二代测序平台中利用分子特异性条形码评估STR滑脱的方法及其应用
二代测序平台中利用分子特异性条形码评估str滑脱的方法及其应用
技术领域
1.本技术涉及二代测序领域,尤其涉及二代测序中利用分子特异性条形码评估str的滑脱的方法及其应用。
背景技术:
2.短串联重复序列(short tandem repeat,str)是以2~6个核苷酸为重复单位,在人类基因组中呈串联重复排列的序列。str基因位点长度一般在100~300bp之间,个体间str核心序列拷贝数目和序列的变化而呈高度多态性。
3.目前,str分型已广泛应用于法医学个体识别和亲子鉴定等领域。
4.经典的str分型原理是利用str两翼区域的保守序列设计位点专一的特异引物,在pcr仪器上进行扩增,最后利用电泳检测扩增产物。基于毛细管电泳平台的str分型技术准确度最高,已经普遍用于人类法医学鉴定,是当前个体识别的金标准。
5.然而在毛细管电泳的str分型图谱中,一个目标等位基因峰(主带)前小一个重复单位的位置往往出现一个信号较弱的影子峰(stutter)或叫影子带,它是由于滑动复制引起的,也可能比主带多一个重复单位。一般影子带的峰面积与主带的峰面积比例小于15%,重复单位越长越容易引起滑脱,影子带峰面积随着滑脱重复单位的增加而变小(主带》主带-1个重复单位》主带-2个重复单位),影子带的存在对混合样本的分型带来了极大的困难。而影子带的形成与pcr扩增过程中str的滑脱密切相关。
6.近年来二代测序(next generation sequencing,ngs)技术发展迅速,具有通量高、集成化、成本低等优势,已在科研和临床诊断等领域取得广泛应用,在法医及农业领域亦具有重要的应用前景。
技术实现要素:
7.本技术提供了一种二代测序平台中利用分子特异性条形码评估str的滑脱的方法及其应用,以解决二代测序平台中如何str滑脱的技术问题。
8.第一方面,本技术提供了一种二代测序中利用分子特异性条形码评估str的滑脱的方法,所述方法包括以下步骤:
9.根据str基因座得到捕获探针;
10.将待测样本的原始dna模板分子分别连接umi接头序列和样品特异性标识码,得到待捕获dna片段,所述待捕获dna片段含有umi接头序列和样品特异性标识码;
11.用所述捕获探针对所述待捕获dna片段进行str捕获,得到目标捕获片段;
12.对所述目标捕获片段进行高通量测序,得到测序数据;
13.根据所述umi接头序列、所述样品特异性标识码和所述待测样本的参考基因组对所述测序数据进行str分型,计算各str基因座的滑脱率;
14.根据所述滑脱率,评估二代测序平台中str的滑脱。
15.可选的,根据所述umi接头序列、所述样品特异性标识码和所述待测样本的参考基因组对所述测序数据进行str分型,计算各str基因座的滑脱率包括:
16.去除所述测序数据中不符合预设标准的序列,并根据所述样品特异的标识码去除污染的测序序列,得到待比对序列;
17.将所述待比对序列与所述待测样本的参考基因组进行比对,得到原始提取序列;
18.在所述原始提取序列中,根据所述umi接头序列,从所述待比对序列中,确定各umi家族的成员及其序列;
19.根据每个所述umi家族的成员及其序列,还原得到每个所述umi家族的原始dna模板序列;
20.对含同一个所述umi接头序列的所述umi家族的所有成员进行str分型,计算每个umi家族的滑脱百分比。
21.可选的,所述对含同一个所述umi接头序列的所述umi家族的所有成员进行str分型,包括:对含同一个所述umi接头序列的所述umi家族的所有成员按照长度多态性进行str分型。
22.可选的,所述不符合预设标准的序列包括:
23.单端读序3端含有预设质量碱基数目超过自身序列1/3碱基数目的序列,所述预设质量碱基为质量值≤20的碱基;
24.序列长度小于100bp的序列。
25.可选的,所述将所述待比对序列与所述待测样本的参考基因组进行比对,得到原始提取序列包括:
26.将所述待比对序列与参考基因组进行比对,保留含str保守基序的序列和含umi接头序列的序列并同时提取str保守基序的序列和umi接头序列,得到原始提取序列。
27.可选的,所述方法还包括:
28.根据所述umi家族的成员个数,确定主要分型和各stutters;
29.根据所述各stutters的成员个数之和与所述umi家族的成员个数,得到str滑脱百分比;
30.根据各stutters的个数和所述主要分型的个数,得到各stutters的比例。
