发展过程中扮演重要角色。dna甲基化是一种重要的表观遗传学修饰,同时也是一种被广泛研究的表观遗传学标记;其作为标记物非常稳定,可通过荧光定量pcr进行非常灵敏的检测,因而有望成为继血清蛋白后新一代的分子标志物。
本研究团队通过对18对非小细胞肺癌样本(肿瘤和匹配的癌旁组织)进行全基因组简化甲基化测序(rrbs)和转录组测序(rna-seq),对两个组学的数据进行整合分析,构建了肺癌dna差异甲基化区域图谱,挖掘异常甲基化基因,并进一步通过机器学习方法筛选出了肺癌诊断的20个dna甲基化的基因指纹(genesignature)。利用这20个基因的dna甲基化指纹能非常有效区分公共数据集tcga肺腺癌和肺鳞癌中的肿瘤和正常组织,这些标志物将有望成为肺癌诊断筛查的重要手段。
技术实现要素:
本发明的目的在于通过高通量甲基化测序数据和rna-seq测序数据的整合分析,进一步结合机器学习方法,提供一组肺癌dna甲基化分子标志物及其在制备用于肺癌早期诊断试剂盒中的应用。
本发明的目的是通过以下技术方案来实现的,一组肺癌dna甲基化分子标志物,所述标志物包括如下20个基因至少一个的甲基化:cdo1,sox17,tcf21,trim58,itga9,cyyr1,clec14a,slit2,znf677,irx2,acvrl1,osr1,adcy8,galnt13,hspb6,irx1,itga5,pcdh17,tbx5和tctex1d1。
进一步地,所示分子标志物通过以下方法获取:
(1)rrbs和rna-seq测序文库构建及测序:首先分别分离出每个病人肺肿瘤组织及其相邻正常肺组织样品中的总dna,用mspi对基因组进行酶切;对产生的dna片段进行末端修复;在末端修复后的3’末端添加碱基a;将dna片段粘性末端a上连接甲基化接头。然后,选择40-220bp大小的dna片段;将选择的片段进行重亚硫酸盐处理,使dna片段中非甲基化的胞嘧啶转化为胸腺嘧啶,经pcr扩增后转变为尿嘧啶;将经过转换的目的片段进行pcr扩增;分离纯化扩增产物,即得到rrbs测序文库。利用标准illumina测序试剂进行rna-seq文库构建。测序文库质检合格之后上机进行双端测序,得到测序原始数据。
(2)测序数据分析:对步骤(1)得到的测序原始数据进行质检。将经过质检的rrbs读段和rna-seq读段分别用bismark和hisat2工具比对到人类参考基因组上,获得全基因组上的甲基化图谱和表达水平信息。
(3)检测差异甲基化区域(dmr)和差异表达基因(deg):对步骤(2)得到的甲基化数据进行聚类分析,然后使用metilene,利用二元分割算法,从肿瘤组织和正常组织中筛选鉴别出差异甲基化区域(dmrs),最后,采用wilcoxon秩和检验方法对候选的dmrs进行统计检验。对于转录本数据,用deseq、edger等r程序包鉴定肿瘤组织和正常组织之间的差异表达基因(deg)。
(4)挖掘肺癌候选甲基化驱动基因:首先将dmrs数据根据基因组信息注释到基因的功能区间,进行注释分类;然后提取启动子区域(tss上游或者下游2kb区域范围)的dmr和相应转录起始位点对应deg的表达数据进行相关性分析,选择负相关关系统计显著的基因作为异常甲基化驱动候选基因。
(5)机器学习筛选肺癌诊断的dna甲基化基因指纹:从tcga公共数据库中下载肺腺癌(luad)和肺鳞癌(lusc)患者的甲基化和表达谱数据,利用这两个数据集对步骤(4)筛选出的甲基化驱动基因的甲基化和表达之间的关联性进行独立验证,进一步筛选出可靠的甲基化驱动基因。
本发明还提供了一种上述dna甲基化分子标志物在制备用于肺癌早期诊断试剂盒中的应用。
进一步地,以筛选出的20个基因的dna甲基化指纹为基础,采用随机森林(rf)方法构建肺癌诊断的数学模型,使用roc曲线及曲线下面积(auc)来评价筛选效果。
