制备聚合物衍生的陶瓷颗粒的方法与流程
根据美国法典第35篇第119条(e)(1)款,本申请要求2015年1月21日提交的美国临时申请序列号62/106094的优先权,所述申请各自的全部公开内容通过引用并入本文。
本发明涉及用于制备小体积形状的聚合物衍生的陶瓷材料的方法和系统。
背景技术:
聚合物衍生的陶瓷(pdc)是从聚合材料的热解衍生出的陶瓷材料,例如通过聚合材料的热解获得。这些材料一般呈固体或半固体状态,其通过固化初始液体聚合物前体(例如pdc前体、pdc前体制剂、前体批料和前体)而获得。经固化但被未热解的聚合物衍生材料可以称为预成型体、pdc预成型体、固化材料以及类似的术语。聚合物衍生的陶瓷可以衍生自许多不同种类的前体制剂,例如起始材料、起始制剂。pdc可以由碳硅烷或聚碳硅烷(si-c)、硅烷或聚硅烷(si-si)、硅氮烷或聚硅氮烷(si-n-si)、碳化硅(sic)、碳硅氮烷或聚碳硅氮烷(si-n-si-c-si)、硅氧烷或聚硅氧烷(si-o)等组成或者衍生自这些材料。
优选的pdc是“聚硅氧碳(polysilocarb)”,例如含有硅(si)、氧(o)和碳(c)的材料。聚硅氧碳材料还可以含有其它元素。聚硅氧碳材料可以由一种或多种聚硅氧碳前体制剂或前体制剂制成。聚硅氧碳前体制剂可以含有:例如,一种或多种官能化硅聚合物,其它聚合物,非硅基交联剂,单体以及潜在的其它成分,例如抑制剂、催化剂、引发剂、改性剂、掺杂剂、填料、增强剂、这些和其它材料的组合和变型以及附加剂。除非另有明确说明,碳氧化硅材料、sioc组合物和类似的术语是指聚硅氧碳材料,并且将包括液体材料、固体未固化材料、固化材料和陶瓷材料。
pdc、pdc制剂和起始材料的示例见于:美国专利申请序列号14/864539、14/634819;美国专利公开第2014/0343220、2014/02244658、2014/0326453、2015/0175750、2008/0095942、2008/0093185、2006/0069176、2006/0004169和2005/0276961号,以及美国专利第8742008、8119057、7714092、7087656、5153295和4657991号,所述文献各自的全部公开内容通过引用并入本文。
通常,除非另有说明,本文所使用的术语“约”意在包含与获得所述值相关的实验或仪器误差的±10%的差异或范围,并且这些差异或范围中较大的是优选的。
如本文所使用,除非另有说明,术语%、重量%和质量%可互换使用,是指第一组分的重量占总重量(例如制剂、混合物、材料或产品)的百分比。如本文所使用,除非另有说明,“体积%”和“%体积”及类似的此类术语是指第一组分的体积占总体积(例如制剂、材料或产品)的百分比。
技术实现要素:
因此,对于小的聚合物衍生的陶瓷和固体,对制备这些体积结构的方法、特别是制备这些结构的预定形状和体积的方法的需求长期存在且日益增长,但是尚未满足。此外,本发明还通过提供本文所教导、公开和要求保护的制品、设备和工艺来解决这些需求。
因此,提供了一种用于通过聚合物衍生的陶瓷前体制备小体积结构的系统,所述系统包括:聚合物衍生的陶瓷前体输送装置,所述装置包括与输送端口流体连通的室,其中,所述室能够输送液体聚合物衍生的陶瓷前体;成形装置,所述成形装置包括具有开口的成形室,所述成形室定义了一个空腔,其中,所述空腔与所述成形室开口流体连通;与所述输送端口流体连通的所述室的开口,由此所述系统能够将液体聚合物衍生的陶瓷以液体形式从输送端口输送到所述空腔中;温度控制装置,所述温度控制装置与所述成形装置热相关,其中,所述空腔能够保持在预定温度;由此,所述系统能够以预定的体积形状向所述空腔提供液体聚合物衍生的陶瓷前体,且其中所述系统能够固化所述空腔中的聚合物衍生的陶瓷前体。
还提供了具有以下特征中的一个或多个特征的系统和方法:其中所述输送装置具有喷嘴;其中所述输送装置具有压力驱动式液滴形成装置;其中所述输送装置具有流动驱动式液滴形成装置;其中所述输送装置具有声式液滴喷射装置;其中所述成型装置具有剪切诱导液滴产生装置;其中所述输送装置具有液滴产生装置;其中所述输送装置包括液滴产生装置,且所述液滴产生装置包括致动器,其中,所述致动器选自下列组成的组:压电式、蓄压器、注射器式、容积式、振动式、电磁式和相变式;其中苏搜成型装置具有液滴产生装置;其中所述成型装置具有液滴产生装置;并且所述液滴产生装置具有致动器,其中所述致动器选自由压电式、蓄压器、气溶胶式、注射器式、容积式、振动式、电磁式和相变式致动器组成的组。
还提供了具有以下特征中的一个或多个特征的系统和方法:其中成型装置具有液滴产生装置,该液滴产生装置选自由压电式、蓄压器、气溶胶式、注射器式、容积式、振动式、电磁式和相变式液滴产生装置组成的组;包括液滴产生装置;包括液滴产生装置;所述液滴产生装置具有致动器;其中所述致动器选自由压电式、蓄压器、注射器式、容积式、注射器式、振动式、电磁式和相变式致动器组成的组;包括液滴产生装置,选自由压电式、蓄压器、注射器式、容积式式、注射器式、振动式、电磁式和相变式致动器组成的组;包括液滴产生装置,其选自由水雾化器、喷气雾化器、雾化器、气辅雾化器、超声喷雾器、超声挤出机、喷墨式装置和烟雾机组成的组;其中,所述端口配置为输送体积小于约0.25立方英寸的前体体积形状;其中,所述端口配置为输送体积小于约500mm3的前体体积形状;其中,所述端口配置为输送体积小于约100mm3的前体体积形状;其中,所述端口配置为输送体积小于约4000立方微米的前体体积形状;其中,所述端口配置为输送体积小于约50立方微米的前体体积形状;其中所述端口配置为输送体积小于约10立方微米的前体体积形状;其中,输送装置配置为输送体积小于约0.25立方英寸的前体体积形状;其中,输送装置配置为输送体积小于约500mm3的前体体积形状;其中,输送装置配置为输送体积小于约100mm3的前体体积形状;输送装置配置为输送体积小于约4000立方微米的前体体积形状;其中,输送装置配置为输送体积小于约50立方微米的前体体积形状;其中,输送装置配置为输送体积小于约10立方微米的前体体积形状;其中,成型装置配置为固化体积小于约0.25立方英寸的前体体积形状;其中,成型装置配置为固化体积小于约500mm3的前体体积形状;其中,所述成型装置配置为固化体积小于约100mm3的前体体积形状;其中,成型装置配置为固化体积小于约4000立方微米的前体体积形状;其中,所述成型装置配置为固化体积小于约50立方微米的前体体积形状;以及其中,所述成型装置配置为固化体积小于约10立方微米的前体体积形状。