31.可选的,所述umi家族的成员个数≥30;所述标识码序列的长度≥12bp,所述umi接头序列的长度≥8bp。
32.可选的,所述捕获探针的核苷酸序列如seqid no.1-45中任意一项所示。
33.可选的,所述高通量测序包括双末端测序或单末端测序。
34.第二方面,本技术提供了一种二代测序中利用分子特异性条形码评估str的滑脱的应用,所述应用包括将第一方面所述的方法用于个体识别和鉴定中。
35.本技术实施例提供的上述技术方案与现有技术相比具有如下优点:
36.本技术实施例提供的该方法,利用标识码序列去除杂质污染序列,利用umi接头序列的分子特异性为每个打断的原始dna模板分子加上唯一的、过量的umi接头,经pcr扩增和目标序列富集后,含有相同umi的str扩增产物形成一个umi家族,通过umi将各个str的每个umi家族的所有成员还原到原始的dna模板分子,比较了还原前后各样本每个str基因座的滑脱率,结果发现经umi还原后的str滑脱率明显低于还原前的,str还原后的滑脱率在所有
样品中都降低到5%以下,甚至为0,说明umi可准确识别pcr过程中str基因座滑脱产生的stutters并将其去除,达到了降低二代测序平台中str滑脱的预期效果。
附图说明
37.此处的附图被并入说明书中并构成本说明书的一部分,示出了符合本发明的实施例,并与说明书一起用于解释本发明的原理。
38.为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单的介绍,显而易见地,对于本领域普通技术人员而言,在不付出创造性劳动性的前提下,还可以根据这些附图获得其他的附图。
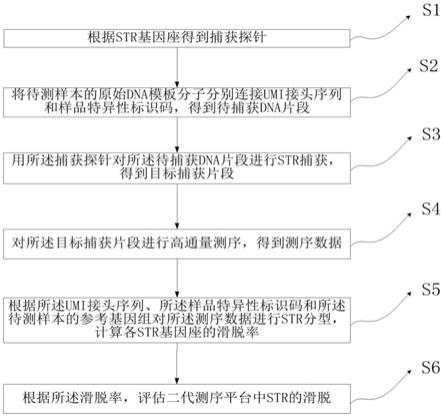
39.图1为本技术实施例提供的一种二代测序中利用分子特异性条形码评估str的滑脱的方法的流程示意图;
40.图2为本技术实施例提供的str高通量文库的构建、测序及数据分析图;
41.图3为本技术实施例提供的分子特异性条形码家族的大小;
42.图4为本技术实施例提供的分子特异性条形码家族的滑脱百分比;
43.图5为本技术实施例提供的每个str基因座的n-1型stutters与主要分型比例的散点图;
44.图6为本技术实施例提供的来源于同一个umi家族的d2s441基因座的基因分型结果;
45.图7为本技术实施例提供的每个str主要分型与stutters之间的皮尔相关系数;
46.图8为本技术实施例提供的th01基因座各umi家族主要分型与stutters的关系;
47.图9为d2s1338基因座中含有同一个umi的家族成员还原为原始dna模板分子的示意图。
具体实施方式
48.为使本技术实施例的目的、技术方案和优点展示的更加明了,下面将结合本技术实施例中的附图,对本技术实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本技术的一部分实施例,而不是全部的实施例。基于本技术中的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所获得的所有其他实施例,都属于本技术保护的范围。
49.下面结合实施例来具体说明本技术方法的优异性。
50.二代测序中利用分子特异性条形码评估str的滑脱的方法,其特征在于,所述方法包括以下步骤:
51.s1.根据str基因座得到捕获探针;
52.本技术实施例中,如果是人的str基因座,可以针对人的22个str基因座的核苷酸序列设计探针以进行str靶标序列的捕获。
53.本技术实施例中,str靶标探针序列的设计:采用常用的24个str基因座设计捕获探针,共设计了45条探针。其中有22个str基因座设计出了45个探针,45个探针的名称依次为str0001-str0045,对应的核苷酸序列依次为seqid no.1-45。平均每个位点设计1-3条探针,详细信息见附表1。
54.表1针对22个str基因座的捕获探针。
55.[0056][0057]
鉴于动物、植物、微生物都具有str基因座,因此本技术的方法可以用于动物、植物、微生物。
[0058]
s2.将待测样本的原始dna模板分子分别连接umi接头序列和样品特异性标识码,得到待捕获dna片段;
[0059]
本技术实施例中,可以利用机械或者酶切方法将待测样本的基因组dna打断成100-500bp大小的dna片段,得到待测样本的原始dna模板分子;然后将这些dna片段的两端分别连接过量的分子特异性条形码序列(umi)和引入样品特异的标识码。
[0060]
