一种制备拟南芥自噬基因突变体的方法及应用
1.本发明涉及生物技术领域,具体地,涉及一种制备拟南芥自噬基因突变体的方法。
背景技术:
2.细胞自噬(autophagy)是真核生物利用溶酶体或液泡对胞内物质和细胞器进行降解的一种进化保守的重要代谢途径。该途径主要通过双层膜结构将待降解底物包裹后,形成自噬小泡(autophagosome),当自噬小泡被运输到液泡附近时,其外膜与液泡膜融合,内膜包裹的底物则释放到液泡中,并被水解酶降解成可循环利用的小分子物质。当真核生物面临营养缺乏或者因分化造成的物质供应不足时,细胞会启动自噬来调节胞内物质的循环利用,以保证其各部分功能的正常运行。自噬对植物的正常生长不可或缺,它几乎参与了植物生长发育的各个阶段,包括种子萌发、个体发育、繁殖和衰老等,尤其在植物应对营养胁迫、抵御病原体侵染和调控细胞程序性死亡中发挥着重要作用。当自噬途径受损时,植物会表现出生长缓慢、过早衰老并对营养物质缺乏敏感等的表型。
3.自噬途径受到一系列自噬相关基因(autophagy-related genes,简称atg基因)的精密调控。其中atg8是自噬途径中的核心蛋白,它通过与磷脂酰乙醇胺(phosphatidylethanolamine,pe)相连而锚定在自噬小泡的双层膜上,参与了自噬小泡的形成与成熟。atg8-pe还起着对接平台的作用,与具有atg8互作基序(atg8 interacting motif,aim)的各种衔接蛋白相互作用,选择性地降解各种自噬底物。尽管atg8在自噬途径中起着重要的作用,但是到目前为止尚未明确其在植物中扮演的具体角色。
4.植物中的atg8是多拷贝基因,在拟南芥中一共有9个拷贝,在玉米中有5个拷贝,在水稻中有6个拷贝。拟南芥的atg8基因家族成员分别为atg8a-i,在进化上可以分成三个亚家族atg8abcd、atg8efg、atg8hi。atg8家族各个拷贝在拟南芥不同组织中均有表达,且不同亚家族的基因表达是特异的。近年来研究表明通过过表达拟南芥自身的atg8基因,或者在拟南芥中异源过表达atg8基因,均会引起拟南芥的自噬水平升高,还能够提高植株的生长发育、种子产量和氮素再利用效率,并且增强植株对非生物胁迫的耐受性。除在拟南芥之外,其他作物中atg8蛋白的功能也逐渐被发现。现有技术通过过表达或敲低水稻osatg8b的转基因材料进行分析,发现osatg8b介导的自噬途径参与了氮素向籽粒的营养循环,并且直接影响了籽粒品质。
5.由于植物中atg8家族存在着多个拷贝,一直缺少直接的遗传材料去详细解析atg8在植物自噬中的功能,尚无以atg8全拷贝缺失的突变体植株模型以供自噬途径的研究。
技术实现要素:
6.本发明的目的是提供一种制备拟南芥自噬基因atg8九突变体的方法。
7.本发明的另一个目的是提供上述制备拟南芥自噬基因atg8九突变体的方法在制备降低植物营养循环的植物模型方面的应用。
8.本发明的再一个目的是提供核苷酸序列如seq id no:1~9所示的拷贝基因的基
因敲除位点和/或核苷酸序列如seq id no:10~18所示序列的sgrna在制备降低植物营养循环利用的拟南芥中的应用。
9.为了实现上述目的,本发明是通过以下方案予以实现的:
10.本发明提供了一种创建atg8基因九重突变体的基因编辑载体的构建方法,包括:
11.(1)首先通过crispr/cas9基因编辑技术构建atg8efg三突变体纯合株系;
12.(2)然后以atg8efg三突变体纯合株系为材料,通过crispr/cas9基因编辑技术构建atg8abcdefg七突变体纯合株系;
13.(3)最后以atg8abcdefg七突变体纯合株系为材料,通过crispr/cas9基因编辑技术构建atg8abcdefghi九突变体纯合株系。
14.本发明提供了一种制备拟南芥自噬基因atg8突变体的方法,在野生型拟南芥中,使用crispr/cas9系统敲除atg8基因的多拷贝基因,所述的拷贝基因为atg8a、atg8b、atg8c、atg8d、atg8e、atg8f、atg8g、atg8h和atg8i,依次对应tair数据库中的locus为at4g21980、at4g04620、at1g62040、at2g05630、at2g45170、at4g16520、at3g60640、at3g06420和at3g15580。
