一种坎地沙坦中间体及坎地沙坦的制备方法与流程
1.本发明涉及一种坎地沙坦中间体制备方法及利用该中间体制备坎地沙坦的方法,属于原料药制备方法的技术领域。
背景技术:
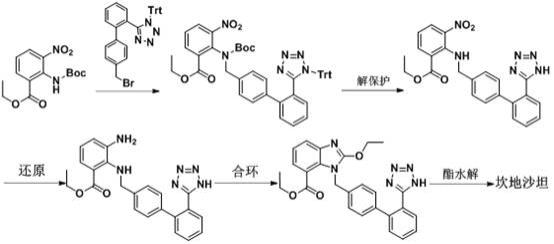
2.坎地沙坦(candesartan,1)因具有高选择性的血管紧张素ii型受体(at1)阻断活性从而表现出很好的临床疗效,临床上广泛的用于高血压的治疗。该药由日本武田公司和瑞典阿斯特拉公司共同开发完成,于1997年在瑞典首先上市,在2003年阿斯利康在全球销售额达到7.5亿美元。坎地沙坦的合成已有大量报道,如:ep 459136,us 5196444 a,cn 1204125c,cn 1207287c等。依据四氮唑结构引入方式不同,典型的合成策略主要有两大类:第一类,是在主体结构形成后直接构建四氮唑。以专利ep 459136,us 5196444 a等为代表,以氰基联苯取代的苯并咪唑衍生物(3)为原料,采用三烷基氯化锡、叠氮化钠与氰基在100℃以上高温长时间反应,得到含四氮唑结构中间体(4),再经水解制得坎地沙坦(1)。该路线作为坎地沙坦/酯合成的最初方案,被国内外许多公司沿用至今。此法制备过程中用到的叠氮化钠是剧毒化合物,且极易爆炸,危险性极高;三烷基叠氮化锡毒性大易附着,对人体和环境危害巨大。第二类,是直接引入四氮唑片段。例如专利cn 102887890a,cn 1800179a,cn 101200455a等,均采用了含有四氮唑中间体作为原料,从而避免剧毒易爆炸等高危因素。典型的合成方法如专利cn 1800179a所述:
该路线以2-叔丁氧羰基氨基-3-硝基苯甲酸乙酯(c3)与n-(三苯基甲基)-5-(4
’‑
溴甲基联苯-2-基)四氮唑(bbtt)为起始原料,依次经n-烷基化、解保护、还原、关环、水解等一系列反应制得坎地沙坦。专利报道使用低级醇与乙酸乙酯或二氯甲烷的混合溶剂可以对叔丁氧羰基和三苯甲基进行解保护。但我们对该工艺进行重复验证时,并未得到目标产品:叔丁氧羰基不能顺利脱除,继而影响到后续步骤无法关环,最终未能得到目标产物。同时我们发现,其他文献(ep459136)报道的在酸性条件下脱除叔丁氧羰基(-boc)的方法在解保护步骤中亦不可行,反应过程中三苯甲基(-trt)保护基由四氮唑迁移至原-boc位置,大量生成副产物(5)。此时体系成分复杂,产物较少、副产物混杂,难以提纯。综上,第一类路线生产安全风险高、对人体环境危害性大,应逐步予以淘汰;第二类路线,现有脱保护合环技术实现性差。绿色安全高效的合成工艺需求迫切。
[0003][0004][0005][0006][0007][0008]
技术实现要素:
[0009]
本发明所要解决的技术问题:本发明提供了一种全新的坎地沙坦的制备方法,即解决了现有技术关于生产安全风险高、对人体危害性大的问题,同时也解决了关于脱保护环合过程出现的问题,实现大规模生产坎地沙坦的新方法。
[0010]
本发明的技术方案如下:首先本发明涉及了一种坎地沙坦的中间体2-[n-[[(2
’‑
(n
’‑
三苯甲基)-四氮唑-5-基)[1,1
’‑
联苯]-4-基]甲基]氨基]-3-硝基苯甲酸乙酯盐酸盐(中间体2)的制备方法:2-叔丁氧羰基氨基-3-硝基苯甲酸乙酯(c3)与n-(三苯基甲基)-5-(4
’‑
溴甲基联苯-2-基)四氮唑(bbtt)在碱和相转移催化剂作用下缩合制得中间体1;中间体1在含水的盐酸乙醇溶液中脱掉叔丁氧羰基(-boc),三苯甲基迁移得到中间体2。
[0011]
