一种去氧胆酸有关物质及其制备方法和检测方法与流程
1.本发明属于化学制药领域,具体而言涉及一种去氧胆酸有关物质h及其制备方法和检测方法。
背景技术:
2.随着我国人均可支配收入和消费水平的提高,人们对医疗美容的需求也日益增多。从美容的角度来看,身体局部脂肪沉积是影响美观的,特别是脸部的脂肪沉积。例如,有双下巴或深颊,会使脸可能看起来更大。为了克服这个问题,以减脂为目的的运动方案和饮食控制方案层出不穷,但效果有限。
3.目前有效的去除颏下脂肪(双下巴)的主要手段有包括脂肪吸除手术、抽脂手术、脂肪吸除手术切口等在内的外科手术疗法。然而,外科疗法的局限性在于它们需要几个星期才能痊愈,某些患者(例如,糖尿病患者)的治愈期更长,并且存在产生副作用的风险,包括出血过多、内部器官损伤、细菌感染、瘢痕或疼痛等。
4.kythera公司开发的去氧胆酸皮下注射制剂在欧洲获批上市,商品名:belkyra,是含去氧胆酸的透明无色、无菌溶液,去氧胆酸的结构式如下:
[0005][0006]
任何影响药物纯度的物质统称为杂质,药物杂质一般包括:有机杂质、无机杂质及残留溶剂。有机杂质主要指由生产工艺引入或物质降解而产生的杂质,有机杂质的化学结构一般与活性成分类似或具渊源关系,故通常又可称之为有关物质。“有关物质”是指“在既定工艺进行生产和正常贮藏过程中可能含有或产生并需要控制的杂质,改变生产工艺时需另考虑增修订有关项目”。
[0007]
出于人用药安全考虑,在药物活性成分的产品商业化之前,国内和国际药品监管机构都会建立很低的杂质质控限度。通常已知杂质的质控限度为0.15%,但未知杂质的质控限度通常会小于0.10%。杂质的研究是药品研发的一项重要内容,它包括选择合适的分析方法,准确地分辨与测定杂质的含量并综合药学、毒理、临床研究的结果确定杂质的合理限度。
[0008]
中间体12(结构如下式12)是去氧胆酸的制备过程中的关键中间体,中间体12经过水解步骤能够制备得到去氧胆酸。因此对中间体12的质量控制,特别是中间体12中基因毒杂质的检测至关重要,目前对中间体12中的基因毒杂质的检测方法鲜有报道。
[0009]
技术实现要素:
[0010]
本发明的目的在于提供一种去氧胆酸有关物质h(结构见下式h)及其制备方法和检测方法。该有关物质h能够用作药品研发过程中的标准品和对照品,该去氧胆酸有关物质h的制备方法简单,纯度高、收率好。该有关物质h检测方法可以检测去氧胆酸中间体12中杂质h的含量和纯度,也可以检测式h化合物本身的纯度,该方法操作灵敏、专属、快速、准确。
[0011][0012]
有关物质h是中间体12生产过程中可能产生的副产物,其环氧结构为基因毒性警示结构,根据ich m7采用ttc法计算该杂质限度,ttc为1.5μg/天,去氧胆酸日剂量为100mg,故去氧胆酸中基因毒杂质限度为15ppm,中间体12到去氧胆酸的收率按40%计算,该杂质在中间体12中限度为6ppm。
[0013]
本发明提供了一种式h化合物或其盐、水合物、立体异构体,式h化合物结构式如下:
[0014][0015]
在一些实施方案中,式h化合物的纯度为95%以上;在一些实施方案中,式h化合物的纯度为97%以上;在一些实施方案中,式h化合物的纯度为98%以上;在一些实施方案中,式h化合物的纯度为99%以上。
[0016]
本发明还提供了一种式h化合物的制备方法,其反应路线如下:
[0017][0018]
进一步地,所述制备方法包括如下步骤:
[0019]
在非质子溶剂的存在下,将中间体9与氧化剂混合,视情况进行搅拌反应,反应完成后,进行柱层析分离,制得式h化合物。
[0020]
在上述式h化合物制备方法的一些实施方案中,所述非质子溶剂选自二氯甲烷、氯仿、四氯化碳中的一种,所述氧化剂为间氯过氧苯甲酸、过氧苯甲酸中的一种。