此外,提供了具有以下特征中的一个或多个特征的系统和方法:其中成型装置具有成形液体。
根据权利要求1所述的系统,其中所述成型装置具有基本上由水和表面活性剂组成的成形液体;其中所述成型装置具有成形液体和混合器;其中所述温度控制装置具有用于提供预定温度分布的控制器;并且,其中所述温度控制装置具有用于提供预定温度分布的控制器,所述温度分布包括第一加热速率、第一保持时间、第二加热速率和第二保持时间。
此外,提供了一种由聚合物衍生的陶瓷前体制备体积结构的系统,所述系统包括:聚合物衍生的陶瓷输送装置,所述装置包括液体聚合物衍生的陶瓷前体、室和端口,其中,所述室能够容纳液体聚合物衍生的陶瓷前体,所述陶瓷前体通过所述端口输送到具有预定体积的体积形状;前体固化装置,所述固化装置包括:空腔、温度控制装置,其中,所述空腔保持在足以固化聚合物衍生的陶瓷前体的体积形状的预定温度,以形成预成型体;与所述空腔流体连通的端口;由此,所述系统能够将液体聚合物衍生的陶瓷前体成形和固化成预定的体积形状结构。
还提供了具有以下特征中的一个或多个特征的系统和方法:其中所述室是管;其中所述端口是喷嘴;其中所述端口位于所述空腔的内部;其中所述空腔容纳成形液体;其中所述空腔容纳具有表面的成形液体,并且端口位于所述表面下方;其中所述体积形状是选自由以下组成的组的形状:球体、丸状、环状、透镜状、圆盘状、面板状、锥形体、截头圆锥形、正方形、矩形、桁架状以及角状;其中体积形状是选自由以下组成的组的形状:通道、中空密封腔室、中空球体、块状、片状、涂层、球形、正方形以及长椭球;以及其中体积形状是选自由以下组成的组的形状:椭圆体、椭球体、卵形、锥形体、多面结构、膜、表层、颗粒、棒状、杆状、角状、柱状、纤维状、短纤维状、管状、杯状、管道以及多面体。
此外,提供了具有以下特征中的一个或多个特征的系统和方法:配置为提供具有预定尺寸的预成型体,并提供至少约90%的预定尺寸的预成型体;配置为提供具有预定尺寸的预成型体,并提供至少约95%的预定尺寸的预成型体;以及,配置为提供具有预定尺寸的预成型体,并提供至少约99%的预定尺寸的预成型体。
此外,提供了具有以下特征中的一个或多个特征的系统和方法:其中所述系统具有挤出机;其中输送装置和成型装置是所述挤出机的部件;以及,其中所述系统具有挤出机;其中所述输送装置和所述固化装置是所述挤出机的部件。
还提供了一种由聚合物衍生的陶瓷前体材料制备体积结构的系统,所述系统包括:聚合物衍生的陶瓷输送装置,所述装置包括与输送端口流体连通的第一室,和一定量的液体聚合物衍生的陶瓷前体;成形和固化装置,所述成形和固化装置包括具有开口的成形室,所述成形室定义一个空腔,其中所述空腔与所述成形室的开口流体连通,并且包含聚合物衍生的陶瓷前体的体积形状;与所述输送端口流体连通的所述室的开口;与所述成形装置热相关的温度控制源,其中所述空腔保持在足以固化所述聚合物衍生的陶瓷前体的体积形状的预定温度;由此,所述系统能够以预定的体积形状将液体聚合物衍生的陶瓷前体材料提供到空腔中,其中所述聚合物衍生的陶瓷前体材料在空腔中固化。
还提供了具有以下特征中的一个或多个特征的系统和方法:其中所述液体聚合物衍生的陶瓷前体选自由硅烷、聚硅烷、硅氮烷、聚硅氮烷、碳硅烷、聚碳硅烷、硅氧烷和聚硅氧烷组成的组;其中所述液体聚合物衍生的陶瓷前体是聚硅氧碳;其中液体聚合物衍生的陶瓷前体是净聚硅氧碳;其中所述液体聚合物衍生的陶瓷前体是增强的聚硅氧碳;其中所述液体聚合物衍生的陶瓷前体是聚硅氧碳;其中所述液体聚合物衍生的陶瓷前体具有聚硅氧碳并且含有氢化物基团;其中所述液体聚合物衍生的陶瓷前体包含聚硅氧碳,不含有溶剂,并且含有氢化物基团;其中所述液体聚合物衍生的陶瓷前体具有聚硅氧碳且含有乙烯基;其中所述液体聚合物衍生的陶瓷前体具有聚硅氧碳但不含溶剂,并且含有乙烯基;其中所述液体聚合物衍生的陶瓷前体具有聚硅氧碳并且含有乙烯基和氢化物基团;其中所述液体聚合物衍生的陶瓷前体具有含有氢化物基团和乙烯基的聚硅氧碳,并且其中氢化物基团与乙烯基的摩尔比为约1.50:1;其中所述液体聚合物衍生的陶瓷前体具有含有氢化物基团和乙烯基的聚硅氧碳,并且氢化物基团与乙烯基的摩尔比为约3.93:1;其中所述液体聚合物衍生的陶瓷前体具有含有氢化物基团和乙烯基的聚硅氧碳,并且其中氢化物基团与乙烯基的摩尔比为约0.08:1至约24.00:1;其中所述液体聚合物衍生的陶瓷前体具有含有氢化物基团和乙烯基的聚硅氧碳,并且其中氢化物基团与乙烯基的摩尔比为约0.08:1至约1.82:1;其中所述液体聚合物衍生的陶瓷前体具有含有氢化物基团和乙烯基的聚硅氧碳,并且其中氢化物基团与乙烯基的摩尔比为约1.12:1至约2.36:1;其中所述液体聚合物衍生的陶瓷前体具有含有氢化物基团和乙烯基的聚硅氧碳,并且其中氢化物基团与乙烯基的摩尔比为约1.75:1至约23.02:1;其中所述液体聚合物衍生的陶瓷前体具有含有氢化物基团和乙烯基的聚硅氧碳,并且其中氢化物基团与乙烯基的摩尔比为约1.50:1至约3.93:1;以及其中所述液体聚合物衍生的陶瓷前体具有含有氢化物基团和乙烯基的聚硅氧碳,并且其中氢化物基团与乙烯基的摩尔比为约1.26:1至约4.97:1;
此外,提供了一种由聚合物衍生的陶瓷前体材料制备小体积结构的系统,所述系统包括:聚合物衍生的陶瓷输送装置,所述装置包括与输送端口流体连通的第一室和一定量的液体聚合物衍生的陶瓷前体;成型装置,所述成型装置包括具有开口的室,并且所述成形室定义一个空腔,其中所述空腔与所述成形室的开口流体连通并且容纳聚合物衍生的陶瓷前体的体积形状;所述室的开口与输送端口流体连通;并且由此所述系统能够将所述液体聚合物衍生的陶瓷前体提供到所述空腔中。