15.优选地,所述的拷贝基因的基因敲除sgrna靶向位点的核苷酸序列如seq id no:1~9所示。
16.更优选地,靶向所述的基因敲除sgrna靶向位点的sgrna的编码基因的核苷酸序列如seq id no:10~18所示。
17.优选地,所述的方法包含以下步骤:
18.s1.使用crispr/cas9系统沉默野生型拟南芥的atg8基因的atg8e、atg8f和atg8g拷贝基因,得到纯合三突变体株系;
19.s2.使用crispr/cas9系统沉默纯合三突变体株系的atg8基因的atg8a、atg8b、atg8c和atg8d拷贝基因,得到纯合七突变体株系;
20.s3.使用crispr/cas9系统沉默纯合七突变体株系的atg8基因的atg8a、atg8b、atg8c和atg8d拷贝基因,得到纯合九突变体株系。
21.更优选地,所述制备拟南芥自噬基因atg8突变体的方法包含以下步骤:
22.s1.使用敲除atg8e、atg8f和atg8g拷贝基因的crispr/cas9载体,所述的crispr/cas9载体包含核苷酸序列如seq id no:10~12所示的sgrna的编码基因;农杆菌法转化野生型拟南芥,经筛选鉴定,得到无cas9背景的atg8efg纯合三突变体株系;
23.s2.使用敲除atg8a、atg8b、atg8c和atg8d拷贝基因的crispr/cas9载体,所述的crispr/cas9载体包含核苷酸序列如seq id no:13~16所示的sgrna的编码基因;农杆菌法转化atg8efg纯合三突变体株系,经筛选鉴定,得到无cas9背景的atg8abcdefg纯合七突变体株系;
24.s3.使用敲除atg8h和atg8i拷贝基因的crispr/cas9载体,所述的crispr/cas9载体包含核苷酸序列如seq id no:17~18所示的sgrna的编码基因;农杆菌法转化atg8abcdefg纯合七突变体株系,经筛选鉴定,得到无cas9背景的atg8abcdefghi纯合九突变体株系。
25.更优选地,crispr/cas9载体为phee401e载体。
26.更优选地,所述筛选为潮霉素筛选。
27.更优选地,所述鉴定为pcr扩增以及测序检测。
28.本发明还提供上述制备拟南芥自噬基因atg8九突变体的方法在制备降低植物营养循环的植物模型方面的应用。
29.核苷酸序列如seq id no:1~9所示的拷贝基因的基因敲除sgrna靶向位点和/或核苷酸序列如seq id no:10~18所示序列的sgrna的编码基因在制备降低植物营养循环利用的拟南芥中的应用也在本发明的保护范围之内。
30.在制备降低植物营养循环利用的拟南芥中的应用也在本发明的保护范围之内。
31.优选地,所述降低植物营养循环利用为使植株对缺氮胁迫敏感和/或对黑暗胁迫敏感。
32.与现有技术相比,本发明具有以下有益效果:
33.本发明通过crispr/cas9基因编辑技术可以在较短时间内达到精确、有效地敲除全部atg8基因的多突变体,为后续研究atg8基因家族的功能奠定坚实基础。本发明还提供了atg8基因在降低植物营养循环利用方面的应用,为培育营养高效利用的植物新品种提供了新的基因资源,在农业生产上具有重要的应用价值。
附图说明
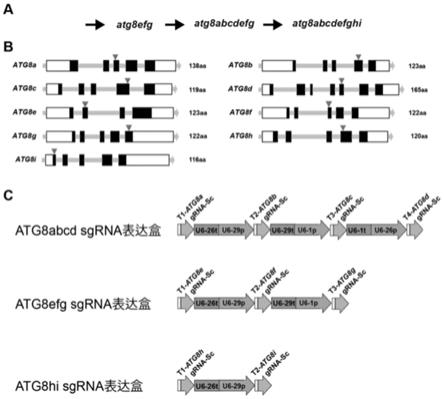
34.图1为通过crispr-cas9基因编辑构建拟南芥atg8abcdefghi九突变体纯合株系技术路线图和atg8基因编辑位点;a为技术路线图;b为atg8基因编辑位点;c为sgrna表达盒示意图。
35.图2为atg8abcdefghi九突变体的鉴定图;a为atg8九突变体在dna水平上的突变情况,框内部分是基因中的pam(protospacer adjacent motif)序列位置;b.atg8纯合突变体突变位点测序峰图;c为atg8efg纯合三突变体株系、atg8abcdefg纯合七突变体及atg8abcdefghi纯合九突变体无cas9背景的鉴定结果。