[0021]
另外,本技术还提供了式h化合物用作标准品或者对照品的应用。本技术还提供了式h化合物在去氧胆酸中间体12或去氧胆酸的杂质检查时作为标准品的用途。
[0022]
本发明还进一步地提供了一种去氧胆酸有关物质h的检测方法,
[0023][0024]
其包括以下步骤:
[0025]
a)将有关物质h对照品溶于溶剂中,配制成一定浓度的有关物质h对照品溶液,对有关物质h对照品溶液浓度记为cs(ng/ml);
[0026]
b)在常压下,采用高效液相色谱-质谱分析方法测定有关物质h对照品溶液中主峰峰面积as;
[0027]
c)将中间体12溶于溶剂中,配制成一定浓度的中间体12的供试品溶液,中间体12的供试品溶液浓度记为ci(mg/ml);
[0028]
d)在常压下,采用高效液相色谱-质谱分析方法测定中间体12供试品溶液中有关物质h的峰面积ai;
[0029]
e)按照以下公式
①
、
②
计算去氧胆酸中间体12中有关物质h的浓度和含量。
[0030]
①
去氧胆酸中间体12中有关物质h的浓度计算公式为:
[0031][0032]
②
去氧胆酸中间体12中有关物质h的含量计算公式为:
[0033][0034]
上式
①
、
②
中:
[0035]
cs:对照品溶液的浓度(ng/ml),ci:样品溶液的浓度(mg/ml),
[0036]
ai:样品溶液中杂质峰面积,as:对照品溶液中主峰峰面积,
[0037]
p:对照品含量(如含量为99.8%,带入0.998进行计算)。
[0038]
在上述有关物质h的检测方法的一些实施方案中,其中,步骤a)和c)中,所述溶剂为乙腈。
[0039]
在上述有关物质h的检测方法的一些实施方案中,其中,步骤a)中,所述有关物质h对照品溶液浓度范围为3.6030~36.0300ng/ml,优选地为18ng/ml。
[0040]
在上述有关物质h的检测方法的一些实施方案中,其中,步骤c)中,所述中间体12供试品溶液浓度ci的范围为1-9mg/ml,优选地为3mg/ml。
[0041]
在上述有关物质h的检测方法的一些实施方案中,其中,所述高效液相色谱-质谱分析法的条件为:高效液相色谱仪、检测器为质谱检测器(ms)、色谱柱用十八烷基硅烷键合硅胶为填充剂,优选的色谱柱为agilent eclipse plus c8色谱柱,进一步优选地色谱柱为agilent eclipse plus c8 3.5μm 4.6
×
100mm或与其效能相当色谱柱;流动相a为水,流动相b为乙腈-甲醇(80:20),流动相c为20mmol/l甲酸铵溶液,按下表1进行线性梯度洗脱;柱温30℃,流速1.0ml/min,进样量20μl。离子源为电喷雾离子源(esi),正离子模式,定量离子为526.2;
[0042]
表1
[0043][0044]
在上述有关物质h检测方法的具体实施方案中,本发明提供了一种去氧胆酸有关物质h的检测方法,
[0045][0046]
其包括以下步骤:
[0047]
a)将有关物质h对照品溶于溶剂中,配制成18ng/ml浓度的对照品溶液,对照品溶液浓度记为cs(ng/ml);
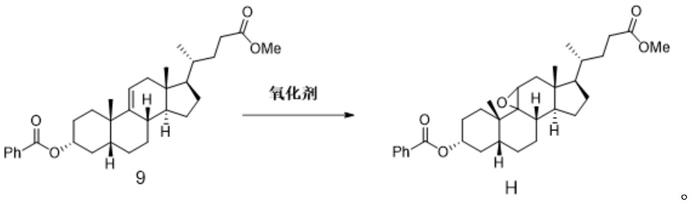
[0048]
b)在常压下,采用高效液相色谱-质谱分析测定有关物质h对照品溶液中主峰峰面积as;
[0049]
c)将中间体12溶于溶剂中,配制成3mg/ml浓度的供试品溶液,供试品溶液浓度记
为ci(mg/ml);
[0050]
d)在常压下,采用高效液相色谱-质谱分析测定中间体12供试品溶液中有关物质h的峰面积ai;
[0051]
e)按照以下公式
①
、
②
计算去氧胆酸中间体12中有关物质h的浓度和含量。
[0052]
①
去氧胆酸中间体12中有关物质h的浓度计算公式为:
[0053][0054]
②
去氧胆酸中间体12中有关物质h的含量计算公式为:
[0055][0056]
上式
①
、
②
中:
[0057]
cs:对照品溶液的浓度(ng/ml),ci:样品溶液的浓度(mg/ml),
[0058]
ai:样品溶液中杂质峰面积,as:对照品溶液中主峰峰面积,
[0059]
p:对照品含量(如含量为99.8%,带入0.998进行计算)。
[0060]
在上述测定方法的一些实施方案中,其中,步骤a)和b)中,所述溶剂为乙腈。
[0061]
在上述测定方法的一些实施方案中,其中,所述高效液相色谱-质谱分析法的条件为:高效液相色谱仪、检测器为质谱检测器(ms)、色谱柱用十八烷基硅烷键合硅胶为填充剂,优选的色谱柱为agilent eclipse plus c8色谱柱,进一步优选地色谱柱为agilent eclipse plus c8 3.5μm 4.6
×
100mm或与其效能相当色谱柱;流动相a为水,流动相b为乙腈-甲醇(80:20),流动相c为20mmol/l的甲酸铵溶液,按上表1进行线性梯度洗脱;柱温30℃,流速1.0ml/min,进样量20μl。离子源为电喷雾离子源(esi),采用正离子模式,定量离子为526.2。
[0062]
有益效果
[0063]
药物中的杂质会不同程度地影响药物的稳定性、安全性和有效性,本发明提供了一种去氧胆酸的全新杂质h以及其制备方法和检测方法,该制备方法操作简单、收率高,通过该方法可以快速得到具有高纯度的式h化合物,从而作为去氧胆酸杂质标准品和对照品。该分析方法能够将该多种性质极其接近的杂质以及去氧胆酸中间体进行有效的分离,对去氧胆酸的质量控制具有重要作用。
[0064]
本发明提供的去氧胆酸有关物质h检测方法在于,配制一定浓度的有关物质h对照品溶液,用高效液相色谱-质谱联用法测定对照品和供试品溶液中有关物质h的峰面积,通过外标法计算有关物质h的含量,灵敏度高、专属性强,从而能准确的定量测定杂质含量。
[0065]
常规检测方法采用紫外检测器,检测限为0.1μg/ml,供试品浓度按3mg/ml计算,检测限约为300ppm,远远达不到6ppm限度所需浓度;而本法检测限为1.8ng/ml,相同供试品浓度下,可检测0.6ppm,限度的10%含量的杂质均能检出。
[0066]
另外,中间体12中含有较多结构类似杂质,存在多个杂质与杂质h具体相同的保留时间、hplc峰重合,常规高效液相色谱-紫外检测器分离较为困难,干扰水平达到300ppm,紫外检测器本身没有分离功能,仅仅是将色谱柱分离洗脱得到的光信号转化为电信号输出,专属性差,无法准确定量该杂质含量;而高效液相色谱-质谱联用技术是在在上述色谱基础上,利用各杂质的分子离子峰或碎片离子峰的不同进行二次分离,专属性好,故能准确定量。
附图说明
[0067]
图1为实施例3中有关物质h对照品溶液的质谱图;
[0068]
图2为实施例3中间体12供试品溶液质谱图;
[0069]
图3为实施例6中有关物质h的峰面积-浓度线性曲线图。
具体实施方式
[0070]
定义和说明
[0071]
除非另有说明,本文所用的下列术语和短语旨在含有下列含义。一个特定的短语或术语在没有特别定义的情况下不应该被认为是不确定的或不清楚的,而应该按照普通的含义去理解。当本文出现商品名时,旨在指代其对应的商品或其活性成分。
[0072]“ich”是人用药品注册技术国际协调会议,英文全称为international conference on harmonization。ich m7为该组织发布的其中一个指导原则,题目是“为限制潜在致癌风险而对药物中dna活性(诱变性)杂质进行的评估和控制”,英文题目为“assessment and control of dna reactive(mutagenic)impurities in pharmaceuticals tolimit potential carcinogenic risk”。