此外,提供了一种由聚合物衍生的陶瓷前体制备小体积结构的系统,所述系统包括:用于输送液体聚合物衍生的陶瓷的装置;成形装置,所述成形装置包括具有开口的成形室,且所述成形室定义一个空腔,其中,所述空腔与所述成形室的开口流体连通;与所述输送装置流体连通的所述室的开口,由此所述输送装置能够将所述液体聚合物衍生的陶瓷输送到所述空腔中;与所述成形装置热相关的温度控制源,其中所述空腔保持在预定温度;由此,所述系统能够以预定形状将液体聚合物衍生的陶瓷前体材料提供到所述空腔中,并以预定形状固定所述聚合物衍生的陶瓷,从而制备体积形状的聚合物衍生的陶瓷预成形体。
此外,提供了一种由聚合物衍生的陶瓷前体制备小体积结构的系统,所述系统包括:聚合物衍生的陶瓷输送装置,所述装置包括与输送端口流体连通的第一室,其中,所述第一室能够容纳液体聚合物衍生的陶瓷前体;用于形成体积形状结构的装置,所述成形装置包括具有开口的成形室,且所述成形室定义一个空腔,其中所述空腔与所述成形室的开口流体连通;与所述输送端口流体连通的所述室的开口;由此所述系统能够将来自所述输送端口的所述液体聚合物衍生的陶瓷以液体形式输送到所述空腔中;与所述成形装置热相关的温度控制源,其中所述空腔保持在预定温度;由此,该系统能够以预定的体积形状将液体聚合物衍生的陶瓷前体材料提供到所述空腔中,且其中聚合物衍生的陶瓷前体材料在空腔中固化。
此外,提供了一种由聚合物衍生的陶瓷前体制备小体积结构的系统,所述系统包括:用于输送液体聚合物衍生的陶瓷的装置;用于形成体积成形结构的装置,所述成型装置包括具有开口的成型室,并且所述成形室定义一个空腔,其中所述空腔与所述成形室的开口流体连通,所述成形室与所述输送端口流体连通,由此所述系统能够将所述液体聚合物衍生的陶瓷以液体形式从输送端口输送到所述空腔中;与成型装置热相关的温度控制源,其中所述空腔被保持在预定温度下;并且由此所述系统能够以预定的体积形式将液体聚合物衍生的陶瓷前体材料提供到空腔中,并且其中聚合物衍生的陶瓷前体材料在空腔中被固化。
另外,还提供了一种由聚合物衍生的陶瓷前体制备小体积结构的系统,所述系统包括:用于形成聚合物衍生的陶瓷前体的小体积形状结构的装置;以及用于将聚合物衍生的陶瓷前体材料的小体积形状结构固化成体积形状的预成型体的装置。
另外,还提供了一种由聚合物衍生的陶瓷前体制备小体积结构的系统,所述系统包括:用于形成聚合物衍生的陶瓷前体的小体积形结构的装置;用于将聚合物衍生的陶瓷前体材料的小体积形状结构固化成体积形状的预成型体的装置;以及用于热解预成型体的装置。
此外,提供了一种由聚合物衍生的陶瓷前体制备小体积结构的系统,所述系统包括:液体容纳容器,该液体容纳容器容纳成形液体;前体输送装置,其包括前体、通道和输送端口,所述通道与输送端口流体连通,由此可以从输送端口输送前体,并且所述输送端口与液体容纳容器流体连通。
还提供了一种由聚合物衍生的陶瓷前体制备小体积结构的方法,所述方法包括:将液体聚合物衍生的陶瓷前体提供到输送装置,所述装置包括与输送端口流体连通的室;使液体前体形成为预定的液体体积形状;并将液体体积形状输送到定义空腔的所述室;以及固化空腔中的液体体积形状以形成聚合物衍生的陶瓷预成型体。
还提供了包括以下特征中的一个或多个特征的方法和系统:其中预成型体具有与所述体积形状相同的形状;其中预成型体具有基本上与体积形状相同的形状;其中预成型体被绿色固化(greencured);其中预成型体被硬固化;其中预成型体被最终固化;包括热解预成型体以形成聚合物衍生的陶瓷;其中输送装置具有喷嘴;并且其中输送装置具有压力驱动式液滴形成装置。
另外,提供了具有以下特征中的一个或多个特征的系统和方法:其中以预定的固化温度分布进行固化;其中以包括第一加热速率、第一保持时间、第二加热速率和第二保持时间的预定固化温度分布进行固化;其中所述体积形状是选自由以下组成的组的形状:球体、丸状、环状、透镜状、圆盘状、面板状、锥形体、截头圆锥形、正方形、矩形、桁架状以及角状;其中所述体积形状是选自由以下组成的组的形状:通道、中空密封腔室、中空球体、块状、片状、涂层、球形、正方形以及长椭球;以及其中所述体积形状是选自由以下组成的组的形状:椭圆体、椭球体、卵形、锥形体、多面结构、膜、表层、颗粒、棒状、杆状、角状、柱状、纤维状、短纤维状、管状、杯状、管道以及多面体。
另外,提供了一种由聚合物衍生的陶瓷前体制备小体积结构的方法,所述系统包括:将液体聚合物衍生的陶瓷形成为液体预定体积形状的步骤;以及将液体预定体积形状固化成具有基本上相同体积形状的预成型体的步骤。
此外,提供了一种由聚合物衍生的陶瓷前体制备小体积结构的方法,所述系统包括:形成聚合物衍生的陶瓷前体的小体积形结构的步骤;将聚合物衍生的陶瓷前体材料的小体积形状结构固化成体积形状的预成型体的步骤;以及热解预成型体的步骤。
另外,提供了一种由聚合物衍生的陶瓷前体制备小体积结构的方法,所述系统包括:形成聚合物衍生的陶瓷前体的小体积净结构;将聚合物衍生的陶瓷前体材料的小体积净结构固化成体积形状的预成型体;以及热解预成型体。
此外,提供了一种由聚合物衍生的陶瓷前体制备小体积结构的方法,所述系统包括:将聚合物衍生的陶瓷前体液体提供到容纳容器;液体容纳容器容纳成形液体;所述前体在与成形液体接触时基本上形成预定的体积形状;以及固化该体积形状以形成预成型体。
另外,提供了一种用于形成聚合物衍生的陶瓷晶片(platelets)的系统,所述系统具有:用于在基底装置上形成液体聚合物衍生的陶瓷前体材料的薄膜的装置;以及用于向液体聚合物衍生的陶瓷的薄膜提供电磁辐射的装置。
还提供了具有以下特征中的一个或多个特征的系统和方法:其中用于形成薄膜的装置是分配头(distributionheader);其中用于形成薄膜的装置是分配辊组件;其中用于形成薄膜的装置是气刀组件;其中所述电磁辐射是白光;以及其中所述电磁辐射是具有至少约100nm的波段的宽带光,其波长为约300nm至约800nm。