本技术实施例中,实验样本采用6个中国汉族无个体的滤纸血样进行dna的提取及定量,使用m48磁珠提取纯化试剂盒(qiagen,美国)对6个中国汉族无关个体的血样进行dna
提取。使用2.0荧光计(invitrogen,美国)对dna进行定量,将定量后的血样dna用于备检。
[0061]
s3.用所述捕获探针对所述待捕获dna片段进行str捕获,得到目标捕获片段;
[0062]
s4.对所述目标捕获片段进行高通量测序,得到测序数据;
[0063]
文库构建、探针捕获与高通量测序:用covariss220超声仪(covaris,woburn,美国)将提取的血样dna进行破碎,以获得100-300bp的dna片段。然后为每个样品dna片段连接含有样品特异dna标识码(tag)和dna模板分子特异的umi接头序列,如图2,其中接头序列是绝对过量的,以保证每个原始的dna分子连接的umi是唯一的。经末端修复和加a尾后,使用genobaits dna文库制备试剂盒(dl002,molbreeding biotechnology co.,ltd,china)构建dna文库。按照文库构建说明书,将每个与illumina测序平台兼容的genobaits文库(100ng)进行合并,再用genobaits dna文库制备试剂(dl001 molbreeding biotechnology co.,ltd,china)进行str靶标捕获。最后在illumina hiseqx-ten(illumina,inc.,sandiego,ca)平台上对富集文库进行双端测序(2
×
150bp)。
[0064]
本技术实施例中,用测序平台的插入片段长度为150bp,除了包含str核心基序及其侧翼序列(本文要求至少20bp长,可以根据自己的需要调整)外,还包含了8bp的umi,12bp的样品特异的标识码,导致探针富集靶标区域的str核心基序长度不超过90bp,序列长的str基因座可能不能被有效富集,可根据研究的需要,加大测序长度,以期所有目标str基因座被富集出来。
[0065]
高通量数据分析:根据构建文库时的样品条形码将总的测序数据进行分库,获得每个样品的原始测序序列;去除不符合预设标准的序列后,再根据tag序列去除污染的序列;然后利用比对软件与人类参考基因组比对,保留含有str保守基序及其侧翼区域(也称边界序列)序列;然后从这些序列中提取str保守基序及umi,以作为我们后续分析的原始提取序列,如图2。
[0066]
6名中国汉族无关个体按照图2进行str基因座的富集、文库构建与数据分析,测序后平均每个样品获得6,503,692条测序序列。每个样本中含有对应tag标签的测序序列被保留下来;然后将其比对到人类参考基因组序列(ftp://hgdownload.cse.ucsc.edu/goldenpath/hg19/chromosomes)上,若序列能比对到参考基因组的str保守基序上,并且保守基序的两翼区域含有指定阈值长度的边界序列(本文要求边界序列是20bp),则认为该序列含有目标str序列,提取该序列包含的umi及str目标序列以进行str分型。所有样品的str的分型结果显示:每个str基因座被富集的靶标序列并不平衡,有些str基因座在6个样品中的富集量均较高,如tpox、th01、d13s317、dys391、d2s441、d16s539、csf1po、d10s1248;而有些位点在所有样品中几乎无富集,如fga、d7s820、vwa、d22s1045、d18s51、d2s1338。
[0067]
s5.根据所述umi接头序列、所述样品特异性标识码和所述待测样本的参考基因组对所述测序数据进行str分型,计算各str基因座的滑脱率;
[0068]
s6.根据所述滑脱率,评估二代测序平台中str的滑脱。
[0069]
本技术实施例中,若没有已公开的参考基因组,也可以是str参考序列,在此统称参考基因组。
[0070]
umi家族大小、str滑脱百分比及stutters和主要分型的关系分析:由于pcr扩增前,每个片段化的dna原始模板分子均连有唯一的umi,因此pcr扩增后含有相同umi的测序