[0073]
本发明中,术语“ttc法”是计算基因毒杂质限度的一种方法,基于ttc计算的可接受日摄入量1.5μg/天被认为是在终生每日暴露情况下可以受到保护的量。
[0074]
本发明中,术语“系统适用性实验”按中国药典规定的含义,色谱系统的适用性实验通常包括理论板数、分离度、灵敏度、拖尾因子和重复性等五个参数。本发明系统适用性评价指标主要灵敏度和重复性。本发明中系统适用性可接受对照为:灵敏度溶液中主峰的信噪比应不小于10,所有对照品溶液峰面积的相对标准偏差应不得大于30%。
[0075]
本发明中,术语“准确度”系指用所建立方法测定的结果与真实值或参比值接近的程度,一般用回收率(%)表示。
[0076]
本发明中,术语“含量”是指有关物质h的质量与中间体12的质量的比值,单位为ppm。
[0077]
本发明中,术语“式h化合物”“有关物质h”、“杂质h”可以互换使用,均是指下式h所示的化合物
[0078]
本发明中,术语“稀释剂”均是指乙腈。
[0079]
本发明中,除另有说明外,所用到的水或所指的水溶液是指分析领域通常所使用分析用水,包括但不限于纯化水、超纯水、去离子水等。
[0080]
本发明中,无论在具体数值之前是否有“约”表示,都是指该具体数值的可以在本领域认可的范围内波动,具体地可以是例如具体数值的绝对值
±
10%、
±
9%、
±
8%、
±
8%、
±
7%、
±
6%、
±
5%、
±
4%、
±
3%、
±
2%或
±
1%内波动。下面会通过实施例具体描述本发明,这些实施例并不意味着对本发明的任何限制。
[0081]
本发明所使用的所有溶剂是市售的,无需进一步纯化即可使用。
[0082]
本发明采用下述缩略词:ms代表质谱;μg/ml代表微克/毫升;ppm代表百万分之一,是重量比;pe代表石油醚;ea代表乙酸乙酯;esi代表电喷雾离子源;r代表线性相关系数,线性相关系数越大,定量能力越好;rsd代表相对对照偏差。
[0083]
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数按重量计算。
[0084]
下面结合具体实施例,进一步阐述本技术。应理解,这些实施例仅用于说明本技术而不用于限制本技术的保护范围。
[0085]
下列实施例中未注明具体条件的试验方法,可按照常规条件或按照制造厂商所建议的条件。除非另行定义,本文所使用的所有专业与科学用语与本领域熟练人员所熟悉的意义相同。
[0086]
实施例1去氧胆酸有关物质h的制备
[0087][0088]
取中间体9(3.00g,6.09mmol),溶于30ml氯仿,加入间氯过氧苯甲酸(2.20g,12.79mmol),室温搅拌反应至反应完全,反应液用50ml饱和亚硫酸钠洗涤,50ml饱和碳酸氢钠溶液洗涤,50ml饱和食盐水洗涤,无水硫酸钠干燥后制沙柱层析分离(v
pe
:v
ea
=20:1),得固体2.97g,纯度为99.87%。
[0089]1h nmr(500mhz,cdcl3)δ8.04(m,2h),7.53(t,j=7.4hz,1h),7.42(m,2h),4.97(d,j=17.8hz,1h),3.67(s,3h),3.20(s,1h),2.35-2.22(m,1h),2.22(m,1h),2.16-2.01(m,6h),1.88(m,2h),1.88-1.77(m,3h),1.74-1.48(m,5h),1.48-1.26(m,2h),1.17(d,j=8.3hz,3h),1.17-1.11(m,3h),0.89(d,3h),0.65(s,3h).