本发明的有益效果是:以本发明标记物为基础,构建肺癌诊断的数学模型;该模型灵敏度高,特异性好,auc可高达0.998,诊断效果良好。综上所述,本发明公开的dna甲基化分子标志物具有良好的诊断指标特性,可以有效用于肺癌诊断,具有较高的临床应用和推广价值。
附图说明
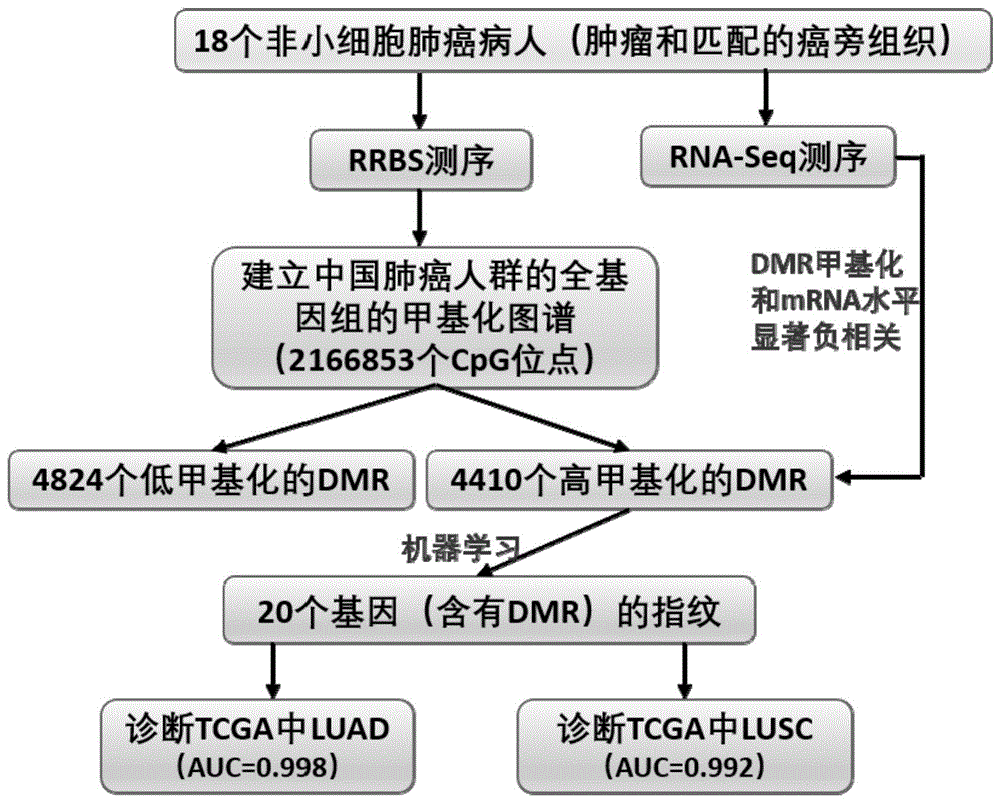
图1是本发明的一个实施例流程图。
图2是肺癌组织与癌旁组织的甲基化图谱特征(a)肺癌组织和癌旁组织中全基因组cpg位点聚类分析图。(b)肺癌组织和癌旁组织中全基因组cpg位点主成分分析图。
图3是20个基因dna甲基化指纹的甲基化和表达图谱特征及其在鉴别tcga中肺肿瘤组织和癌旁正常组织中的表现。(a)18对非小细胞肺癌及其癌旁组织的甲基化和mrna表达谱;(b)tcga肺腺癌中roc分析;(c)tcga肺鳞癌中roc分析。20基因的甲基化指纹能准确区分肿瘤和正常组织。
具体实施方式
下面通过具体实施例子对本发明作进一步阐述,应该说明的是,下述说明仅是为了解释本发明,并不对其内容进行限定。
如图1所示,本发明分子标志物的获取方法如下:
1、测序文库构建及高通量测序:收集18个i期非小细胞肺癌样本,分别分离出每个病人肺肿瘤组织及其相邻正常肺组织样品中的总dna,用mspi对基因组进行酶切;然后对产生的dna片段进行末端修复;在末端修复后的3’末端添加碱基a;将dna片段粘性末端a上连接甲基化接头;然后选择40-220bp大小的dna片段;将选择的片段进行重亚硫酸盐处理,这样可以将dna片段中非甲基化的胞嘧啶转化为胸腺嘧啶,经pcr扩增后转变为尿嘧啶;将经过转换的目的片段进行pcr扩增;分离纯化扩增产物,即得到rrbs测序文库。利用标准illumina测序试剂进行rna-seq文库构建。测序文库质检合格之后上机进行高通量双端测序,通过检测荧光信号来确定测得的碱基及顺序,得到fastq文件。fastq文件包含了读段的全部信息,包括测序仪器的基本信息、碱基排列,以及读段中碱基的质量得分等。
2、测序数据分析:针对测序的质量控制,将使用fastqc等软件提供的测试方法来评估读段质量。我们采用专门处理rrbs数据的整合软件trim_galore,去除rrbs测序数据中低质量的读段或对读段进行剪裁。完成质量控制后,我们使用bismark,把每个片段序列转换处理后比对到参考基因组(hg19),得到每个样本全基因组单个cpg位点的甲基化水平。我们只保留测序深度大于5并且至少在10对样本中都存在的cpg位点,这样共得到2574098个cpg位点,然后去除与snp重合的位点(snp142)和x,y染色体上的位点后,结果剩下2166853个cpg位点。利用k-nearest算法推断cpg位点的甲基化缺失值。然后,对36个样本的cpg位点进行样本聚类分析和主成分分析。从构建的系统树图看,这些cpg位点可以把非小细胞肺癌和正常组织分成不同两类(图2a)。主成分分析也观察到非小细胞肺癌和正常组织之间明显地分离(图2b),这些结果表明非小细胞肺癌和正常组织间有不同的甲基化模式。另外,与正常组织比较,非小细胞肺癌样本之间有较大的变异(图2b),显示不同肿瘤样本之间存在异质性的甲基化模式。对于转录本数据,我们使用hisat2工具比对到人类参考基因组上,并使用stringtie把联配后序列组装构建一个精简的转录本集合;然后进一步量化转录本的表达水平。