另外,提供了一种由液体聚合物衍生的陶瓷前体形成固化聚合物衍生的陶瓷的方法,所述方法具有:在基底上形成液体聚合物衍生的陶瓷前体的薄层的步骤;以及用电磁辐射固化该薄层的步骤。
附图说明
图1是根据本发明的过程和成型系统的实施例的示意图。
图1a是根据本发明的成型系统的分布成型板的实施例的透视图。
图2是根据本发明的过程和成型系统的实施例的示意图。
图3是根据本发明的冲击射流成型系统和过程的实施例的示意图。
图4是根据本发明的共线型高压喷射成型系统和过程的实施例的示意图。
图5是根据本发明的超声高频成型系统和过程的实施例的示意图。
图6是根据本发明的超声喷嘴成型系统和过程的实施例的示意图。
图7是根据本发明的喷雾反射成型系统和过程的实施例的示意图。
图8是根据本发明的成型台成型系统和过程的实施例的示意图。
图9是根据本发明的泡沫成型系统和过程的实施例的示意图。
图10是根据本发明的喷雾热解成型系统和过程的实施例的示意图。
图11是根据本发明的喷雾固化系统和过程的实施例的流程图。
图11a是根据本发明的在喷雾固化方法中使用的固化室的实施例的示意图,该固化室提供并流和逆流的组合。
图11b是根据本发明的在喷雾固化方法中使用的固化室的实施例的示意图,该固化室提供并流。
图12是根据本发明的注塑机及用于模制小pdc固化预成型体的方法的实施例的横截面图。
图12a是根据本发明的图12的注塑机在模制周期的一个阶段的实施例的横截面图。
图12b是根据本发明的图12的注塑机在模制周期的一个阶段的的实施例的横截面图。
图12c是根据本发明的图12的注塑机的横截面图,示出了模制部从注射部和模具开口轴向移动以喷射模制的微固化pdc预成型体。
图12d是根据本发明的图12的实施例的部件的局部放大视图,示出了阀构件在液体pdc部与注塑机的注射部之间关闭的实施例。
图12e是根据本发明的图12的实施例的部件的局部放大视图,示出了阀构件在液体pdc部与注射部之间打开的实施例,以使预定喷射体积的pdc制剂流入注射部。
图12f是根据本发明的图12的实施例的部件的局部放大视图,示出了在用pdc制剂从pdc制剂部开始填充树脂流动通道过程中的注射销的位置的实施例。
图12g是根据本发明实施例的图12的实施例的部件的局部放大视图,示出了阀构件在制剂部与注射部之间的定位的实施例。
图13是根据本发明实施例的轧型成型系统和过程的实施例的透视图。
图13a是图13的实施例的辊的一部分的放大透视图。
图14是根据本发明的溶液形成系统和过程的实施例的示意图。
图15是根据本发明的溶液形成系统和过程的实施例的示意图。
图16是根据本发明的溶液形成系统和过程的实施例的透视图。
图17是根据本发明的溶液形成系统和过程的实施例的工艺流程图。
图18是根据本发明的表示聚结的实施例的曲线图。
图19是根据本发明的表示范德华力的实施例的曲线图。
图20是根据本发明的表示自由能随分离变化的曲线的实施例的曲线图。
图21是根据本发明的表示聚结动力学路径的实施例的图表。
图22是根据本发明的表示液滴尺寸随粘度变化的实施例的曲线图。
图23是根据本发明的表示等温和非等温过程的实施例的曲线图。
图24是根据本发明的表示两种不同表面活性剂浓度的粒度的实施例的图表。
图25是根据本发明的表示颗粒分布随时间变化的实施例的曲线图。
图26是根据本发明的表示速度和物质吸附的实施例的曲线图。
具体实施方式
大体上,本发明涉及由pdc前体制备小体积形状的方法、系统、设备和过程,并且涉及提供小体积形状的pdc预成型体和聚合物衍生的陶瓷。特别地,本发明的实施例还通过pdc前体制备具有良好的、高且超高均匀度和可复制性的小形状。本发明的实施例还以高生产率和长运行时间大量地制备pdc前体、pdc预制成型体、pdc塑料、pdc固化材料以及聚合物衍生的陶瓷的体积形状。
通常,由本发明实施例制成的体积形状小,例如其横截面为从约2英寸至0.01微米(μm),小于约1英寸,小于约3英寸,小于约3/4英寸,小于约1/3英寸,小于约5000微米,小于约4000微米,小于约2000微米,小于约1000微米,小于约500微米,小于约100微米,小于约10微米,小于约1微米,小于约0.5微米以及小于约0.1微米。体积形状的体积可以为从约4.25立方英寸至约0.0004立方微米,小于约0.25立方英寸,小于约525立方毫米,小于约100立方毫米,小于约50立方毫米,小于约4000立方微米,小于约2000立方微米,小于约100立方微米,小于约50立方微米,小于约0.5立方微米以及小于约0.00005立方微米。通过本发明实施例制成的小体积形状可分别重小于约30克,小于约15克,小于约10克,小于约1克,小于约0.5克,小于约0.1克,小于约0.01克,小于约0.0001克,小于约0.00001克,小于约10-8克,小于约10-10克以及小于约10-15g。对于一种或多种物理性质,例如形状、尺寸、重量、粗糙度、密度、孔隙率、强度、电气性能、导电性、光学性能、热力学、离子性能等以及它们的组合和变型,通过本发明实施例制备的小体积形状可以是大体均匀的,可以是完全无规的,可以在预定范围内。
所述体积形状可以是任何形状,包括:例如球体、薄片、片状、片屑、丸状、环状、透镜状、圆盘状、面板状、锥形体、截头圆锥形、正方形、矩形、桁架状、角状、通道、中空密封腔室、中空球体、块状、片状、涂层、球形、正方形、长椭球、椭圆体、椭球体、卵形、锥形体、多面结构、膜、表层、颗粒、棒状、杆状、角状、柱状、纤维状、短纤维状、管状、杯状、管道、多面体(例如八面体、十二面体、截半二十面体、菱形三十面体和棱柱)以及这些和其它更复杂工程和建筑形状的组合和变型。
通常,聚合物衍生的陶瓷及其固化预成型体可以是任何体积形状,并且优选地是任何预定的体积形状。固化预成型体可以具有与陶瓷相同的形状或不同的体积形状。因此,前体批料可以成形为例如薄片、片状、片屑、球形、球体、正方形、长椭球、椭圆体、椭球体、卵形、锥形体、杆状、箱状、多面结构以及多面体(例如八面体、十二面体、截半二十面体、菱形三十面体和棱柱)以及其它此类结构,用于固化和热解或者在固化和热解时成形为这些形状。聚合衍生陶瓷可以制成任何颗粒形状,其用作或建议用作例如颜料、附加剂、研磨料、填料和水力压裂支撑剂。球形结构是目前优选的支撑剂形状的示例。