优选的,本步中中间体1制备过程使用的碱为无机碱,优选为碳酸钾,碳酸钾的投
料方式为分批加入。
[0012]
优选的,本步中相转移催化剂为四丁基溴化铵(tbab)。
[0013]
优选的,本步中通过控制盐酸乙醇溶液中的氯化氢含量和水含量,可以将中间体1完全转化并直接从体系中析出,一步获得高收率高纯度的中间体2。
[0014]
其次本发明还涉及了一种通过中间体2制备坎地沙坦的方法:中间体2在氯化亚锡作用下被还原脱保护得到中间体3;中间体3与原碳酸四乙酯反应得到中间体4;中间体4在碱的作用下水解制得坎地沙坦。
[0015]
优选的,本步中通过氯化亚锡的使用,可以一步实现中间体2的还原与脱保护。
[0016]
发明效果:本发明提供了一种坎地沙坦新的制备方法,具有以下优点:1)该方法避免了传统工艺使用剧毒易爆的叠氮化钠和腐蚀污染的三烷基氯化锡的弊端,提供了一条全新的更为绿色安全、简便高效的合成路线;2)通过控制盐酸乙醇溶液中的氯化氢含量和水含量,可以将中间体1完全转化并直接从体系中析出,一步获得高收率高纯度的中间体2;3)中间体2可以不先脱保护再还原合环,而是在氯化亚锡合水合物的作用下一步实现还原和脱保护,而其他常规氢化还原等方式则没有该效果。
[0017]
具体实施方式:以下对本发明的原理和特征进行描述,所举实施例用于进一步解释本发明,但不限制本发明的范围。
[0018]
实施例1第一步,缩合-保护基迁移:中间体2的制备向1l四口瓶中依次投入c3(56.2g,0.181mol)、bbtt(103.9g,0.186mol)、tbab(2.81g,5%g/g)、无水碳酸钾(62.4g,0.453mol)及丙酮(400g),开启搅拌和加热,回流反应12h。tlc(石油醚:乙酸乙酯=4:1)监控反应完成。趁热抽滤,减压蒸馏回收丙酮,得深色厚油。向残留物中加入二氯甲烷、水各200ml,搅拌洗涤,静置分液弃去水相,减压蒸馏回收二氯甲烷,得深色厚油中间体1。向其中加入配置好预冷的盐酸乙醇溶液(氯化氢含量25%,水含量18%)256g,于20~30℃搅拌反应20h,反应液中析出大量亮黄色固体。hplc监控原料剩余量《1.0%。抽滤,滤饼用无水乙醇淋洗, 45℃鼓风干燥12h得110g亮黄色固体粉末。中间体2纯度96.85.0%。收率为83.9%。
[0019]
中间体2:esi-ms(m/z):709.2517[m+na]+;1h nmr(600 mhz, dmso-d6) δ:8.09 ppm(m, 2h),7.81 ppm(d, 1h, j=7.8 hz),7.62 ppm(t, 1h,j=7.2 hz),7.55 ppm(t, 1h,j=7.2 hz),7.48 ppm(d, 1h, j=7.2 hz),7.32 ppm(m, 9h),7.14 ppm(d, 2h, j=7.8 hz),7.05 ppm(d, 2h, j=8.4 hz),6.85 ppm(m, 7h),4.25 ppm(m, 2h),4.05 ppm(d, 2h, j=4.2 hz),1.26 ppm(t, 3h, j=6.6 hz )。
[0020]