36.图3为atg8abcdefghi九突变体纯合株系的atg8蛋白表达量图;m:protein marker;wt:野生型;atg7-2:自噬突变体;2m:atg8hi突变体;3m:atg8efg突变体;4m:atg8abcd突变体;5m:atg8efghi突变体;7m:atg8abcdefg突变体;9m:atg8abcdefghi突变体;h3:组蛋白h3为蛋白内参。
37.图4为纯合atg8abcdefghi九突变体的氮胁迫表型;a为野生型(col-0)、自噬突变体atg7-2和atg8abcdefghi九突变体先在ms固体培养基上培养7天,再转移到含氮和缺氮的液体培养基中分别培养一周,然后拍照记录表型;b为图a中的叶绿素含量统计。
38.图5为纯合atg8abcdefghi九突变体的黑暗胁迫胁迫表型;a为野生型(col-0)、自噬突变体atg7-2、atg8abcdefg七突变体和atg8abcdefghi九突变体先播种在不含蔗糖的ms固体培养基上长日照生长两周,再转移到黑暗中培养10天,最后在长日照下恢复12天,拍照记录表型;b为图a中的存活数量统计。
具体实施方式
39.下面结合说明书附图及具体实施例对本发明作出进一步地详细阐述,所述实施例只用于解释本发明,并非用于限定本发明的范围。下述实施例中所使用的试验方法如无特殊说明,均为常规方法;所使用的材料、试剂等,如无特殊说明,为可从商业途径得到的试剂
和材料。
40.实施例1拟南芥九重突变体atg8abcdefghi的创制
41.根据图1a所示技术路线图,采用crispr-cas9基因编辑技术构建拟南芥atg8abcdefghi九突变体纯合株系。具体步骤如下:
42.1、atg8efg三突变体的创制
43.(1)从拟南芥tair数据库中获取atg8e(at2g45170)、atg8f(at4g16520)、atg8g(at3g60640)的基因组序列,根据基因组序列中的cds序列通过网站http://skl.scau.edu.cn/确定crispr/cas9基因编辑位点(基因编辑位点如图1b所示)。
44.atg8e基因编辑位点位于第3个外显子,序列为gaaaagagaaaagctgaagc(seq id no.1);
45.atg8f基因编辑位点位于第2个外显子,序列为gatccaggtgattgttgaga(seq id no.2);
46.atg8g基因编辑位点位于第5个外显子,序列为gatgagaataaggaagaaga(seq id no.3)。
47.将基因编辑位点与tair数据库进行blast序列比对,确定基因编辑位点的特异性。
48.以基因编辑位点为sgrna的靶向位点,分别设计sgrna的编码基因,序列如下:
[0049][0050][0051]
(2)根据上述基因编辑位点,设计引物用于构建编辑atg8efg基因的crispr/cas9载体,引物序列如下:
[0052]
atg8efg-dt1-primer-f:atatatggtctcgattgaaaagagaaaagctgaagcgtt;
[0053]
atg8efg-dt1-primer-r:atattattggtctcaatctcttagtcgactctaccaat;
[0054]
atg8efg-dt2t3-primer-f:atattattggtctcaagatgatccaggtgattgttgagagtt;
[0055]
atg8efg-dt2t3-primer-r:attattggtctcgaaactcttcttccttattctcatc。
[0056]
(3)atg8e sgrna表达盒及atg8fg sgrna表达盒的构建
[0057]
用hipure plasmid kits质粒提取试剂盒(美基,p1001-03c)分别提取pcbc-dt1t2质粒(与“doi:10.1186/s12870-014-0327-y”中的“pcbc-dt1t2”相同)、pcbc-dt2t3质粒(与“doi:10.1186/s12870-014-0327-y”中的“pcbc-dt2t3”相同)。
[0058]
以pcbc-dt1t2质粒dna为模板使用上述引物atg8efg-dt1-primer-f和atg8efg-dt1-primer-r用phanta max super-fidelity dna polymerase高保真酶(诺唯赞,p505-d3)通过pcr进行片段扩增,利用atg8g sgrna和pcbc-dt1t2质粒,生成含有atg8esgrna(seq id no.10)的atg8e sgrna表达盒(如图1c所示)。