除非另有明确说明,球体和球形应该是指并包括占“完美球体(perfectsphere)”内总体积的至少约90%的任何结构,即沿着结构表面的所有点具有相等距离的半径。基本上完美的球体占完美球体内总体积的至少约98%。大体上完美的球体占完美球体内总体积的至少约95%。
术语“已知形状(90%完美)”“大体上完美的形状(完美形状的95%)”以及“基本上完美(完美形状的98%)”适用于其它已知的被定义的几何形状(并且将用于本文中,除非另有说明)。因此,“已知的几何形状”(例如“立方体”)意味着该形状的总体积的至少约90%在完全已知的被定义的几何形状的范围之内,例如,对于立方体,其具有相等长度、宽度和高度的六个边,所有的边以直角连接。“大体上完美的形状”如“大体上完美的立方体”占完美立方体的形状的至少95%,而“基本上完美的形状”如“基本上完美的立方体”占完美立方体的形状的至少95%。
所述系统、装置和方法的实施例能够制备相同类型的高度无规尺寸的颗粒,例如所有的形状都是大体上完美的球体,但具有无规和变化的体积,以制备具有高度无规粒度的高度无规形状,例如具有不同体积的许多不同形状以及它们的组合和变型。
关于类型、体积或两者,所述系统、装置和方法的实施例优选地能够制备高度均匀的形状。因此,例如,所述方法的实施例所制备的球体在目标尺寸的至少90%之内,是目标尺寸的至少95%以及目标尺寸的至少99%或更多。例如,所述方法的实施例可以制备球形珠粒、球型珠粒、基本上完美的球形珠粒和大体上完美的球形珠粒,这些各自可以在10目的范围内占其尺寸的至少约90%,在10目的范围内占其尺寸的至少约95%,在10目的范围内占其尺寸的至少约98%,以及在10目的范围内占其尺寸的至少约99%。此外,例如,所述方法可以制备球形珠粒、球型珠粒、基本上完美的球形珠粒和大体上完美的球形珠粒,这些各自可以在5目的范围内占其尺寸的至少约90%,在5目的范围内占其尺寸的至少约95%,在5目的范围内占其尺寸的至少约98%,以及在5目的范围内占其尺寸的至少约99%。优选地,在不需要过滤、分选或筛选固化形状且不需要过滤、分选或筛选热解形状的情况下,在制备陶瓷和固化预成型体的体积形状时获得这些水平的均匀度。除了能够严格控制粒度分布之外,本方法的实施例能够制备大量高度均匀的预定形状,例如,所制备的形状的至少约90%、至少约95%和至少约99%符合目标或预定形状。例如,由前体批料制成的至少约98%的珠粒(例如支撑剂)可以基本上呈球形。
通常,前体制剂最初是液体,或者如果不是,则将其液化。然后将该液体前体制剂固化,以形成固体或半固体材料(例如塑料),其也称为预成型体或固化预成型体。然后将预成型体热解成陶瓷。
应当理解,本说明书中标题的使用是出于清楚、参考的目的,并不以任何方式进行限制。因此,标题下描述的方法组成和公开内容应在包括各种示例的本说明书的完整内容的背景下阅读。本说明书中标题的使用不应限制本发明的保护范围。
获得聚硅氧碳前体的一般过程
通常,聚合物衍生的陶瓷前体制剂、特别是聚硅氧碳前体制剂通常可以通过三种类型的过程来制备,但也可以使用其它过程以及这些过程的变型和组合。这些过程通常包括将前体组合而形成前体制剂。一种类型的过程通常包括:以优选为基本上不发生化学反应的不含溶剂的过程(例如“混合过程”)将前体材料混合在一起。另一种类型的过程通常包括化学反应(例如“反应型过程”),以形成特定的(例如定制的)前体制剂,该前体制剂可以是单体、二聚体、三聚体和聚合物。第三类过程具有在无溶剂环境中的两种或更多种组分的化学反应(例如“反应共混型过程”)。通常,在混合过程中,基本上所有的、优选为所有的化学反应在后续加工过程中发生,例如在固化、热解或两者的过程中。
应当理解,反应型过程、反应共混型过程和混合型过程的这些术语是为了方便起见而使用并用作简短的参考。这些术语不是也不应被视为限制。例如,反应型过程可用于产生一种前体材料,然后在与另一种前体材料的混合过程中使用。在美国专利公开第2014/0343220、2014/0274658、2014/0126453和2015/0175750号中公开和教导了这三种过程和pdc前体制剂,所述文献各自的全部公开内容通过引用并入本文。
在本说明书中,另外还在其相应的标题下描述了这些过程类型。应当理解,对一个标题下的一个过程的教导和对其它标题下的其它过程的教导可适用于彼此,并且适用于本说明书中的其它章节、实施例和教导,反之亦然。一种类型过程的起始或前体材料可以用于另一种类型的过程。此外,应当理解,这些标题下描述的过程应当在包括各种示例和实施例的本说明书的完整内容的背景下阅读。
应当理解,可以使用这些过程的组合和变型获得前体制剂、中间体、终产品和最终产品。根据产品的特定工艺和所需特征,一种过程类型的前体和起始材料可以用于另一种过程。来自混合型过程的制剂可以用作反应型过程或反应共混型过程中的前体或组分。类似地,可以在混合型过程和反应共混过程中使用来自反应型过程的制剂。类似地,可以在混合型过程和反应型过程中使用来自反应共混型过程的制剂。因此,优选地,可以将来自其它过程的最佳性能和特征组合和利用,以提供有成本效益且高效的过程和最终产品。这些过程提供了很大的灵活性,从而为中间产品、终产品和最终产品创建定制特征,因此,任何这些过程及其组合都可以提供特定的预定产品。在选择哪种类型的过程作为优选时,可以考虑成本、可控性、保存期限、规模扩大、制备容易度等因素。
除了可商购获得外,前体还可以通过烷氧基化类的过程(例如乙氧基化过程)的方式来制备。前体材料也可以通过乙炔反应路径获得。通常有几种已知的途径将乙炔加入si-h。因此,例如,四甲基环四硅氧烷可以在催化剂的存在下与乙炔反应生成四甲基四乙烯基环四硅氧烷。然后,可以将该产物开环并聚合形成直链乙烯基甲基硅氧烷。可替代地,可以通过使甲基二氯硅烷(通过直接处理或rochow法获得)与乙炔反应来制备典型的乙烯基硅烷。然后,可以纯化这些单体(因为可能存在扰乱)以形成乙烯基甲基二氯硅烷。然后乙烯基单体可以通过水解聚合而形成许多具有各种链长度的环状和直链硅氧烷,包括:例如各种环四硅氧烷(例如d4')和各种环戊硅氧烷(例如d5')。