13
c nmr(125mhz,cdcl3)δ174.6(1c,c
24
),166.2(1c,c
26
),132.6(1c,c
30
),130.9(1c,c
27
),129.6(2c,c
28,c32
),128.2(2c,c
29,c31
),74.4,67.5,56.2,51.48,51.4,45.0,41.8,40.6,40.1,36.4,35.62,35.0,34.7,33.5,31.0,30.8,28.9,28.2,28.2,26.7,24.2,23.94(1c,c
21
),17.9(1c,c
19
),14.0(1c,c
18
).ms m/z:531.1[m+na]
+
。
[0090]
实施例2去氧胆酸有关物质h的制备
[0091][0092]
取中间体9(3.00g,6.09mmol),溶于30ml二氯甲烷,加入过氧苯甲酸(1.77g,12.79mmol),室温搅拌反应至反应完全,反应液用50ml饱和亚硫酸钠洗涤,50ml饱和碳酸氢
钠溶液洗涤,50ml饱和食盐水洗涤,无水硫酸钠干燥后制沙柱层析分离(v
pe
:v
ea
=20:1),得固体3.00g,纯度为99.83%。
[0093]
实施例3本发明的有关物质h的检测方法
[0094]
1.仪器与分析条件
[0095]
安捷伦高效液相色谱仪连接ab sicx三重四级杆质谱,色谱柱为agilent eclipse plus c8 4.6
×
100mm,3.5um,流动相a为水,流动相b为乙腈-甲醇(80:20),流动相c为20mmol/l甲酸铵溶液,按下表2进行线性梯度洗脱;柱温30℃,流速1.0ml/min,进样量20μl。离子源为电喷雾离子源(esi),正离子模式,定量离子为526.2。稀释剂为乙腈。
[0096]
表2
[0097][0098]
2.对照品溶液和供试品溶液的配制
[0099]
对照品溶液的配制:准确称量有关物质h对照品36mg,置200ml量瓶中,加适量稀释剂使溶解并稀释至刻度,摇匀。精密移取1.0ml至100ml量瓶中,加稀释剂稀释至刻度,摇匀。精密移取1.0ml至100ml量瓶中,加稀释剂稀释至刻度,摇匀即得。
[0100]
灵敏度溶液的配制:转移2ml对照品溶液到10ml量瓶中,加稀释剂稀释至刻度,摇匀即得。
[0101]
供试品溶液的配制:准确称量中间体12样品60mg,置20ml量瓶中,加适量稀释剂使溶解并稀释至刻度,摇匀即得。
[0102]
3.实验过程
[0103]
取溶剂20μl,注入高效液相色谱仪,记录色谱图,溶剂应无干扰。
[0104]
取灵敏度溶液20μl,注入高效液相色谱仪,记录色谱图,灵敏度溶液中主峰的信噪比应不小于10。
[0105]
取供试品溶液20μl注入高效液相色谱仪,记录色谱图。按照以下公式
①
、
②
计算去氧胆酸中间体12中有关物质h的浓度和含量。
[0106]
①
去氧胆酸中间体12中有关物质h的浓度计算公式为:
[0107][0108]
②
去氧胆酸中间体12中有关物质h的含量计算公式为:
[0109][0110]
上式
①
、
②
中:
[0111]
cs:对照品溶液的浓度(ng/ml),ci:样品溶液的浓度(mg/ml),
[0112]
ai:样品溶液中杂质峰面积,as:对照品溶液中主峰峰面积,
[0113]
p:对照品含量(如含量为99.8%,带入0.998进行计算)。
[0114]
实施例4本发明有关物质h的检测方法的专属性和系统适用性考察试验
[0115]
本实验的目的在于确认实施例1的杂质h的检测方法是否符合系统适用性的要求,系统适用性考察实验的可接受对照为:6针对照品溶液有关物质h峰面积的rsd应≤30.0%,灵敏度溶液中主峰的信噪比不小于10。
[0116]
1.仪器及分析条件:同实施例1.