第二步,还原脱保护-合环:中间体4的制备依次将中间体2(36.2g,0.05mol)、氯化亚锡合二水(39.6g,0.175mol)、乙酸乙酯(290g)、盐酸羟胺(1.74g,0.025mol)、水(18.0g)投入1l四口反应瓶中。升温至30℃,保温反应20h。反应液由浑浊变为澄清,颜色加深。tlc监控(乙酸乙酯:石油醚=1:1)反应完成。降至室温,缓慢投入碳酸氢钠固体(41.9g,0.50mol),加毕继续搅拌0.5h,调节溶液ph保持在4~6。抽滤,淋洗,减压蒸除溶剂,得棕色厚油中间体3。向其中加入原碳酸四乙酯(28.8g,0.15mol),甲苯(25g),于25~35℃保温反应20h,析出大量固体。tlc监控反应终点。向反应瓶中加入甲苯(75g),搅拌分散均匀,随后移入冷井中降温至5~10℃,保温打浆2h。抽滤,滤饼用少量甲苯淋洗。50~60℃鼓风干燥至恒重。得类白色固体粉末19.7g。收率为84.2%左右,中间体4主峰纯度96.31%。
[0021]
第三步,水解:坎地沙坦的制备向100ml单口瓶中依次投入中间体4(4.69g,0.01mol)、甲醇(14.0ml),室温搅拌混匀,随后加入预配的氢氧化钠溶液(2.40g氢氧化钠溶于14.0ml水中),升温至45℃,保温反应2h,得澄清溶液。减压蒸除甲醇。向残液中补加14.0ml水,降温至0~10℃后用1m盐酸溶液调ph=4~5,此时析出大量白色固体。保温养晶1h。抽滤,用水淋洗至流出液ph接近中性为止, 60~80℃鼓风干燥至恒重,得白色固体粉末3.98g。本步收率为90.2%。坎地沙坦主峰纯度96.11%。
[0022]
本路线总收率为:63.7%。
[0023]
实施例2第一步,缩合-保护基迁移:中间体2的制备向1l四口瓶中依次投入c3(50g,0.161mol)、bbtt(98.8g,0.177mol)、tbab(2.5g,5%g/g)、无水碳酸钾(55.5g,0.403mol)及丙酮(300g),开启搅拌和加热,回流反应10h。tlc(石油醚:乙酸乙酯=4:1)监控反应完成。趁热抽滤,减压蒸馏回收丙酮,得深色厚油。向残留物中加入二氯甲烷、水各200ml,搅拌洗涤,静置分液弃去水相,减压蒸馏回收二氯甲烷,得深色厚油中间体1。向其中加入配置好预冷的盐酸乙醇溶液(氯化氢含量25%,水含量20%)228.5g,于20~30℃搅拌反应24h,析出大量亮黄色固体。hplc监控原料剩余量《1.0%。抽滤,滤饼用无水乙醇淋洗, 45℃鼓风干燥12h得98.3g亮黄色固体粉末。中间体2纯度94.42%。收率为84.37%。
[0024]
第二步,还原脱保护-合环:中间体4的制备依次将中间体2(36.2g,0.05mol)、氯化亚锡合二水(45.3g,0.20mol)、乙酸乙酯(250g)、盐酸羟胺(1.74g,0.025mol)、水(18.0g)投入1l四口反应瓶中。升温至40℃,保温反应20h。反应液由浑浊变为澄清,颜色加深。tlc监控(乙酸乙酯:石油醚=1:1)反应完成。降至室温,缓慢投入碳酸氢钠固体(41.9g,0.50mol),加毕继续搅拌0.5h,调节溶液ph保持在4~6。抽滤,淋洗,减压蒸除溶剂,得棕色厚油中间体3。向其中加入原碳酸四乙酯(23.1g,
0.12mol),甲苯(25g),于40℃保温反应20h,析出大量固体。tlc监控反应终点。向反应瓶中加入甲苯(75g),搅拌分散均匀,随后移入冷井中降温至0~10℃。保温打浆2h。抽滤,滤饼用少量甲苯淋洗。50~60℃鼓风干燥至恒重。得类白色固体粉末18.8g。收率为80.2%左右。中间体4主峰纯度94.98%。
[0025]
第三步,水解:坎地沙坦的制备向100ml单口瓶中依次投入中间体4(4.69g,0.01mol)、甲醇(14.0ml),室温搅拌混匀,随后加入预配的氢氧化钾溶液(1.68g氢氧化钾溶于14.0ml水中),升温至45℃,保温反应4h,得澄清溶液。减压蒸除甲醇。向残液中补加14.0ml水,降温至0~10℃后用冰乙酸溶液调ph=4~5,此时析出大量白色固体。保温养晶1h。趁冷抽滤,用水淋洗至流出液ph接近中性为止, 60~80℃鼓风干燥至恒重,得白色固体粉末3.56g。本步收率为80.8%。坎地沙坦主峰纯度96.66%。