序列可形成一个umi家族(aumi family)。本技术中umi家族的大小(size)指的是一个umi家族中的所有成员个数,我们统计了不同umi家族大小的频数(出现的次数)分布;同时对每个umi家族中每个成员进行基因分型,鉴于我们研究的是二代测序平台中str的滑脱,因此本文只考虑每个成员的长度多态性,暂时不考虑其序列多态性。把每个umi家族中长度相同的基因型合并,并统计其成员个数,其中成员数目最多的基因型为主要分型。如一个umi家族中所有成员的原始基因分型结果为:(aagg)13有5个成员,(aagg)12(atgg)1有2个成员,(aagg)12有4个成员,把长度相同的(aagg)13和(aagg)12(atgg)1这两个基因型合并统计后共有7个成员,(aagg)13作为该umi家族的主要分型,类似于毛细管电泳的主带,这里我们只考虑其长度,其它的长度的成员均作为stutters,类似于毛细管电泳的滑脱峰(影子带)。每个umi家族的str滑脱百分比用该家族stutters个数与所有成员个数的百分比表示。统计了不同umi家族大小(图3)的str滑脱百分比的分布(图4)和每个str基因座的所有umi家族数目及其滑脱百分比的分布。
[0071]
根据每个str基因座的滑脱情况,统计了比对到每个str基因座至少拥有30个成员的umi家族数目。结果发现th01、tpox、d13s317、d16s539、d2s441、csf1po和d10s1248基因座的umi家族数目均大于1000个,而d7s820基因座没有这样的umi家族,d18s51、d22s1045、d2s1338、fga及vwa基因座umi家族数目均小于50个。该结果与比对到每个str基因座的原始测序序列数目的分析结果相一致。分析了比对到每个str基因座的所有umi家族的str滑脱百分比,结果显示当str基因座的umi家族数目较多时,该str基因座的滑脱百分比相对较低,如th01、tpox基因座;而当str基因座的umi家族数目较少时,则该str基因座的滑脱百分比相对较高,如d18s51、d22s1045。
[0072]
此外,统计了每个umi家族的主要分型(n_0)与增加1-2个串联重复单位(n+1,n+2)或减少1-4个串联重复单位的stutters(n-1,n-2,n-3,n-4)的关系。stutters是指str进行pcr扩增时由于dna聚合酶的滑脱所形成的产物,一般比主要分型(n_0)少一个重复单位(n-1),也有增加或减少一定重复单位的。stutters比例指的是一个umi家族中每种类型的stutters成员数目与主要分型成员数目的比值。本文主要统计了每个umi家族中增加1-2个串联重复单位(n+1,n+2)或减少1-4个串联重复单位的stutters(n-1,n-2,n-3,n-4)与主要分型(n_0)成员数目的比值,并要求每个str基因座至少有60个umi家族(6个样本合并分析)。结果显示每个str基因座n-1型stutters较多,n+1与n-2的次之,其他类型的更低。从n-1型stutters与其主要分型比值的散点图上(图5),发现n-1型stutters与主要分型比值介于0-1之间,尽管比值接近于1的stutters,在各umi家族中占的比例极低,但是说明有些str基因座可能在pcr扩增的第一轮就发生了滑脱,如样品2中d2s441位点的一个umi家族共有30个成员(umi条形码为:aatcagag),其中48bp长度的成员有15个,44bp长度的成员也有15个(图6),若根据成员数目最多的原则来确定每个umi家族主要分型,则无法确定该umi家族的主要分型。另外我们发现当umi家族数目大于500个时,除了d10s1248位点外,其它位点的stutters与主要分型的比值都降到了0.15以下。
[0073]
本实例还分析统计了成员数目相对较多的stutters(n-1,n+1,n-2)与主要分型(n_0)之间的关系。所有str基因座主要分型与stutters之间的皮尔相关系数,如图7所示。相对于一些str基因座,如d19s433(0.16)、d13s317(0.21),有些str基因座的主要分型与n-1型stutters的皮尔相关系数较高,呈明显的正相关,如dys391(0.69)、tpox(0.67)。n-1与
n-2的stutters的皮尔相关系数总体比主要分型与n-1型stutters的皮尔相关系数低些,但比主要分型与n+1的stutters的皮尔相关系。图8中展示了th01基因座主要分型与各stutters的关系。对角线上展示的为主要分型及stutters的序列覆盖情况。散点图上为主要分型及stutters的序列数目。r为主要分型与其他stutters的皮尔相关系数。红线表示通过局部加权散点平滑方法处理后的stutters和主要分型序列读数之间的趋势图。由图8可知,该str基因座的n-1型stutters与主要分型的皮尔相关系数较高,n-2与n-1型stutters的皮尔相关系数次之,呈明显的正相关,而其他stutters之间或者stutters与主要分型之间没有明显相关性。