[0117]
2.溶液配制:参照实施例1方法配制对照品溶液、灵敏度溶液及供试品溶液。
[0118]
3.实验过程:
[0119]
取稀释剂20μ1,注入高效液相色谱仪,记录色谱图,由色谱图可知,溶剂无干扰,方法专属性强。
[0120]
取灵敏度溶液20μl,注入高效液相色谱仪进行灵敏度试验,记录色谱图。灵敏度溶液中主峰的信噪比为51。
[0121]
取20μ1对照品溶液注入高效液相色谱仪,连续进样6针考察进样精密度,具体结果见下表3。
[0122]
表3系统进样精密度结果
[0123]
进样次数123456rsd峰面积9460009390009440009040009360009220001.7%
[0124]
通过上述实验和结果可知:灵敏度试验中,灵敏度溶液中主峰的信噪比为51,不小于10,方法灵敏;精密度试验中,6针对照品溶液的主峰面积的rsd为1.7%,远低于30.0%,进样精密度良好。
[0125]
实施例5本发明有关物质h的检测方法的检测限和定量限
[0126]
本实验的目的在于:确定该方法条件下,被测物可被准确定量的最低限度(定量限)和可被检测到的最低限度(检测限)。
[0127]
1.仪器及分析条件同实施例1。
[0128]
2.溶液配制
[0129]
定量限溶液:精密称取有关物质h对照品36.47mg,置200ml量瓶中,加稀释剂溶解并稀释至刻度,摇匀。精密移取1.0ml上述溶液至100ml量瓶中,加稀释剂稀释至刻度,摇匀。精密移取1.0ml至100ml量瓶中,加稀释剂稀释至刻度,摇匀。转移2.0ml到10ml量瓶中,加稀释剂稀释至刻度,摇匀,即得。
[0130]
检测限溶液:转移5ml定量限溶液至10ml量瓶中,用稀释剂定容,摇匀,即得。
[0131]
3.实验过程及结果
[0132]
系统适用性按实施例2方法验证合格后,依次取连续取6次定量限和3次检出限溶液各20μl,注入高效液相色谱仪,记录色谱图,记录峰面积,考察定量限和检出限的信噪比及定量限溶液峰面积的相对标准偏差,具体结果见下表4、表5。
[0133]
3针检出限溶液有关物质h峰平均信噪比为14,大于3,通过;
[0134]
6针定量限溶液有关物质h峰平均信噪比为35,大于10,通过;
[0135]
6针定量限溶液有关物质h峰的峰面积的%rsd为3,小于30,通过。
[0136]
表4:有关物质h的定量限结果
[0137][0138]
表5:有关物质h的检出限结果
[0139][0140]
通过上述实验和结果可知,本发明所述有关物质h检测方法的检出限浓度为1.8ng/ml,为限度水平的10%,本发明所述检测方法的定量限浓度为3.6ng/ml,为限度浓度的20%,综上可知,本发明所述有关物质h的检测方法能准确、灵敏地检出杂质h。
[0141]
实施例6本发明有关物质h的检测方法的线性试验
[0142]
本实验的目的在于:确定在预先设定的范围内,被测溶液的测定结果与浓度之间的线性关系。
[0143]
1.仪器及分析条件同实施例1。
[0144]
2.溶液配制
[0145]
线性储备液:精密量取有关物质h对照品36mg,置200ml量瓶中,加稀释剂稀释至刻度,摇匀。精密移取1.0ml上述溶液至100ml量瓶中,加稀释剂稀释至刻度,摇匀。即得。
[0146]
200%线性溶液:精密转移2.0ml线性储备液于100ml量瓶中,加稀释剂稀释至刻度,摇匀。
[0147]
100%线性溶液:精密转移50.0ml200%溶液于100ml量瓶中,加稀释剂稀释至刻度,摇匀。记为100%。
[0148]
80%线性溶液:精密转移8.0ml100%溶液于10ml量瓶中,加稀释剂稀释至刻度,摇匀。记为80%。
[0149]
50%线性溶液:精密转移5.0ml100%溶液于10ml量瓶中,加稀释剂稀释至刻度,摇匀。记为50%。
[0150]
20%线性溶液:精密转移2.0ml100%溶液于10ml量瓶中,加稀释剂稀释至刻度,摇匀。记为20%。
[0151]
3.实验过程及结果