[0026]
本路线总收率为:54.7%。
[0027]
实施例3:第一步,缩合-保护基迁移:中间体2的制备向1l四口瓶中依次投入c3(50g,0.161mol)、bbtt(98.8g,0.177mol)、tbab(2.5g,5%)、无水碳酸钾(60g,0.483mol)及乙腈(350g),开启搅拌和加热,回流反应12h。tlc(石油醚:乙酸乙酯=4:1)监控反应完成。趁热抽滤,减压蒸馏回收乙腈,向残留物中加入二氯甲烷、水各200ml,搅拌洗涤,静置分液弃去水相,减压蒸馏回收二氯甲烷,得深色厚油。向其中加入配置好预冷的盐酸乙醇溶液(氯化氢含量25%,水含量15%)228.5g,于20~30℃搅拌反应20h,析出亮黄色固体。hplc监控原料剩余《1.0%。抽滤,滤饼用无水乙醇淋洗, 45℃鼓风干燥12h得78.9g黄色固体粉末。收率67.71%;纯度92.72%。
[0028]
第二步,还原脱保护-合环:中间体4的制备将中间体2(36.2g,0.05mol)、氯化亚锡合二水(39.6g,0.175mol)、乙酸乙酯(250g)、盐酸羟胺(1.74g,0.025mol)、水(18.0g)投入1l四口反应瓶中,于30℃保温反应24h。tlc监控(乙酸乙酯:石油醚=1:1)反应完成。缓慢投入碳酸氢钾固体(50.1g,0.50mol),加毕继续搅拌0.5h,溶液ph=4~6。抽滤,淋洗;减压蒸除溶剂,得棕色厚油。向其中加入原碳酸四乙酯(28.8g,0.15mol),甲苯(25g),于10-20℃保温反应24h,析出大量固体。tlc监控反应终点。向反应瓶中加入甲苯(75g),搅拌分散均匀,降温至0~5℃保温1h。抽滤,滤饼用少量甲苯淋洗。50~60℃鼓风干燥至恒重。得类白色固体粉末20.5g。收率87.2%;纯度95.6%。
[0029]
第三步,水解:坎地沙坦的制备向100ml单口瓶中依次投入中间体4(4.69g,0.01mol)、甲醇(14.0ml),室温搅拌混匀,随后加入预配的氢氧化钾溶液(1.68g氢氧化钾溶于14.0ml水中),升温至40℃,保温反应4h,得澄清溶液。减压蒸除甲醇。向残液中补加14.0ml水,降温至0~10℃后用冰乙酸溶液调ph=4~5,此时析出大量白色固体。保温养晶1h。抽滤,用水淋洗至流出液ph接近中性为止, 60~80℃鼓风干燥至恒重,得白色固体粉末3.42g。本步收率为77.62%。坎地沙坦主峰纯度96.49%。
[0030]
本路线总收率为:45.8%。
[0031]
实施例4:第一步,缩合-保护基迁移:中间体2的制备
向1l四口瓶中依次投入c3(50g,0.161mol)、bbtt(98.8g,0.177mol)、tbab(2.5g,5%)、无水碳酸钾(55.5g,0.403mol)及丙酮(300g),开启搅拌和加热,回流反应9h。tlc(石油醚:乙酸乙酯=4:1)监控反应完成。趁热抽滤,减压蒸馏回收丙酮,向残留物中加入二氯甲烷、水各200ml,搅拌洗涤,静置分液弃去水相,减压蒸馏回收二氯甲烷,得深色厚油。向其中加入配置好预冷的盐酸乙醇溶液(氯化氢含量18%,水含量25%)228.5g,于20~30℃搅拌反应18h,析出大量亮黄色固体。hplc监控原料剩余《1.0%。抽滤,滤饼用无水乙醇淋洗, 45℃鼓风干燥12h得97.1g亮黄色固体粉末。收率83.3%;纯度94.35%。