[0059]
以pcbc-dt2t3质粒dna为模板使用上述引物atg8efg-dt2t3-primer-f和atg8efg-dt2t3-primer-r用phanta max super-fidelity dna polymerase高保真酶(诺唯赞,p505-d3)通过pcr进行片段扩增,利用atg8e sgrna、atg8g sgrna和pcbc-dt2t3质粒,生成含有atg8esgrna(seq id no.11)和atg8g sgrna(seq id no.12)的atg8fg sgrna表达盒(如图1c所示)。
[0060]
上述引物部分序列与pcbc-dt1t2或pcbc-dt2t3载体匹配,可以以这些载体为模版,扩增sgrna的骨架部分;这些引物部分序列与特异atg8的靶标位点相同,从而引入靶标序列。
[0061]
(4)将atg8e sgrna表达盒及atg8fg sgrna表达盒克隆到phee401e载体
[0062]
phee401e载体是一个植物表达的双元载体,除了表达crispr/cas9酶外,还包含在植物表达所需的其他元件和转基因筛选标记。
[0063]
用hipure plasmid kits质粒提取试剂盒(美基,p1001-03c)提取phee401e质粒(与“doi:10.1186/s13059-015-0715-0”中“additional file 5”的“phee401e”相同),使用基于bsaⅰ酶切和连接的goldengate克隆方法,以“边切边连”的方法将atg8e sgrna表达盒及atg8fg sgrna表达盒组装到phee401e中,获得phee401e-atg8efg基因编辑载体。
[0064]
(5)转化大肠杆菌dh5α
[0065]
将连接产物用热激法于42℃转化大肠杆菌dh5α,菌液涂布在含有50mg/ml kan抗生素的lb固体培养基上,倒置于37℃培养箱中培养12~16h。
[0066]
(6)载体鉴定
[0067]
挑取平板上长出的单菌落,摇菌扩繁。以菌液为模板进行pcr鉴定。
[0068]
(7)将构建好的phee401e-atg8efg基因编辑载体用冻融法导入农杆菌gv3101中。
[0069]
(8)以野生型拟南芥的种子为t0代种子,利用农杆菌,通过沾花法的转化方法将phee401e-atg8efg导入野生型拟南芥。获得转基因t1代atg8efg三突变的种子,t1代均为杂合子。
[0070]
(9)t1代atg8efg纯合三突变体株系的筛选及鉴定
[0071]
将t1代种子铺种在含有30μg/ml潮霉素的ms培养基上,筛选出t1代转基因阳性的幼苗,苗期14天时移栽到穴盘。
[0072]
在苗期30天左右时,使用如表1所示的引物,以2
×
taq master mix(dye plus)酶(诺唯赞,p112-03)通过pcr扩增靶点两侧的基因组序列并测序,然后与野生型基因组序列比较,确定靶点已经被crispr/cas9敲除。atg8efg三突变体株系鉴定结果如图2a和图2b中atg8e、atg8f、atg8g所示。
[0073]
表1鉴定atg8efg纯合三突变体株系所用的引物
[0074][0075]
(10)获得无cas9背景的atg8efg纯合三突变体株系
[0076]
通过t1代atg8efg纯合三突变体株系自交,单株收种,获得t2代种子。
[0077]
将t2代种子铺种在含有低浓度的潮霉素(15μg/ml)的ms培养基上,选择阳性苗:阴性苗符合3:1分离比的培养基皿。根据cas9背景和潮霉素抗性连锁的规律,将不含潮霉素抗性的阴性苗移栽,通过自交,单株收种,获得t3代种子。
[0078]
将t3代种子铺种在含有30μg/ml潮霉素的ms培养基上,选择全是阴性苗的培养皿,获得无cas9背景的atg8efg纯合三突变体株系(如图2c所示)。
[0079]
2、atg8abcdefg七突变体的创制
[0080]
