一种富马酸酰胺类化合物或其药学上可接受的盐及其制备方法和应用
1.本发明属于有机合成技术领域,尤其涉及一种富马酸酰胺类化合物或其药学上可接受的盐及其制备方法和应用。
背景技术:
2.干扰素基因刺激物(sting)是细胞溶质dna传感途径中的一种细胞内衔接蛋白,可介导针对病原体入侵的先天免疫反应。它充当来自细菌来源的环状二核苷酸或由cgas(环状gmp-amp合酶)产生的2',3'-cgamp(2',3'-环状gmp-amp)的细胞溶质传感器。因此,cgas-sting通路在维持宿主先天免疫反应的稳态中起着重要作用。刺激后,sting可以触发ifn(i型干扰素)和其他促炎介质(例如细胞因子)的产生。sting的过度激活与许多自身免疫和炎症性疾病的发病机制有关,包括aicardi-goutieres综合征(ags)、系统性红斑狼疮(sle)等,因此,sting被认为是治疗炎症的极好靶点。在过去几年中,对sting抑制剂的研究得到了加强。迄今为止,经过不断努力,一些靶向sting的小分子抑制剂逐渐浮出水面。根据作用机制,这些sting抑制剂可分为两大类:(1)占据环二核苷酸(cdn)结合位点的内源性sting激动剂的竞争性拮抗剂;(2)靶向sting n端跨膜区半胱氨酸(cys88和cys91)棕榈酰化的小分子抑制剂。
3.上述靶向sting的小分子取得了一些成功,但因为传统的小分子抑制剂是“占用驱动的”,抑制剂治疗后sting信号的适应性抵抗和重新激活可能会发生。
4.因此,有必要提供一种活性更好的靶向sting的降解剂。
技术实现要素:
5.本发明旨在至少解决现有技术中存在的上述技术问题之一。为此,本发明提供了一种富马酸酰胺类化合物或其药学上可接受的盐。
6.本发明还提供了所述富马酸酰胺类化合物或其药学上可接受的盐的制备方法。
7.本发明还提供所述富马酸酰胺类化合物或其药学上可接受的盐的应用。
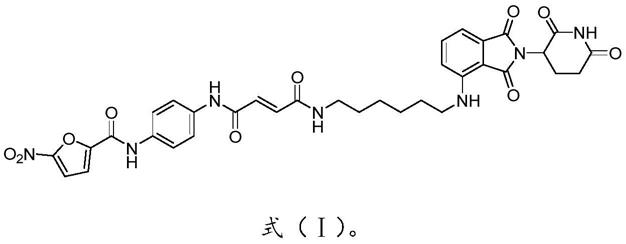
8.本发明的第一方面提供了一种富马酸酰胺类化合物或其药学上可接受的盐,所述富马酸酰胺类化合物或其药学上可接受的盐的结构式如式(ⅰ)所示:
[0009][0010]
本发明关于富马酸酰胺类化合物或其药学上可接受的盐的技术方案中的一个技
术方案,至少具有以下有益效果:
[0011]
本发明提供的富马酸酰胺类化合物能够通过泛素-蛋白酶体途径有效地降解thp-1细胞系中的sting蛋白,dc50为3.2μm,在顺铂-aki小鼠模型中显示出高效的的体内抗炎功效,并有效保护小鼠免受顺铂诱导的肾脏损伤。这是因为发明人选择了特定α,β-不饱和羰基linker,使得富马酸酰胺类化合物具有优异的活性。
[0012]
本发明的第二方面提供一种富马酸酰胺类化合物或其药学上可接受的盐的制备方法,包括如下步骤:
[0013]
将化合物4、化合物8、2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯和n,n-二异丙基乙胺在第三有机溶剂中反应,即得式(ⅰ)化合物;所述化合物4和化合物8的结构式如下:
[0014][0015]
根据本发明的一些实施方式,所述化合物4通过如下步骤制得:
[0016]
s1.将4-氟异苯并呋喃-1,3-二酮、2,6-二氧哌啶-3-氯化铵和醋酸钠在醋酸中反应,即得化合物2a;
[0017]
s2.将化合物2a、(6-氨基己基)氨基甲酸叔丁酯和n,n-二异丙基乙胺溶于第一有机溶剂中,加热反应即得化合物3,在酸性条件下反应得到化合物4;所述化合物2a和化合物3的结构式如下:
[0018][0019]
根据本发明的一些实施方式,所述化合物8通过如下步骤制得:
[0020]
s3.在0~5℃条件下,将5-硝基呋喃-2-碳酰氯加入到化合物5a和n,n-二异丙基乙胺的溶剂中反应,即得化合物6,在酸性条件下继续反应得到化合物7;
[0021]
