一种2-甲基-5-溴嘧啶的制备方法与流程
一种2
‑
甲基
‑5‑
溴嘧啶的制备方法
技术领域
1.本发明涉及一种2
‑
甲基
‑5‑
溴嘧啶的制备方法,属于有机合成技术领域。
背景技术:
[0002]2‑
甲基
‑5‑
溴嘧啶,cas:7752
‑
78
‑
5,英文名:5
‑
bromo
‑2‑
methyl pyri midine,嘧啶类化合物在生命活动中是一类很重要的物质,它作为新药分子设计和合成的基本砌块早已经引起人们的关注。
[0003]5‑
溴嘧啶的衍生物因具有显著的化学治疗、生物化学等活性,作为药物中间体已合成出大量具有生物活性的核苷类药而在制药工业和基因工程方面具有广阔的发展前景。其中wo2018/229543,2018,a2报道了5
‑
溴
‑2‑
甲基嘧啶作为药物中间体合成一种补体因子d抑制剂,可治疗补体相关疾病,如自身免疫性疾病,炎性疾病和神经退行性疾病。2
‑
甲基
‑5‑
溴嘧啶广泛应用于stille等偶联反应中,从而得到新的具有生理活性的药物。
[0004]
然而关于2
‑
甲基
‑5‑
溴嘧啶,文献中有二甲基锌或者三甲基铝与5
‑
溴
‑2‑
碘嘧啶反应,由于二甲基锌或者三甲基铝属于易制爆类化合物,在空气中极其容易自燃,并且收率较低(36%左右),该工艺不适合规模生产,其中二甲基锌与5
‑
溴
‑2‑
碘嘧啶反应(wo 2014/75392,2014,a1)化学反应式为:
[0005]
三甲基铝与5
‑
溴
‑2‑
碘嘧啶反应(wo2014/98831,2014,a1)化学反应式为:
[0006]
专利wo2011/130628,2011,a1报道2
‑
甲基
‑5‑
溴嘧啶
‑4‑
羧酸通过脱羧合成5
‑
溴
‑2‑
甲基嘧啶,收率61%。由于原材料2
‑
甲基
‑5‑
溴嘧啶
‑4‑
羧酸较为昂贵,原料不易得,不利于工业化生产。
[0007]
上述方法均是原料昂贵,或者原料易燃易爆,收率低等确定,因此有必要针对这一技术问题进行解决,来降低其生产成本,以满足日益增长的市场需求。
技术实现要素:
[0008]
为了克服上述技术缺陷,本发明公开了一种2
‑
甲基
‑5‑
溴嘧啶的制备方法。该方法包括:a)以2
‑
氨基
‑5‑
溴嘧啶为原料,经重氮化后,桑德迈尔反应或席曼反应;以2
‑
羟基
‑5‑
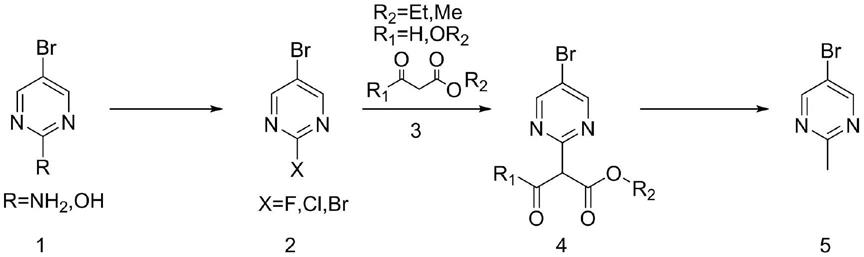
溴嘧啶为原料,经过卤代试剂反应;得到化合物2;b)化合物3在亲核试剂条件下与化合物2发生取代反应,以得到化合物3;c)最后化合物4在碱性、高温条件下反应得到式5化合物。该方法原料易得,成本低廉,流程简便,具备潜在的工业化放大前景。
[0009]
本发明所述一种2
‑
甲基
‑5‑
溴嘧啶的制备方法,包括如下步骤:包括如下步骤:
[0010]
a):将2
‑
氨基
‑5‑
溴嘧啶与酸混合,加入亚硝酸钠水溶液,随后与氟硼酸反应得到氟硼酸重氮盐,加热下分解,得到2
‑
氟
‑5‑
溴嘧啶;将2
‑
氨基
‑5‑
溴嘧啶与酸混合,依次加入溴素和亚硝酸钠水溶液反应得到2,5
‑
二溴嘧啶;将2
‑
羟基
‑5‑
溴嘧啶与三氯氧磷溶于有机溶剂中,在三乙胺存在下反应得到2
‑
氯
‑5‑
溴嘧啶;
[0011]
b):将化合物3溶于有机溶剂中,加入碱溶液和化合物2反应得到化合物4,最后加入盐酸水解或加入碳酸钾/dmso反应得到2
‑
甲基
‑5‑
溴嘧啶。反应方程式表示为:
[0012][0013]
进一步地,在上述技术方案中,步骤a)所述酸为硫酸、氢溴酸,重氮盐反应温度在
‑
4℃~4℃。
[0014]