为获得atg8abcdefg七突变体,以鉴定获得的无cas9背景的atg8efg突变体株系为母本,进行新一轮的crispr/cas9基因编辑。
[0081]
(1)从拟南芥tair数据库中获取atg8a(at4g21980)、atg8b(at4g04620)、atg8c(at1g62040)和atg8d(at2g05630)基因组序列,以与上述相同的方法确定crispr/cas9基因编辑位点(图1b)。
[0082]
atg8a基因编辑位点位于第3个外显子,序列为gattgtatgtaggtgattg(seq id no.4);
[0083]
atg8b基因编辑位点位于第4个外显子,序列为gccaatttgtgtacgttgtg(seq id no.5);
[0084]
atg8c基因编辑位点位于第4个外显子,序列为gttgtgtgtttaccagttgg(seq id no.6);
[0085]
atg8d基因编辑位点位于第5个外显子,序列为tgatgtcccggatattgat(seq id no.7)。
[0086]
将基因编辑位点与tair数据库进行blast序列比对,确定基因编辑位点的特异性。
[0087]
以基因编辑位点为sgrna的靶向位点,分别设计sgrna的编码基因,序列如下:
[0088][0089]
(2)根据上述基因编辑位点,设计引物用于构建编辑atg8abcdefg基因的crispr/cas9载体,引物序列如下:
[0090]
atg8abcddt1-primer-f:atatatggtctcgattgacttcttcttgtcaatgtcgtt;
[0091]
atg8abcddt1-primer-r:atattattggtctcaatctcttagtcgactctaccaat;
[0092]
atg8abcddt2-primer-f:tgacttcttcttgtcaatgtcgttttagagctagaaatagc;
[0093]
atg8abcddt2-primer-r:atattattggtctcatcactacttcgtctctaaccat;
[0094]
atg8abcddt3t4-primer-f:atattattggtctcaagatgttgtgtgtttaccagttgggtt;
[0095]
atg8abcddt3t4-primer-r:attattggtctcgaaacctgcaataaaaggataacac。
[0096]
(3)atg8a sgrna表达盒、atg8b sgrna表达盒和atg8cd sgrna表达盒的构建
[0097]
用hipure plasmid kits质粒提取试剂盒(美基,p1001-03c)分别提取pcbc-dt1t2质粒、pcbc-dt2t3质粒、pcbc-dt3t4质粒(与“doi:10.1186/s12870-014-0327-y”中的“pcbc-dt3t4”相同)。
[0098]
以提取的pcbc-dt1t2质粒dna为模板,使用上述引物atg8abcddt1-primer-f和atg8abcddt1-primer-r,用phanta max super-fidelity dna polymerase高保真酶(诺唯赞,p505-d3)通过pcr进行片段扩增,利用atg8a sgrna整合至pcbc-dt1t2质粒中,生成含有atg8a sgrna(seq id no.13)的atg8a sgrna表达盒(如图1c所示)。
[0099]
以提取的pcbc-dt2t3质粒dna为模板,使用上述引物atg8abcddt2-primer-f和atg8abcddt2-primer-r,用phanta max super-fidelity dna polymerase高保真酶(诺唯
赞,p505-d3)通过pcr进行片段扩增,利用atg8b sgrna和pcbc-dt2t3质粒,生成含有atg8b sgrna(seq id no.14)的atg8b sgrna表达盒(如图1c所示)。
[0100]