s4.将化合物7、4-环戊烯-1,3-二酮和n,n-二异丙基乙胺溶解在第二有机溶剂中加热反应,即得化合物8;
[0022]
所述化合物5a、化合物6和化合物7的结构式如下:
[0023]
[0024]
所述(6-氨基己基)氨基甲酸叔丁酯的cas号为51857-17-1。
[0025]
所述4-环戊烯-1,3-二酮的cas号为930-60-9。
[0026]
根据本发明的一些实施方式,所述第一溶剂、第二溶剂和第三溶剂分别独立的选自n,n-二甲基甲酰胺、乙酸乙酯、四氢呋喃中的至少一种。
[0027]
根据本发明的一些实施方式,步骤s2中,所述加热的温度为80~100℃。
[0028]
根据本发明的一些实施方式,步骤s2中,所述反应时间为10~24h。
[0029]
根据本发明的一些实施方式,步骤s4中,所述加热的温度25~35℃。
[0030]
根据本发明的一些实施方式,所述在酸性条件下是指将溶剂的ph值调成小于7即可。包括但不限于在溶剂中加入盐酸、硫酸或硝酸。
[0031]
根据本发明的一些实施方式,所述第三溶剂反应的时间为8~18h。
[0032]
本发明的第三方面提供所述富马酸酰胺类化合物或其药学上可接受的盐在制备sting降解剂中的应用。
[0033]
本发明的第四方面还提供了一种药物组合物,所述组合物含有本发明的酰胺类化合物或其药学上可接受的盐,和药学上可接受的赋形剂。
[0034]
一般术语
[0035]
本发明中所述“药学上可接受的”是指在施用于动物或人类时不产生不良反应、过敏反应或其他不利反应的分子实体和组合物,如本发明中的“药学上可接受的赋形剂”包括任何和所有溶剂、分散介质、包衣、抗细菌剂和抗真菌剂、等渗剂和吸收延迟剂等,此类赋形剂用于药学活性物质的使用是本领域所熟知的。
[0036]
本发明中所述“药学上可接受的盐”包括碱加成盐和酸加成盐。
[0037]
药学上可接受的碱加成盐可以用金属或胺(例如碱金属和碱土金属或有机胺)来形成。化合物的药学上可接受的盐也可以用药学上可接受的阳离子来制备。适合的药学上可接受的阳离子是本领域技术人员所熟知的并且包括碱金属阳离子、碱土金属阳离子、铵阳离子和季铵阳离子。碳酸盐或碳酸氢盐也是可能的。用作阳离子的金属的是钠、钾、镁、铵、钙或三价铁等。适合的胺的包括异丙胺、三甲胺、组氨酸、n,n'-二苄基乙二胺、氯普鲁卡因、胆碱、二乙醇胺、二环己胺、乙二胺、n-甲基葡糖胺和普鲁卡因。
[0038]
药学上可接受的酸加成盐包括无机酸盐或有机酸盐。适合的酸盐包括盐酸盐、甲酸盐、乙酸盐、柠檬酸盐、水杨酸盐、硝酸盐、磷酸盐。其他适合的药学上可接受的盐是本领域技术人员所熟知的并且包括例如甲酸、乙酸、柠檬酸、草酸、酒石酸或扁桃酸、盐酸、氢溴酸、硫酸或磷酸;与有机羧酸、磺酸、磺酸基酸或磷酸基酸或n-取代的氨基磺酸,例如乙酸、三氟乙酸(tfa)、丙酸、乙醇酸、琥珀酸、马来酸、羟基马来酸、甲基马来酸、富马酸、苹果酸、酒石酸、乳酸、草酸、葡糖酸、葡糖二酸、葡糖醛酸、柠檬酸、苯甲酸、肉桂酸、扁桃酸、水杨酸、4-氨基水杨酸、2-苯氧基苯甲酸、2-乙酰氧基苯甲酸、扑酸、烟酸或异烟酸的盐;以及与氨基酸,例如在自然界中参与蛋白质合成的20种α氨基酸,例如谷氨酸或天冬氨酸,以及与苯乙酸、甲磺酸、乙磺酸、2-羟基乙磺酸、乙烷1,2-二磺酸、苯磺酸、4-甲基苯磺酸、萘2-磺酸、萘1,5-二磺酸、2-磷酸甘油酸或3-磷酸甘油酸、葡萄糖6-磷酸、n-环己基氨基磺酸(用于环己氨磺酸盐的形成),或与其他酸性有机化合物,例如抗坏血酸的盐。
[0039]
含有本发明所述的药物组合物能以常规方式来制造,例如通过常规混合、溶解、造粒、糖衣丸制备、磨细、乳化、囊封、捕集或冻干方法。适当的配制品取决于所选的施用途径。
附图说明
[0040]
图1是本发明实施例1制备的式(ⅰ)化合物对sting降解的浓度和时间关系图;
[0041]