混合型过程
前体材料可以是甲基含氢硅氧烷或取代和改性的甲基含氢硅氧烷主链附加剂、反应性单体、硅氧烷主链附加剂与硅烷改性剂或有机改性剂的反应产物以及其它类似类型的材料,如硅烷基材料、硅氮烷基材料、碳硅烷基材料、苯酚/甲醛基材料以及它们的组合和变型。优选地,所述前体在室温下为液体,尽管它们可以是熔融的固体或者是可溶于其它一种前体的固体。
在器皿中将前体混合在一起,优选地在室温下混合。优选地,将少量溶剂(例如水、有机溶剂、极性溶剂、非极性溶剂、己烷、thf、甲苯)加入到前体材料混合物中,更优选地,不含溶剂。优选地,每种前体材料与其它前体材料是可混溶的,例如它们可以以任何相对量或任何比例混合,并且将不会分离或沉淀。此时,“前体混合物”或“聚硅氧碳前体制剂”已经完成(注意,如果仅使用单一前体,该材料将简称为“聚硅氧碳前体”或“聚硅氧碳前体制剂”或“制剂”)。尽管已经完成,但也可以将填充剂和增强剂加入到制剂中。在制剂的优选实施例中,在被固化之前,当混合制剂时或者当在器皿中将制剂保持在预浸材料上或保持一段时间时,在制剂内基本上不发生化学反应(例如交联或聚合),更优选地,不发生化学反应。
可以在以下多种气氛和条件下混合前体:例如空气,惰性气体,氮气,氩气,流动气体,静态气体,减压,加压,环境压力以及它们的组合和变型。
此外,可以将诸如环己烷、1-乙炔基-1-环己醇(可购自aldrich公司)、八甲基环四硅氧烷和四甲基四乙烯基环四硅氧烷的抑制剂加入到聚硅氧碳前体制剂中,例如抑制性聚硅氧碳前体制剂。应当注意的是,四甲基四乙烯基环四硅氧烷可以同时作为反应物和反应阻滞剂(例如抑制剂),这取决于存在的量和温度,例如,在室温下它是阻滞剂,而在升高温度下它是反应物。此外,这时也可以在加工过程中将其它材料加入到聚硅氧碳前体制剂中,例如填充型聚硅氧碳前体制剂,包括本说明书中所述的或本领域已知的填料,如sic粉末、炭黑、砂、聚合物衍生的陶瓷颗粒、颜料、颗粒、纳米管、晶须或其它材料。此外,具有抑制剂和填充剂两者的制剂将被认为是抑制性填充型聚硅氧碳前体制剂。
可以使用催化剂或引发剂,并且可以在固化之前,在形成前体制剂或将前体制剂制成结构时(或在这之前、不久之前或之前的早些时间)加入催化剂或引发剂。催化作用有助于加快和促进前体制剂的固化,以形成预成型体。
在加入催化剂后将前体制剂保持用于固化的时间段被称为“贮存期(potlife)”,例如在使用之前,经催化的制剂可以在其保持器皿中保留多长时间。根据具体的制剂、是否使用抑制剂(如果使用的话,使用的量)、储存条件(例如温度、低o2气氛)和潜在的其它因素,前体制剂的贮存期可以是例如约5分钟至约10天,约1天至约6天,约4至5天,约30分钟,约15分钟,约1小时至约24小时以及约12小时至约24小时。
催化剂可以是任何基于铂(pt)的催化剂,例如,可以将其稀释到以下范围:约百万分之0.01(ppm)pt至约250ppmpt,约0.03ppmpt,约0.1ppmpt,约0.2ppmpt,约0.5ppmpt,约0.02至0.5ppmpt,约1ppm至200ppmpt,并且对于一些应用和实施例优选为约5ppm至50ppmpt。所述催化剂可以是过氧化物基催化剂,例如在高于90℃,浓度为0.1%至3%过氧化物以及约0.5%和2%的过氧化物的情况下,其半衰期为10小时;它可以是一种有机类过氧化物;它可以是能够与si-h键、si-oh键或不饱和碳键反应的任何有机金属催化剂,这些催化剂可以包括:二月桂酸二丁基锡,辛酸锌,过氧化物,例如钛、锆、铑、铱、钯、钴或镍的有机金属化合物。所述催化剂还可以是任何其它的铑、铼、铱、钯、镍和钌类或基于这些金属的催化剂。可以使用这些和其它催化剂的组合和变型。催化剂可以购自arkema公司,商品名称:luperox,例如luperox231;购自johnsonmatthey公司,商品名称:karstedt’s催化剂、ashby’s催化剂、speier’s催化剂。
此外,可以使用这些和其它催化剂的定制和特定组合,使得它们与特定制剂相匹配,并且以这种方式选择性地和特异性地催化特定成分的反应。此外,可以使用这些类型的匹配催化剂-制剂体系来提供预定的产品特征,例如固化结构和陶瓷的孔结构、孔隙率、密度、密度分布、高纯度、超高纯度以及其它形态或特征。
在用于制备前体制剂的混合型过程中,优选地仅在制备起始材料的过程中、固化过程和热解过程中发生化学反应或分子重排。在制备起始材料或前体时发生或使用了化学反应(例如聚合、还原、缩合、取代)。在通过混合型过程制备聚硅氧碳前体制剂时,优选地,不发生或基本上不发生化学反应和分子重排。相比于制备聚合物衍生的陶瓷的现有方法,本混合型过程的这些实施例提供了显著的优点,在制备前体制剂的过程中无需并且不使用聚合反应或其它反应。优选地,在这些混合类型的制剂和过程的实施例中,聚合、交联或其它化学反应主要在固化过程中发生,优选为基本上、更优选为仅在固化过程中发生。
所述前体可以是硅氧烷主链附加剂,如甲基含氢硅油(mh),其通式如下所示:
mh可以具有约400mw至约10000mw,约600mw至约3000mw的分子量(“mw”,可以测量为重量平均分子量,以amu为单位或以g/mol计),并且可以具有优选为约20cps至约60cps的粘度。甲基硅氧烷单元“x”的百分比可以为1%至100%。二甲基硅氧烷单元“y”的百分比可以为0%至99%。可以使用该前体来提供交联结构的主链以及固化预成型体和陶瓷材料的其它特征和特性。该前体还可以通过与不饱和碳化合物反应而改性,以制备新的或另外的前体。通常,甲基含氢硅油流体(mhf)具有最少量的“y”,更优选地,“y”实际上为零。
所述前体可以是硅氧烷主链附加剂,如乙烯基取代的聚二甲基硅氧烷,其通式如下所示:
该前体可以具有约400mw至约10000mw的分子量(mw),并且可以具有优选为约50cps至约2000cps的粘度。甲基乙烯基硅氧烷单元“x”的百分比可以为1%至100%。二甲基硅氧烷单元“y”的百分比可以为0%至99%。优选地,x为约100%。可以使用该前体来降低交联密度并且提高韧性,以及提供固化预成型体和陶瓷材料的其它特征和特性。