以提取的pcbc-dt3t4质粒dna为模板,使用上述引物atg8abcddt3t4-primer-f和atg8abcddt3t4-primer-r,用phanta max super-fidelity dna polymerase高保真酶(诺唯赞,p505-d3)通过pcr进行片段扩增,利用atg8c sgrna、atg8d sgrna和pcbc-dt3t4质粒,生成含有atg8c sgrna(seq id no.15)和atg8d sgrna(seq id no.16)的atg8cd sgrna表达盒(如图1c所示)。
[0101]
(4)以与上述相同的方法,将atg8a sgrna表达盒、atg8b sgrna表达盒和atg8cd sgrna表达盒组装到phee401e中,构建编辑atg8abcd基因的phee401e-atg8abcd基因编辑载体,转化大肠杆菌dh5α及载体鉴定。将构建好的phee401e-atg8abcd基因编辑载体用冻融法导入农杆菌gv3101中。
[0102]
(5)以atg8efg纯合三突变体的种子为t0代种子,利用农杆菌,将phee401e-atg8abcd导入atg8efg纯合三突变体。获得转基因t1代atg8abcdefg七突变体的种子,t1代均为杂合子。
[0103]
(6)t1代atg8abcdefg纯合七突变体株系的筛选及鉴定
[0104]
将t1代种子铺种在含有30μg/ml潮霉素的ms培养基上,筛选出t1代转基因阳性的幼苗,苗期14天时移栽到穴盘。
[0105]
在苗期30天左右时,使用如表2所示的引物,以2
×
taq master mix(dye plus)酶(诺唯赞,p112-03)通过pcr扩增靶点两侧的基因组序列并测序,然后与野生型基因组序列比较,确定靶点已经被crispr/cas9敲除。atg8abcdefg七突变体株系鉴定结果如图2a和图2b中atg8a、atg8b、atg8c、atg8d所示。
[0106]
表2鉴定atg8abcdefg纯合七突变体株系所用的引物
[0107][0108]
(7)获得无cas9背景的atg8abcdefg纯合七突变体株系
[0109]
通过t1代atg8abcdefg纯合七突变体株系自交,单株收种,获得t2代种子。
[0110]
将t2代种子铺种在含有低浓度的潮霉素(15μg/ml)的ms培养基上,选择阳性苗:阴性苗符合3:1分离比的培养基皿。根据cas9背景和潮霉素抗性连锁的规律,将不含潮霉素抗性的阴性苗移栽,通过自交,单株收种,获得t3代种子。
[0111]
将t3代种子铺种在含有30μg/ml潮霉素的ms培养基上,选择全是阴性苗的培养皿,获得无cas9背景的atg8abcdefg纯合七突变体株系(图2c)。
[0112]
3、atg8abcdefghi九突变体的创制
[0113]
为获得atg8abcdefghi九突变体,以鉴定获得的无cas9背景的atg8abcdefg突变体株系为母本,进行新一轮的crispr/cas9基因编辑。
[0114]
(1)从拟南芥tair数据库中获取atg8h(at3g06420)和atg8i(at3g15580)基因组序列,以与上述相同的方法确定crispr/cas9基因编辑位点(图1b)。
[0115]
atg8h基因编辑位点位于第4个外显子,序列为tggtccaacagtcatgtctcg(seq id no.8);
[0116]
atg8i基因编辑位点位于第1个外显子,序列为gttcaaggaacaatacacgt(seq id no.9)。
[0117]
将基因编辑位点与tair数据库进行blast序列比对,确定基因编辑位点的特异性。
[0118]
以基因编辑位点为sgrna的靶向位点,分别设计sgrna的编码基因,序列如下:
[0119][0120]
(2)根据上述基因编辑位点,设计引物用于构建编辑atg8abcdefg基因的crispr/cas9载体,引物序列如下:
[0121]
atg8hi-dt1-primer-f:atatatggtctcgattggtccaacagtcatgtctcggtt;
[0122]
atg8hi-dt2-primer-r:attattggtctcgaaacacgtgtattgttccttgaacaa。
[0123]
(3)atg8hi sgrna表达盒的构建
[0124]