图2是本发明实施例1制备的式(ⅰ)化合物、对比例1~4化合物的降解效果图;
[0042]
图3是本发明实施例1制备的式(ⅰ)化合物体内抗炎效果图;
[0043]
图4是本发明实施例1制备的式(ⅰ)化合物的核磁共振氢谱图;
[0044]
图5是本发明实施例1制备的式(ⅰ)化合物的核磁共振碳谱图;
[0045]
图6是本发明实施例1制备的式(ⅰ)化合物的质谱图。
具体实施方式
[0046]
下面将对本发明实施例中的技术方案进行清楚、完整地描述,但本发明的实施方式不限于此。
[0047]
本发明所采用的试剂、方法和设备,如无特殊说明,均为本技术领域常规试剂、方法和设备。
[0048]
以下实施例及对比例中采用的原料如下:
[0049]
sting小分子抑制剂c-170:购自invivochem;
[0050]
pomalidomide:购自invivochem;
[0051]
mg132:购自上海麦克林生化科技有限公司;
[0052]
bafilomycin:购自大连美仑生物技术有限公司;
[0053]
所有细胞系均购自美国类型培养物收集中心(atcc);
[0054]
薄层层析硅胶板使用青岛gf254硅胶板,薄层色谱法(tlc)使用的硅胶板采用的规格是0.15mm~0.20mm,薄层层析分离纯化产品采用的规格是0.4mm~0.5mm;
[0055]
柱层析一般使用烟台黄海硅胶200~300目硅胶为载体。
[0056]
化合物的结构是通过核磁共振(nmr)来确定的。nmr位移以(ppm)的单位给出。nmr的测定是用(bruker avance iii 400)核磁仪,测定溶剂为氘代二甲基亚砜(dmso-de),氘代氯仿(cdc13),氘代甲醇(cd3od),内标为四甲基硅烷(tms);
[0057]“dc
50”是指降解50%蛋白时的剂量。
[0058]
实施例1
[0059]
实施例1提供一种富马酸酰胺类化合物或其药学上可接受的盐,所述富马酸酰胺类化合物或其药学上可接受的盐的结构式如式(ⅰ)所示:制备方法如下,合成路线如下:
[0060][0061]
化合物4的制备
[0062]
s1.将化合物2 4-氟异苯并呋喃-1,3-二酮(5g,30.12mmol)、2,6-二氧哌啶-3-氯化铵(4.96g,30.12mmol)和naoac(2.69g,36.14mmol)在acoh(100ml)中的溶液回流12小时。
将反应混合物倒入冰水(1000ml)中并过滤,得到化合物2a(7.49g,90%产率).1h nmr(400mhz,dmso)δ11.14(s,1h),7.95(td,j=7.9,4.5hz,1h),7.79(d,j=7.3hz,1h),7.73(t,j=8.9hz,1h),5.15(dd,j=12.9,5.4hz,1h),2.89(ddd,j=17.2,13.9,5.4hz,1h),2.67-2.51(m,2h),2.14-1.97(m,1h).
[0063]
s2.将化合物2a(1.0g,3.62mmol)、(6-氨基己基)氨基甲酸叔丁酯(0.94g,4.34mmol)和n,n-二异丙基乙胺(0.7g,5.43mmol)溶于n,n-二甲基甲酰胺中,90℃反应12小时,将反应混合物倒入水中(100ml),并用乙酸乙酯(3x 80ml)萃取。合并的有机相在减压下干燥和浓缩。粗产物通过柱层析(pe/ea=6:1~2:1)纯化,得到即得化合物3(0.39g,23%产率)。1h nmr(400mhz,cdcl3)δ8.18(s,1h),7.56-7.48(m,1h),7.14(dd,j=7.9,4.4hz,1h),7.01(d,j=8.5hz,1h),6.42(t,j=5.8hz,1h),4.94(dd,j=12.1,5.3hz,1h),3.50-3.43(m,2h),3.42-3.34(m,2h),2.99-2.82(m,2h),2.82-2.71(m,2h),1.47(s,9h).