所述前体可以是硅氧烷主链附加剂,如乙烯基取代的和乙烯基封端的聚二甲基硅氧烷,乙烯基取代的和氢封端的聚二甲基硅氧烷,烯丙基封端的聚二甲基硅氧烷,乙烯基封端的聚二甲基硅氧烷(“vt”),硅烷醇(羟基)封端的聚二甲基硅氧烷,硅烷醇(羟基)封端的乙烯基取代的二甲基硅氧烷和氢(氢化物)封端的聚二甲基硅氧烷,二苯基封端的硅氧烷,单苯基封端的硅氧烷,二苯基二甲基聚硅氧烷,乙烯基封端的二苯基二甲基聚硅氧烷以及羟基封端的二苯基二甲基聚硅氧烷。
各种环硅氧烷可以用作制剂中的反应性分子。它们可以通过以下命名系统或通式来描述:dxd*y,其中“d”表示二甲基甲硅烷氧基单元,“d*”表示取代的甲基硅烷氧基单元,其中“*”基团可以是乙烯基、烯丙基、氢化物基团、羟基、苯基、苯乙烯基、烷基、环戊二烯基或其它有机基团,x为0-8,y为>=1,x+y为3-8。
前体批料还可以含有非硅基交联剂(即非硅基交联剂与硅氧烷主链附加剂的反应产物)以及它们的组合和变型。非硅基交联剂旨在并在固化过程中提供交联能力。例如,可以使用的非硅基交联剂包括:环戊二烯(cp)、甲基环戊二烯(mecp)、二环戊二烯(“dcpd”)、甲基二环戊二烯(medcpd)、三环戊二烯(tcpd)、戊间二烯、二乙烯基苯、异戊二烯,降冰片二烯、乙烯基降冰片烯、丙烯基降冰片烯、异丙烯基降冰片烯、甲基乙烯基降冰片烯、双环壬二烯、甲基双环壬二烯、丙二烯、4-乙烯基环己烯、1,3-庚二烯、环庚二烯、1,3-丁二烯、环辛二烯以及它们的异构体。通常,可以使用含有两个(或更多个)不饱和c=c键的任何烃作为交联剂,不饱和c=c键可以与前体中的si-h、si-oh或其它si键反应。含有氧、氮和硫的一些有机材料也可以用作交联部分。
这种前体还可以用在双固化体系中;以这种方式,双固化可以允许在单一制剂中使用多种固化机制。例如,可以使用缩合型固化和加成型固化。这进一步使得能够具有复杂的固化分布,例如可以通过分离型固化提供初始固化并通过分离型固化提供最终固化。
所述前体可以是反应性单体。该反应性单体将包括诸如四甲基四乙烯基环四硅氧烷(“tv”)的分子,其通式如下所示:
可以使用该前体来提供支化剂、三维交联剂以及用于固化预成型体和陶瓷材料的其它特征和特性(还需注意,在某些制剂中(例如高于2%)和某些温度(例如约室温至约60℃)下,该前体可以作为交联的抑制剂,例如可以抑制氢化物与乙烯基的交联。)
前体可以是反应性单体,例如三乙烯基环四硅氧烷、二乙烯基环四硅氧烷、三乙烯基氢化环四硅氧烷(trivinylmonohydridecyclotetrasiloxane)、二乙烯基二氢化环四硅氧烷(divinyldihydridecyclotetrasiloxane)以及六甲基环四硅氧烷。
所述前体可以是硅烷改性剂,如乙烯基苯基甲基硅烷、二苯基硅烷、二苯基甲基硅烷和苯基甲基硅烷(其中的一些可以用作封端基或端基)。这些硅烷改性剂可以提供扩链剂和支化剂。它们还改善了韧性,改变了折射率,并且提高了固化材料的高温固化稳定性,以及由此提高了固化材料的强度等。所述前体(如二苯基甲基硅烷)可以用作封端剂,其也可以改善韧性,改变折射率,并且提高固化材料的高温固化稳定性,以及由此提高固化材料的强度。
所述前体可以是硅烷改性剂与乙烯基封端的硅氧烷主链附加剂的反应产物。所述前体可以是硅烷改性剂与羟基封端的硅氧烷主链附加剂的反应产物。所述前体可以是硅烷改性剂与氢化物封端的硅氧烷主链附加剂的反应产物。所述前体可以是硅烷改性剂与tv的反应产物。所述前体可以是硅烷的反应产物。考虑到位阻现象,所述前体可以是硅烷改性剂与环硅氧烷的反应产物。所述前体可以是部分水解的正硅酸四乙酯(tertraethylorthosilicate),如tes40或silbond40。所述前体也可以是购自通用电气公司(generalelectriccompany)(威尔顿,康涅狄格州(wilton,conn))的甲基四硅氧烷,如sr-350。所述前体也可以是苯甲基硅氧烷,例如购自wackerchemieag的604。所述前体也可以是购自瓦克化学股份公司(wackerchemieag)的甲基苯基乙烯基硅氧烷,如h62c。
所述前体还可以从以下中选择:
因此,除了前述类型的前体之外,可以预期的是,前体还可以是以下通式的化合物:
其中封端基e1和e2选自诸如三甲基硅(-si(ch3)3)、二甲基硅羟基(-si(ch3)2oh)、二甲基氢化硅(-si(ch3)2h)、二甲基乙烯基硅(-si(ch3)2(ch=ch2))、(-si(ch3)2(c6h5))和二甲基烷氧基硅(-si(ch3)2(or))的基团。r基团r1、r2、r3和r4可以全部不同,或者一个或多个可以相同。因此,例如r2与r3相同,r3与r4相同,r1与r2不同而r3与r4相同等等。r基团选自诸如以下的基团:氢化物(-h)、甲基(me)(-c)、乙基(-c-c)、乙烯基(-c=c)、烷基(-r)(cnh2n+1)、烯丙基(-c-c=c)、芳基(`r)、苯基(ph)(-c6h5)、甲氧基(-o-c)、乙氧基(-o-c-c)、硅烷氧基(-o-si-r3)、烷氧基(-o-r)、羟基(-o-h)、苯乙基(-c-c-c6h5)以及甲基苯基乙基(-c-c(-c)(c6h5))。
通常,聚硅氧碳制剂的配制实施例可以具有例如约0%至50%的mh,约20%至约99%的mh,约0%至约30%的硅氧烷主链附加剂,约1%至约60%的反应性单体,约30%至约100%的tv以及约0%至约90%的硅氧烷主链附加剂与硅烷改性剂或有机改性剂反应产物的反应产物。
在混合制剂时,应该用足够的时间使前体被有效地混合和分散。通常,约15分钟至1小时的混合就足够了。一般情况下,前体制剂相对地且基本上对剪切不敏感,因此泵或混合的类型不是关键的。还需注意的是,在较高粘度制剂中可能需要额外的混合时间。在混合过程中,制剂的温度应优选地保持在约45℃以下,优选地保持在约10℃(注意,这些混合条件适用于经预催化的制剂)。