[0152]
系统适用性按实施例2方法验证合格后,依次取20%、50%、80%、100%、200%各浓度的线性溶液20μl,注入高效液相色谱仪,记录色谱图,记录峰面积,以峰面积a为纵坐标,浓度(ng/ml)为横坐标进行线性回归,相关系数r应≥0.990。线性结果见下表6。
[0153]
表6线性结果
[0154]
样品浓度(ng/ml)峰面积a20%线性溶液3.603020200050%线性溶液9.007552700080%线性溶液14.4120735000100%线性溶液18.0150925000200%线性溶液36.03001910000线性方程y=52196.4557x+13512.7660r=0.9988
[0155]
通过上述试验和结果可知,杂质h在3.6030~36.0300ng/ml浓度范围内与峰面积呈良好线性关系,线性相关系数r为0.9988。
[0156]
实施例7本发明有关物质h的检测方法准确度考察试验
[0157]
本实验的目的在于:确定该色谱方法在预期线性范围内的测定结果与真实值的差异,从而确认是否该方法可以获得准确的测试结果。
[0158]
1.仪器及分析条件同实施例1。
[0159]
2.溶液配制
[0160]
100%水平对照品溶液(即对照品溶液):精密量取有关物质h对照品36.11mg于200ml量瓶中,加稀释剂稀释至刻度,摇匀。精密移取2.0ml上述溶液至200ml量瓶中,加稀释剂稀释至刻度,摇匀,记为对照品储备液。精密移取2.0ml对照品储备液至200ml量瓶中,加稀释剂稀释至刻度,摇匀,记为对照品溶液(即100%水平对照品溶液)。
[0161]
200%水平对照品溶液:精密转移2.0ml对照品储备液于加入适量稀释剂的100ml量瓶中,加稀释剂稀释至刻度,摇匀,即得。
[0162]
20%水平的对照品溶液(即灵敏度溶液):转移20.0ml对照品溶液到100ml量瓶中,加稀释剂稀释至刻度,摇匀,即得。
[0163]
准确度溶液的配置:
[0164]
未加标溶液:精密称量60mg中间体12样品至20ml量瓶中,加稀释剂稀释使样品全部溶解,定容至刻度线,摇匀即得,配制双份,标记为未加标溶液-01、未加标溶液-02。
[0165]
20%水平加标溶液:精确称量60mg中间体12样品至20ml量瓶中,加20%水平的对照品溶液稀释使样品全部溶解,定容至刻度,摇匀即得,配制三份。
[0166]
100%水平加标溶液;精确称量59.45mg中间体12样品至20ml量瓶中,加100%水平的对照品溶液稀释使样品全部溶解,定容至刻度,摇匀,即得,配制六份。
[0167]
200%水平加标溶液:精确称量59.33mg中间体12样品至20ml量瓶中,加200%水平的对照品溶液稀释使样品全部溶解,定容至刻度,摇匀,即得200%水平加标溶液;
[0168]
3.实验过程及结果
[0169]
系统适用性按实施例2方法验证合格后,取20%水平加标溶液,100%水平加标溶液和200%水平加标溶液各进样20μ1,计算含量及回收率,各浓度水平项下回收率的平均值应为84%~91%,回收率的rsd均≤5.0%。准确度结果见下表7。
[0170][0171]
公式中:
[0172]cm
是加标溶液中有关物质h的含量,单位ppm
[0173]cc
是未加标溶液中有关物质h的平均含量,单位ppm
[0174]cg
是加标溶液中有关物质h的理论加样量,单位ppm
[0175]cg
=(有关物质h加样量/中间体12样品量)
×
106[0176]
表7准确度结果
[0177][0178]
通过试验可知,杂质h各浓度项下回收率的平均值为84%~91%,回收率的rsd分别为4.5%、3.5%、1.0%,准确度良好,证明本方法对去氧胆酸的测定准确可行。
[0179]
以上,对本发明的实施方式进行了说明。但是,本发明不限定于上述实施方式。凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1