以提取的pcbc-dt1t2质粒dna为模板,使用上述引物atg8hi-dt1-primer-f和atg8hi-dt2-primer-r,用phanta max super-fidelity dna polymerase高保真酶(诺唯赞,p505-d3)通过pcr进行片段扩增,利用atg8h sgrna、atg8i sgrna和pcbc-dt1t2质粒,生成含有atg8h sgrna(seq id no.17)和atg8i sgrna(seq id no.18)的atg8hi sgrna表达盒(如图1c所示)。
[0125]
(4)以与上述相同的方法,将atg8hi sgrna表达盒组装到phee401e中,构建编辑atg8hi基因的phee401e-atg8hi基因编辑载体,转化大肠杆菌dh5α及载体鉴定,将构建好的phee401e-atg8hi基因编辑载体用冻融法导入农杆菌gv3101中。
[0126]
(5)以atg8abcdefg纯合七突变体株系的种子为t0代种子,利用农杆菌,将phee401e-atg8hi导入atg8abcdefg纯合七突变体。获得转基因t1代atg8abcdefghi九突变的种子,t1代均为杂合子。
[0127]
(6)t1代atg8abcdefghi纯合九突变体株系的筛选及鉴定
[0128]
将t1代种子铺种在含有30μg/ml潮霉素的ms培养基上,筛选出t1代转基因阳性的幼苗,苗期14天时移栽到穴盘。
[0129]
在苗期30天左右时,使用如表3所示的引物,以2
×
taq master mix(dye plus)酶(诺唯赞,p112-03)通过pcr扩增靶点两侧的基因组序列并测序,然后与野生型基因组序列比较,确定靶点已经被crispr/cas9敲除。atg8abcdefghi九突变体株系鉴定结果如图2a和图2b中atg8h和atg8i所示。
[0130]
表3鉴定atg8abcdefghi纯合九突变体株系所用的引物
[0131][0132][0133]
(7)获得无cas9背景的atg8abcdefghi纯合九突变体株系
[0134]
通过t1代atg8abcdefghi纯合九突变体株系自交,单株收种,获得t2代种子。
[0135]
将t2代种子铺种在含有低浓度的潮霉素(15μg/ml)的ms培养基上,选择阳性苗:阴性苗符合3:1分离比的培养基皿。根据cas9背景和潮霉素抗性连锁的规律,将不含潮霉素抗性的阴性苗移栽,通过自交,单株收种,获得t3代种子。
[0136]
将t3代种子铺种在含有30μg/ml潮霉素的ms培养基上,选择全是阴性苗的培养皿,获得无cas9背景的atg8abcdefghi纯合九突变体株系(图2c)。
[0137]
实施例2蛋白免疫印迹杂交鉴定拟南芥九重突变体atg8abcdefghi的atg8蛋白表达情况
[0138]
1、实验方法
[0139]
(1)蛋白提取;将野生型拟南芥、自噬突变体atg7-2拟南芥(abrc拟南芥保藏中心;货号:gabi_655b06)、atg8hi突变体(以野生型种子为t0代种子,利用农杆菌,将phee401e-atg8hi导入野生型种子获得)、实施例1制得的atg8efg纯合三突变体、atg8abcd突变体(以野生型种子为t0代种子,利用农杆菌,将phee401e-atg8abcd导入野生型种子获得)、atg8efghi突变体(以atg8efg为t0代种子,利用农杆菌,将phee401e-atg8hi导入atg8efg获得)、实施例1制得的atg8abcdefg纯合七突变体、实施例1制得的atg8abcdefghi纯合九突变体的种子,在培养皿上生长1周左右。称取拟南芥整株幼苗样品约100mg,放进1.5ml的离心管中。用研磨棒研磨样品至匀浆,加入蛋白提取液,旋涡震荡5min,再用沸水煮5~10min。然后室温离心16,000
×
g,5min。将上清液转移到新的1.5ml离心管中,获得蛋白样品。
[0140]
(2)蛋白质免疫印迹法检测atg8突变体株系中atg8的表达:取上述蛋白样品20μl,于80v电压跑胶30min;后换成120v电压至跑胶结束;用10v恒压转膜过夜。最后用商品化atg8抗体(abcam,ab77003)检测突变体株系中的atg8的表达量。以组蛋白h3为蛋白内参(abcam,ab1791)。
[0141]
(3)蛋白质免疫印迹鉴定结果分析:
[0142]
2、实验结果
[0143]
如图3所示,与野生型拟南芥相比,atg8hi二突变体、atg8efg三突变体和atg8efghi五突变体中atg8蛋白的表达量基本没有差异,而atg8abcd四突变体和atg8abcdefg七突变体中atg8蛋白的表达量明显减少,atg8abcdefghi九突变体中几乎检测不到atg8蛋白。表明atg8abcdefghi九突变体中的atg8完全被敲除。
[0144]
实施例3拟南芥atg8abcdefghi纯合九突变体缺氮胁迫的表型分析
[0145]
1、实验方法
[0146]
(1)将野生型拟南芥、自噬突变体atg7-2和实施例1制得的atg8abcdefghi纯合九突变体的种子分别用75%的乙醇浸泡灭菌10min,灭菌后的种子于4℃冰箱春化2天。
[0147]
(2)将春化后的种子点播于含0.7%琼脂糖的1/2ms固体培养基中,置于22℃光照培养箱中萌发培养7天。
[0148]
(3)配置ms液体培养基(1l):称取4.3g ms固体培养基(sigma,m5519-1l)、0.4g的mes(生工生物工程(上海)股份有限公司,a100169)、10g的蔗糖、用ddh2o定容至1l,最后用2mm koh调节ph值至5.7。
[0149]
配置ms-n液体培养基(1l):量取100ml的10
×
ms-n溶液(sigma,m0529-1l)、3ml的1m cacl2、1ml的1m mgso4、5ml的1m kcl、1.25ml的1m kh2po4,称取0.4g的mes、10g的蔗糖,用ddh2o定容至1l,最后用2mm koh调节ph值至5.7。
[0150]
(4)将萌发的种子转移到ms、ms-n液体培养基中再生长一周,用相机拍照并记录结果。
[0151]
2、实验结果
[0152]
atg7是一个修饰atg8的酶,与atg8属于不同的基因家族。现有技术公开了(phillips ar,et al.the atg12-conjugating enzyme atg10 is essential for autophagic vesicle formation in arabidopsis thaliana.genetics.2008;178(3):1339-1353.),当atg7缺失时,atg8不发挥功能,因此通常通过缺失atg7来间接研究atg8的功能。atg7-2是拟南芥atg7基因的缺失突变体,是一种经典的自噬突变体,该突变体对缺氮胁迫和黑暗胁迫敏感。
[0153]
如图4a和4b所示,经缺氮胁迫处理后,atg8abcdefghi纯合九突变体叶片黄化、叶绿体含量降低并出现大量死亡,表现出对缺氮胁迫的敏感表型。
[0154]
实施例4拟南芥atg8abcdefghi纯合九突变体黑暗胁迫处理的表型分析
[0155]
1、实验方法
[0156]
(1)将野生型拟南芥、自噬突变体atg7-2、实施例1制得的atg8abcdefg纯合七突变体和atg8abcdefghi纯合九突变体的种子分别用75%的乙醇浸泡灭菌10min,消毒后的种子于4℃冰箱春化2天。
[0157]
(2)配置ms-c固体培养基(1l):称取4.43g的ms和0.5g的mes,用ddh2o定容至1l,用2mm koh调节ph值至5.7,最后加入7g琼脂粉。
[0158]
(3)将春化后的种子点播于在不含有1%(w/v)蔗糖的ms-c固体培养基上,然后置于22℃光照培养箱中萌发培养14天,转移到黑暗条件下生长10天,然后恢复光照,10~12天后用相机拍照并记录结果。
[0159]
2、实验结果
[0160]
如图5a和图5b所示,经黑暗处理处理后,自噬突变体atg7-2、atg8abcdefg纯合七突变体和atg8abcdefghi纯合九突变体均大量死亡,表现出对黑暗胁迫处理的敏感表型。
[0161]
最后所应当说明的是,以上实施例仅用以说明本发明的技术方案而非对本发明保护范围的限制,对于本领域的普通技术人员来说,在上述说明及思路的基础上还可以做出其它不同形式的变化或变动,这里无需也无法对所有的实施方式予以穷举。凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1