[0064]
将化合物3(0.35g,0.84mmol)溶解于etoac(5ml)中,然后向反应中添加4m盐酸二氧六环溶液(0.42ml,1.68mmol)。反应在室温下搅拌6小时。然后减压蒸除溶剂得到化合物4(0.22g,74%产率)。
[0065]
化合物8的制备
[0066]
将1,4-二氨基苯(5g,46.3mol)溶解在无水dcm(25ml)中,然后缓慢添加boc酸酐(10.01g,46.3mol)。0℃下搅拌6小时,直到原料完全消耗。减压蒸除溶剂,然后通过柱层析(pe/etoac=50:1)纯化粗产物,得到白色固体化合物5a(7.2g,75%产率)。1hnmr(400mhz,dmso)δ8.76(s,1h),7.06(d,j=7.6hz,2h),6.46(d,j=8.7hz,2h),4.72(s,2h),1.44(s,9h)。
[0067]
s3.在0~5℃下,将5-硝基呋喃-2-碳酰氯(1.01g,5.76mmol)加入到含有化合物5a(1.00g,4.80mmol)和dipea(1.24mg,9.6mmol)的无水dcm(10ml)溶液中。室温搅拌3小时。抽滤并用少量dcm洗涤,得到黄色固体化合物6(1.42g,85%产率)。1h nmr(400mhz,dmso)δ10.53(s,1h),9.35(s,1h),7.80(d,j=3.9hz,1h),7.62-7.58(m,3h),7.44(d,j=8.7hz,2h),1.48(s,9h).
[0068]
将化合物6溶解于etoac(5ml)中,然后向反应中添加4m盐酸二氧六环溶液(0.42ml,1.68mmol)。反应在室温下搅拌6小时。得到化合物7。1h nmr(400mhz,dmso)δ10.27(s,1h),7.79(d,j=3.7hz,1h),7.55(d,j=3.7hz,1h),7.35(d,j=8.4hz,2h),6.56(d,j=8.4hz,2h),5.06(s,2h).
[0069]
s4.将化合物7(1.5g,6.1mmol)、4-环戊烯-1,3-二酮(0.71g,7.3mmol)和n,n-二异丙基乙胺(0.94g,7.3mmol)溶解在无水四氢呋喃(15ml)中加热回流反应4h,减压蒸除溶剂,残留物添加25ml水。缓慢滴加稀盐酸将ph值调节至3-4,过滤所得沉淀物,用水洗得到得化合物8(1.5g,0.71mmlol)(1.54g,85%产率)。1h nmr(400mhz,dmso)δ13.12(s,1h),10.64(s,1h),10.48(s,1h),7.81(d,j=3.5hz,1h),7.71(d,j=8.7hz,2h),7.65(s,1h),7.63(s,2h),6.48(d,j=12.1hz,1h),6.32(d,j=12.0hz,1h).
[0070]
式(ⅰ)化合物的制备
[0071]
将制备的化合物4(0.11g,0.29mmol)、化合物8(0.1g,0.29mmol)、2-(7-氮杂苯并三氮唑)-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(0.12g,0.32mmol)和n,n-二异丙基乙胺(0.11g,0.86mmol)在n,n-二甲基甲酰胺(2ml)中反应,室温下搅拌10小时后,将反应混合物倒入水
(20ml)中并用etoac(3
×
20ml)萃取。合并的有机相并用饱和食盐水洗涤,经无水硫酸钠干燥。减压浓缩溶剂,残余物经柱层析(dcm/meoh=150:1~50:1)纯化,即得式(ⅰ)化合物(0.17g,产率47%)。1h nmr(400mhz,dmso)δ11.45(s,1h),11.09(s,1h),10.60(s,1h),8.70(t,j=5.3hz,1h),7.82(d,j=3.9hz,1h),7.69(d,j=9.0hz,2h),7.65
–
7.61(m,3h),7.59
–
7.54(m,1h),7.08(d,j=8.6hz,1h),7.01(d,j=7.0hz,1h),6.53(t,j=5.6hz,1h),6.27(s,2h),5.05(dd,j=12.9,5.3hz,1h),3.31
–
3.26(m,2h),3.16(dd,j=12.4,6.4hz,2h),2.92
–
2.81(m,1h),2.68
–
2.55(m,2h),2.05
–
1.99(m,1h),1.58(s,2h),1.47(d,j=6.0hz,2h),1.36(s,4h).