进一步地,在上述技术方案中,步骤a)所述加热下分解温度为120
‑
125℃。
[0015]
进一步地,在上述技术方案中,步骤a)所述2
‑
氯
‑5‑
溴嘧啶制备中,有机溶剂选自甲苯或二甲苯,反应温度为80
‑
85℃。
[0016]
进一步地,在上述技术方案中,步骤b)所述有机溶剂选四氢呋喃或2
‑
甲基四氢呋喃。
[0017]
进一步地,在上述技术方案中,步骤b)所述碱溶液选自20%叔丁醇钾/四氢呋喃溶液或40%叔戊醇钠/甲苯溶液。
[0018]
进一步地,在上述技术方案中,步骤b)所述化合物2、碱和化合物3摩尔比为1:1.1
‑
1.15:1.1
‑
1.15。
[0019]
进一步地,在上述技术方案中,步骤b)所述反应温度在0~80℃。
[0020]
进一步地,在上述技术方案中,步骤b)化合物4采用盐酸水解脱羧时,反应温度100
‑
105℃。
[0021]
进一步地,在上述技术方案中,步骤b)化合物4采用碳酸钾/dmso时,反应温度110
‑
120℃。
[0022]
进一步地,在上述技术方案中,步骤b)所述2
‑
甲基
‑5‑
溴嘧啶提纯方法乙醇重结晶或者乙酸乙酯重结晶。
[0023]
发明有益效果
[0024]
与文献报道的合成方法相比,本发明具有如下有益效果:本发明采用不释怀原料不同合成路线得到产品,原料易得,避免使用易燃易爆的试剂或昂贵的原料,降低成本,方便了工业化放大生产。
[0025]
其中2
‑
氟
‑5‑
溴嘧啶采用乙腈为溶剂,重氮化过程减少废水的生成。
具体实施方式
[0026]
下面通过具体实例对本发明进行进一步说明。这些实施例应理解为仅用于说明本发明而不用于限制本发明的保护范围。在阅读了本发明记载的内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等效变化和修改同样落入本发明权利要求所限定的范围。
[0027]
实施例1
[0028]
氮气保护下,向反应瓶内投入34.8g(0.2mol)2
‑
氨基
‑
5溴嘧啶和105ml乙腈搅拌溶清,降温并控制
‑
4℃~4℃,滴加60%硫酸100ml,控制温度
‑
4℃~4℃滴加50%亚硝酸钠水溶液30.4g(0.22mol),滴加完毕后,该温度下反应3小时,控制温度
‑
4℃~4℃加入48%氟硼酸水溶液40.2g(0.22mol),滴加完毕后反应4小时,tlc检测几乎无原料剩余,此时有固体析出,过滤,滤饼用冷水淋洗。
[0029][0030]
将滤饼投入另一反应瓶内,加入200ml二甲苯,升温至120℃反应8小时,tlc检测无氟硼酸重氮盐,加入5%碳酸氢钠水溶液调节ph=7.5
‑
8.0,分层,有机相浓缩至不流液,得到2
‑
氟
‑5‑
溴嘧啶29.45g,收率83.2%,hplc:97.8%。1hnmr(400mhz,cdcl3):9.19(s,2h).
[0031]
实施例2
[0032][0033]
氮气保护下,向反应瓶内投入34.8g(0.2mol)2
‑
氨基
‑
5溴嘧啶和48%hbr水溶液180ml,控制0
‑
4℃加入溴素48.0g(0.3mol),形成黄色的悬浮液。控制0
‑
4℃逐滴加入50%亚硝酸钠的水溶液55.2g(0.4mol)。添加后,将混合物0
‑
4℃搅拌3h,hplc检测原料小于3%,然后倒入冰100ml中。用30%氢氧化钠水溶液中和并调节ph=10
‑
11,并用二氯甲烷萃取,有机相浓缩至不流液,然后用100ml乙酸乙酯:石油醚=1:4重结晶,得到白色固体2,5
‑
二溴嘧啶34.83g,收率73.2%,hplc:99.5%。1hnmr(400mhz,cdcl3):8.66(s,2h).