3、差异甲基化区域(dmr)和差异表达基因(deg)鉴定:我们使用metilene软件鉴别肿瘤组织中候选的差异甲基化区域dmr。metilene整合了二元分割算法,能够快速,高效在多个样本中鉴别出差异甲基化区域。然后,我们采用配对的wilcoxon秩和检验对这些候选的dmr进行统计检验,运用benjamini-hochberg方法控制多重检验的假阳性(fdr,falsediscoveryrate),选择甲基化差异大于0.1且fdr<0.05的区域作为进一步分析的dmr。最终,我们在全基因组水平共鉴定9234个dmrs,其中非小细胞肺癌中高甲基化dmrs4410个(包含97594个cpg位点),低甲基化dmrs4824个(包含65009个cpg位点);与低甲基化dmrs相比,高甲基化dmrs的长度较短,但包括更多的cpg位点。对于转录本数据,我们使用deseq、edger等r程序包鉴定肿瘤组织和正常组织之间的差异表达基因(deg)。
4、挖掘肺癌候选甲基化驱动基因:首先将dmrs数据根据基因组信息注释到基因的功能区间,进行注释分类;然后提取启动子区域(tss上游或者下游2kb区域范围)的dmr和相应转录起始位点对应deg的表达数据进行相关性分析,选择负相关关系统计显著的基因作为异常甲基化驱动候选基因。该过程共鉴定出190个基因的表达与其启动子区域dmrs呈显著负相关,其中106个高甲基化基因的mrna表达下降。对上述筛选出来的基因集进行go生物过程富集分析,明确候选基因相关的生物学功能,细胞组成,生物学过程;以及利用kegg数据库进行信号通路富集分析,确定相关基因参与的最主要生化代谢途径和信号转导途径。
5、机器学习筛选肺癌诊断的dna甲基化基因指纹:我们从tcga数据库中下载肺腺癌(luad)和肺鳞癌(lusc)患者的甲基化和表达谱数据,验证鉴定的190个候选甲基化驱动基因的甲基化和表达之间是否具有关联性,进一步筛选出可靠的甲基化驱动基因。我们从tcga中提取了其中133个基因的甲基化和表达谱数据(另外57个基因没有infinium450k芯片探针覆盖),分析了他们之间的相关性,结果发现其中81(60.9%)个基因的甲基化和表达间也有显著负相关,显示较好的一致性。这其中,31个基因是同时在luad和lusc数据集中都有的,包括20个高甲基化低表达基因和11个低甲基化高表达基因。由于高甲基化抑制基因表达是经典的调控范式,我们选取了20个高甲基化低表达的基因作为最终的肺癌甲基化诊断指纹(表1,图3a)。筛选差异甲基化基因后,我使用roc曲线及曲线下面积(auc)来评价筛选效果。针对tcga中的luad(tumor:460;normal:32)和lusc(tumor:371;normal:41)两个数据集,我们分别各自将它们随机分成等量的训练组(50%)和检验组(50%),采用随机森林(rf)方法构建肺癌诊断的数学模型;结果显示这20个基因的dna甲基化指纹能非常有效区分tcga中luad(图3b,auc为0.998)和lusc(图3c,auc为0.992)与正常组织。其中12个基因已在肺癌中报道,另外8个是肺癌中新的异常甲基化驱动基因,包括adcy8,galnt13,hspb6,irx1,itga5,pcdh17,tbx5和tctex1d1;我们采用焦磷酸测序的方法在另外独立的23对非小细胞肺癌与癌旁组织样本中验证了它们甲基化水平的差异,且这8个新鉴定的基因的甲基化指纹能有效区分该独立数据集的癌和癌旁,auc值高达0.965。这些结果表明我们提供的这组dna甲基化分子标志物是一种非常有潜力的肺肿瘤诊断标志物。
表1dna甲基化指纹中的20个基因的dmr信息
本发明筛选的分子标志物在制备用于早期肺癌诊断的试剂盒中的应用,以筛选出的20个基因的dna甲基化指纹为基础,利用焦磷酸甲基化测序或者捕获二代测序分析肿瘤组织样本、气管镜活检样本或血液样本,采用随机森林(rf)方法构建肺癌诊断的数学模型,使用roc曲线及曲线下面积(auc)来评价筛选效果;该模型灵敏度高,特异性好,auc可高达0.998,诊断效果良好。
上述实施例用来解释说明本发明,而不是对本发明进行限制,在本发明的精神和权利要求的保护范围内,对本发明作出的任何修改和改变,都落入本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!