13
c nmr(101mhz,dmso)δ173.21,170.51,169.35,167.70,165.22,163.36,154.73,152.13,148.46,146.82,136.66,135.82,133.80,133.19,132.59,131.55,129.19,121.63,121.05,120.01,117.55,116.74,113.89,110.77,109.43,55.31,48.95,42.19,39.14,31.38,29.09,29.04,26.57,26.44,22.56.hrms m/z:calcd for c34h33n7o10[m+h]
+
700.2289,found 700.2357.hplc:tr 15.442min,purity 99.115%.
[0072][0073]
对比例1
[0074]
选择sting小分子抑制剂c-170,结构式为;
[0075]
对比例2
[0076]
选择pomalidomide,结构式为:
[0077]
对比例3
[0078]
选择mg132,结构式为:
[0079]
对比例4
[0080]
选择bafilomycin,结构式为:
[0081]
性能测试
[0082]
将实施例1制备的式(ⅰ)化合物、对比例1~4的化合物进行体外降解试验,步骤如下:
[0083]
(1)细胞培养:人单核细胞白血病细胞(thp-1)在含有10%胎牛血清(gibco,usa),0.05mmβ-巯基乙醇(macklin,chn)和1%青霉素链霉素(gibco,usa)的rpmi 1640(gibco,usa)中培养和维持。在37℃下,将细胞保持在95%空气/5%二氧化碳的增湿培养箱中。
[0084]
(2)western blot实验:将细胞均匀接种到6孔板中,以不同浓度的式(ⅰ)以及对比例1~4的化合物孵育。用含有1%蛋白酶抑制剂和1%磷酸酶抑制剂的ripa裂解缓冲液收集全细胞裂解液。蛋白质浓度通过bca分析进行定量,等量的蛋白质通过10%sds-page电泳,转移到聚偏二氟乙烯转移膜(pvdf,0.45μm)上,并在4℃下针对不同的靶抗体孵育过夜。主要抗体包括β-tublin抗体(fd0064,fdbio science)、抗sting抗体(ab181125,abcam)。蛋白质表达水平标准化为β-tublin。使用多功能成像分析系统(美国fluorchem r)暴露条带并记录图像。使用imagej软件评估每个靶带的灰度。
[0085]
将实施例1制备的式(ⅰ)化合物进行体内抗炎疗效研究
[0086]
体内抗炎疗效研究:8周龄c57bl/6j雄性小鼠购自广州南方医科大学实验动物技术开发有限公司,经24小时昼夜循环、自由饮食和饮水后,随机分为4组:对照组、顺铂诱导模型组(25mg/kg)、低剂量治疗组(1,30mg/kg)和高剂量治疗组(1,60mg/kg).小鼠在顺铂注射前1小时腹腔注射式(ⅰ)化合物,并每天在同一时间连续给药。顺铂治疗后72小时后,眼球取血,离心分离血清,使用血清生化自动分析仪测定肌酐(creatinine)和尿素氮(bun)浓度。
[0087]
从图1看,上述体内外实验表明,本发明所述化合物1能够通过泛素-蛋白酶体途径有效地降解thp-1细胞系中的sting蛋白,且降解50%蛋白时的剂量dc
50
=3.2μm。
[0088]
从图2看,其降解效果比对比例1的c-170抑制剂、对比例2的pomalidomide、对比例3和对比例4的化合物更显著。另外,bafilomycin属于溶酶体体抑制剂,将式(ⅰ)化合物和bafilomycin混合孵育验证了降解过程并不是通过溶酶体途径降解。
[0089]
另外,在图2中,mg132的浓度是100nm是通过预试验确定的最终浓度。分别初筛了10μm、1μm和100nm。结果发现,10μm和1μm两个浓度的作用之后细胞出现明显的死亡,细胞活力下降,从而导致western blot实验不准确。最终选择对thp-1细胞活力没有影响的100nm作为试验浓度。
[0090]
从图3看,在顺铂-aki小鼠模型中,分别设置了对照组、顺铂诱导模型(25mg/kg)、低剂量治疗组(1,30mg/kg)和高剂量治疗组(1,60mg/kg),实施例1制备的式(ⅰ)化合物在顺铂-aki小鼠模型中显示出高效的体内抗炎功效,并有效保护小鼠免受顺铂诱导的肾脏损伤。其中当剂量为60mg/kg时,式(ⅰ)化合物的的抗炎效果更好。
[0091]
从图4~6看,通过核磁共振氢谱图、碳谱图和质谱图已经证明合成了式(ⅰ)化合物。
[0092]
显然,本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求
的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1