[0034]
实施例3
[0035][0036]
氮气保护下,向反应瓶内投入2
‑
羟基
‑5‑
溴嘧啶35g(0.2mol)、三氯氧磷61.3g(0.4mol)和甲苯200ml投入到反应釜内。升温至35℃,滴加三乙胺40.5g(0.4mol),滴加结束后升温80~85℃下搅拌反应6小时,取样hplc检测原料小于2%,降温,减压浓缩除去大部分甲苯和三氯氧磷,将反应液投入到10℃水中淬灭,用20%碳酸钠水溶液调节ph=8~9,加入
二氯甲烷萃取,减压浓缩至不流液,得到2
‑
氯
‑5‑
溴嘧啶33.8g,收率87.4%,hplc:95.9%。1hnmr(400mhz,cdcl3):8.74(s,2h).
[0037]
实施例4
[0038][0039]
氮气保护下,向反应瓶内投入乙酰基乙酸乙酯13g(0.1mol)和四氢呋喃100ml混合,降温至
‑
5℃,并控制温度在
‑
5~5℃下滴加20%叔丁醇钾四氢呋喃溶液61.7g(0.11mol),滴毕后0℃反应0.5小时,随后滴加含2
‑
氟
‑5‑
溴嘧啶17.7g(0.1mol)的四氢呋喃30ml的混合溶液,滴毕后缓慢升温至室温,反应2小时,取样hplc检测原料小于0.5%。
[0040]
加入40%醋酸水溶液淬灭,分层,有机相浓缩至不流液,加入碳酸钾55.3g(0.4mol)和100ml二甲基亚砜,升温至110℃,反应4小时,取样hplc检测中间体剩余3%,降温至室温,加入100ml水,分层,水相甲基叔丁基醚萃取,合并有机相,碳酸氢钠水洗三次,有机相减压浓缩除掉甲基叔丁基醚,随后加入乙醇重结晶,得到2
‑
甲基
‑5‑
溴嘧啶14.12g,hplc:99.6%,收率81.6%。1hnmr(400mhz,cdcl3):8.79(s,2h),2.78(s,3h).
[0041]
实施例5
[0042][0043]
氮气保护下,向反应瓶内投入丙二酸二乙酯16g(0.1mol)和四氢呋喃100ml混合,降温至
‑
5℃,并控制温度在
‑
5~5℃下滴加20%叔丁醇钾/四氢呋喃溶液61.7g(0.11mol),滴毕后0℃反应0.5小时,随后滴加含2,5
‑
二溴嘧啶23.8g(0.1mol)/四氢呋喃30ml混合溶液,滴毕后缓慢升温至室温,反应1小时,取样hplc检测原料小于1.5%。
[0044]
加入40%醋酸水溶液淬灭,分层,有机相浓缩至不流液,加入30%盐酸300ml混合,升温至105℃回流,反应12小时,hplc检测中间体剩余1.5%,降温至室温,加入20%碳酸钠调节ph=8.0
‑
9.0,加入乙酸乙酯萃取,分层,水相用乙酸乙酯萃取,合并有机相减压浓缩剩余50ml,重结晶得到2
‑
甲基
‑5‑
溴嘧啶12.4g,hplc:99.3%,收率:71.5%。
[0045]
实施例6
[0046][0047]
氮气保护下,向反应瓶内投入乙酰基乙酸甲酯11.6g(0.1mol)和2
‑
甲基四氢呋喃
100ml混合,降温至
‑
5℃,并控制温度在
‑
5~5℃下滴加40%叔戊醇钾/甲苯溶液30.3g(0.11mol),滴毕后0℃反应0.5小时,随后滴加含2
‑
氯
‑5‑
溴嘧啶19.3g(0.1mol)/2
‑
甲基四氢呋喃30ml混合溶液,滴毕后缓慢升温至室温,反应2小时,取样hplc检测原料小于0.5%。
[0048]
加入40%醋酸水溶液淬灭,分层,有机相浓缩至不流液,加入碳酸钾69.1g(0.5mol)和130ml二甲基亚砜,升温至110℃,反应3小时,取样hplc检测中间体剩余2%,降温至室温,加入100ml水,分层,水相甲基叔丁基醚萃取,合并有机相,用8%碳酸氢钠水洗三次,有机相减压浓缩除掉甲基叔丁基醚,随后加入乙醇重结晶,得到2
‑
甲基
‑5‑
溴嘧啶14.81,hplc:99.7%,收率:85.6%。
[0049]
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明披露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1