[0032]
第二步,还原脱保护-合环:中间体4的制备将中间体2(36.2g,0.05mol)、氯化亚锡合二水(39.6g,0.175mol)、乙酸乙酯(250g)、盐酸羟胺(1.74g,0.025mol)、水(18.0g)投入1l四口反应瓶中,于40℃保温反应20h。tlc监控(乙酸乙酯:石油醚=1:1)反应完成。缓慢投入碳酸氢钾固体(50.1g,0.50mol),加毕继续搅拌0.5h,溶液ph=4~6。抽滤,淋洗;减压蒸除溶剂,得棕色厚油。向其中加入原碳酸四乙酯(23.1g,0.12mol),甲苯(25g),于10-20℃保温反应24h,析出大量固体。tlc监控反应终点。向反应瓶中加入甲苯(75g),搅拌分散均匀,降温至0~5℃保温1h。抽滤,滤饼用少量甲苯淋洗。50~60℃鼓风干燥至恒重。得类白色固体粉末19.5g。收率83.3%;纯度96.6%。
[0033]
第三步,水解:坎地沙坦的制备向100ml单口瓶中依次投入中间体4(4.69g,0.01mol)、甲醇(14.0ml),室温搅拌混匀,随后加入预配的氢氧化钾溶液(1.68g氢氧化钾溶于14.0ml水中),升温至40℃,保温反应4h,得澄清溶液。减压蒸除甲醇。向残液中补加14.0ml水,降温至0~10℃后用冰乙酸溶液调ph=4~5,此时析出大量白色固体。保温养晶1h。抽滤,用水淋洗至流出液ph接近中性为止, 60~80℃鼓风干燥至恒重,得白色固体粉末3.51g。本步收率为79.66%。坎地沙坦主峰纯度95.43%。
[0034]
本路线总收率为:55.3%。
[0035]
对照例1:第一步,缩合-保护基迁移:中间体2的制备向1l四口瓶中依次投入c3(50g,0.161mol)、bbtt(98.8g,0.177mol)、tbab(2.5g,5%)、无水碳酸钾(55.5g,0.403mol)及丙酮(300g),开启搅拌和加热,回流反应10h。tlc(石油醚:乙酸乙酯=4:1)监控反应完成。趁热抽滤,减压蒸馏回收丙酮,向残留物中加入二氯甲烷、水各200ml,搅拌洗涤,静置分液弃去水相,减压蒸馏回收二氯甲烷,得深色厚油。向其中加入市售盐酸乙醇(氯化氢含量32%,水含量<0.5%)228.5g,于20~30℃搅拌反应24h,析出微量固体。抽滤,45℃鼓风干燥过夜,得4.8g浅黄色固体粉末。收率4.1%;纯度12.02%。
[0036]
对照例2:第一步,缩合-保护基迁移:中间体2的制备向1l四口瓶中依次投入c3(50g,0.161mol)、bbtt(98.8g,0.177mol)、tbab(2.5g,5%)、无水碳酸钾(55.5g,0.403mol)及丙酮(300g),开启搅拌和加热,回流反应10h。tlc(石油醚:乙酸乙酯=4:1)监控反应完成。趁热抽滤,减压蒸馏回收丙酮,向残留物中加入二氯甲烷、水各200ml,搅拌洗涤,静置分液弃去水相,减压蒸馏回收二氯甲烷,得深色厚油。向其中加入配置好预冷的盐酸乙醇溶液(氯化氢含量10%,水含量30%)228.5g,于20~30℃搅拌反应24h,监测原料大量剩余。补加盐酸乙醇溶液(氯化氢含量10%,水含量30%)181.5g,升温50℃
继续反应12h。抽滤,滤饼用少量无水乙醇淋洗,45℃鼓风干燥过夜得76.7g浅黄色固体粉末。收率65.8%;纯度47.40%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1