[0074]
本技术的方法,可以识别和去除二代测序平台pcr过程中str滑脱产生的stutters,达到降低str滑脱的效果,利于混合样本的精准分型。
[0075]
本技术的stutters产物,是dna聚合酶的滑脱产物;理论上该umi家族的所有成员只能分型出一种等位基因,若有别的分型则是由于滑脱造成的。提取每个str基因座每个umi家族成员的保守序列,按照图9示例对每个umi家族进行还原。每个str基因座未经还原的原始测序序列和还原后原始dna模板分子,分别按照长度多态性进行基因分型,并计算统计每个str基因座的滑脱率。由于每个umi家族的所有成员均来源于同一个原始的dna模板分子,含有相同的umi,因此利用umi和本团队开发的计算机程序可以将属于该umi家族的所有成员还原到原始的dna模板分子,通过比较还原前的原始测序序列与还原后的原始dna模板分子的基因分型结果来研究每个样本中各str基因座的滑脱情况。考虑到单个个体的每个str基因座一般最多存在着两种等位基因,当然也有存在三种等位基因分型的,本文目的主要是评估二代测序中的str的滑脱,因此暂不考虑这种分型结果。参考前人对每个str基因座纯合性和杂合性判定的原则,本文将每个基因座排名第一的等位基因与该基因座所有分型等位基因的比值≥0.7时,则认为该str基因座的等位基因为纯合子,只有一种等位基因(主要等位基因),其余的等位基因我们认为均为滑脱造成的;若排名第一和第二的等位基因与该基因座所有分型等位基因的比值≥0.35时,认为该str基因座存在两种等位基因为杂合子,排名第一的称为主要等位基因,排名第二的为次等位基因,其余等位基因均为pcr扩增过程中的滑脱产物。滑脱率即为每个str基因座stutters占所有等位基因分型的百分比。本技术文件通过分析测序序列经umi还原前及还原后各样本中每个str基因座的滑脱率,以评估umi能否达到降低二代测序平台中的str的滑脱的效果。
[0076]
分析结果表明还原后除了d12s391基因座在一个样品中滑脱率为12.239外,其它str基因座的滑脱率均降低到了5%以下,有些基因座甚至降低到0;而未经还原的原始测序序列,每个str基因座的滑脱率均比还原后的高。如:d10s1248,d3s1358还原前的平均滑脱率都在17%以上,经umi还原到原始dna模板分子后,滑脱率平均降低了14%。若把6个样品当作6个生物重复,我们发现每个str基因座还原前后的基因分型结果一致,而滑脱率均被显著降低,说明我们利用umi把高通量测序序列还原到原始的dna模板分子的方法对降低str滑脱率有明显的效果。
[0077]
本技术文件的又一目的:考虑str的长度多态性,还可考虑其序列多态性,因此相对于传统的ce,利用umi将测序数据还原为原始的dna模板分子,在混合分型拆分中将更具有潜在的优势。
[0078]
需要说明的是,在本文中,诸如“第一”和“第二”等之类的关系术语仅仅用来将一
个实体或者操作与另一个实体或操作区分开来,而不一定要求或者暗示这些实体或操作之间存在任何这种实际的关系或者顺序。而且,术语“包括”、“包含”或者任何其他变体意在涵盖非排他性地包含,从而使得包括一系列要素的过程、方法、物品或者设备不仅包括那些要素,而且还包括没有明确列出的其他要素,或者还包括为这种过程、方法、物品或者设备所固有的要素。在没有更多限制的情况下,由语句“包括一个
……”
限定的要素,并不排除在包括所述要素的过程、方法、物品或者设备中还存在另外的相同要素。
[0079]
以上所述仅是本发明的具体实施方式,使本领域技术人员能够理解或实现本发明。对这些实施例的多种修改对本领域的技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其他实施例中实现。因此,本发明将不会被限制于本文所示的这些实施例,而是要符合与本文所申请的原理和新颖特点相一致的最宽的范围。
[0080]
[0081]
[0082]
[0083]
[0084]
[0085]
[0086]
[0087]
[0088]
[0089]
[0090]
[0091]
[0092]
[0093]
[0094]
[0095]
[0096]
[0097]
[0098]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1