反应型过程
在反应型过程中,一般在溶剂的存在下,通常使用化学反应将一种、两种或更多种前体组合,以形成基本上由单一聚合物组成的前体制剂,然后可以将其催化、固化和热解。该过程提能够形成定制的前体制剂,当固化定制的前体制剂时,可以提供具有独特和期望特征的塑料,如耐高温性、阻燃性、阻滞性、强度以及其它特征。也可以热解固化材料而形成具有独特特征的陶瓷。反应型过程允许通过选择掺入到构成前体制剂的聚合物中的官能团来达到最终产品中不同类型的官能度的预定平衡,例如苯基,其通常不用于陶瓷但有益于为塑料提供高温能力,以及苯乙烯,其通常不能为塑料提供高温特征但为陶瓷提供益处。
通常,用作前体制剂的定制聚合物通过在缩合反应中使前体反应来制备,以形成聚合物前体制剂。然后,通过水解反应将该前体制剂固化成预成型体。所述缩合反应形成如下所示类型的聚合物:
其中聚合单元中的r1和r2可以是氢化物(-h)、甲基(me)(-c)、乙基(-c-c)、乙烯基(-c=c)、烷基(-r)(cnh2n+1)、不饱和烷基(-cnh2n-1)、环状烷基(-cnh2n-1)、烯丙基(-c-c=c)、丁烯基(-c4h7)、戊烯基(-c5h9)、环戊烯基(-c5h7)、甲基环戊烯基(-c5h6(ch3))、降冰片烯基(-cxhy,其中x=7-15,y=9-18)、芳基(`r)、苯基(ph)(-c6h5)、环庚烯基(-c7h11)、环辛烯基(-c8h13)、乙氧基(-o-c-c)、硅烷氧基(-o-si-r3)、甲氧基(-o-c)、烷氧基(-o-r)、羟基(-o-h)、苯乙基(-c-c-c6h5)甲基苯基乙基(-c-c(-c)(c6h5))以及乙烯基苯基乙基(-c-c(c6h4(-c=c)))。r1和r2可以相同或不同。定制的前体聚合物可以具有几个不同的聚合单元,例如a1、a2、an,并且可以包括多达10个、20个或更多个单元,或者可以仅包含单个单元,例如,由反应过程制成的mhf可以仅具有单个单元。
实施例可包括前体,该前体包括三乙氧基甲基硅烷、二乙氧基甲基苯基硅烷、二乙氧基甲基氢化硅烷、二乙氧基甲基乙烯基硅烷、二甲基乙氧基乙烯基硅烷、二乙氧基二甲基硅烷、乙氧基二甲基苯基硅烷、二乙氧基二氢硅烷、三乙氧基苯基硅烷、二乙氧基氢化三甲基硅氧烷、二乙氧基甲基三甲基硅氧烷、三甲基乙氧基硅烷、二苯基二乙氧基硅烷、二甲基乙氧基氢化硅氧烷等以及这些和其它前体(包括本说明书中陈述的其它前体)的组合和变型。
链端单元si端1和si端2可以来自二甲基乙氧基乙烯基硅烷、乙氧基二甲基苯基硅烷和三甲基乙氧基硅烷的前体。另外,如果适当地控制聚合过程,则可以从用于提供聚合物的重复单元的前体中获得羟基端帽。
通常,将前体与乙醇(或其它材料,用于吸收热量,例如提供热质量)、过量的水和盐酸(或其它质子源)一起加入到器皿中。将该混合物加热至达到其活化能,之后的反应通常为放热。通常,在该反应中,水与前体单体的硅的乙氧基反应而形成羟基(其中乙醇为副产物)。一旦形成,该羟基与另一种前体单体的硅上的乙氧基开始反应,从而引起聚合反应。继续进行该聚合反应,直到构建期望的链长。
优选地,在来自反应型过程的聚合物前体制剂的固化过程中使用催化剂。可以使用与固化来自混合型过程的前体制剂所用聚合物相同的聚合物。应当注意,通常与混合型制剂不同,固化反应型聚合物时不一定需要催化剂。还可以使用抑制剂。然而,如果不使用催化剂,反应时间和速率将会变慢。来自反应过程的固化材料的固化和热解基本上与来自混合过程和反应混合过程的固化材料的固化和热解相同。
反应型过程可以在以下多种气氛和条件下进行:例如,空气,惰性气体,氮气,氩气,流动气体,静态气体,减压,环境压力,加压以及它们的组合和变型。
反应共混型过程
在反应共混型过程中,前体在不存在溶剂的情况下反应形成前体制剂。例如,反应共混型过程的实施例具有由mhf和二环戊二烯(“dcpd”)制备的前体制剂。利用反应共混过程产生mhf/dcpd聚合物,并将该聚合物用作前体制剂(可以单独使用该聚合物以形成固化或热解产物,或者将其作为混合或反应过程中的前体)。已知分子量和氢化物当量质量的mhf;“p01”(p01是溶于四乙烯基环四硅氧烷的2%pt(0)四乙烯基环四硅氧烷络合物,用四乙烯基环四硅氧烷将其稀释20倍至0.1%pt(0)络合物)。以这种方式,每1%的催化剂负载量,提供10ppmpt。使用了40至90%的mhf起始原料(0.20重量%,已知的活性当量)和10至60%的二环戊二烯(纯度≥83%)。在该过程的实施例中,带有混合器的可密封反应器皿可用于该反应。在空气中(尽管可以使用其它类型的气氛),在密封器皿中进行该反应。优选地,反应在大气压下进行,但是可以使用更高和更低的压力。此外,反应共混型过程可以在以下多种类型的气氛和条件下进行:空气,惰性气体,氮气,氩气,流动气体,静态气体,减压,环境压力,加压以及它们的组合和变型。
固化和热解
前体制剂(包括来自上述类型过程的聚硅氧碳前体制剂)以及其它制剂可被固化形成固体、半固体或塑料类材料。通常,将前体制剂分散、成形或以其它方式形成为预成型体,所述预成型体将包括任何体积结构或形状,包括薄膜和厚膜。在固化时,可以通过初始固化来处理聚硅氧碳前体制剂,以提供部分固化的材料,部分固化的材料也可以称为预成型体、绿色材料(greenmaterial)或绿色固化体(greencure)(并非意指关于材料颜色的任何事物)。然后可以进一步固化该绿色材料。因此,可以使用一个或多个固化步骤。所述材料可被“最终固化”,即固化到这样的点:在该点,材料具有用于其预期目的而必需的物理强度和其它性能。固化的量可以达到最终固化(或“硬固化”),即在该点,所有或基本上所有的化学反应已经停止(例如,通过材料中不存在反应性基团或者反应性基团的减少随时间呈平稳状态来测定)。因此,所述材料可以根据其预期用途和目的而被固化至不同的程度。例如,在一些情况下,最终固化和硬固化可以是一样的。固化条件如气氛和温度可以影响固化材料的组成。
在将前体制剂制成结构或预成型体时,前体制剂(例如聚硅氧碳制剂)可以例如使用以下技术形成:喷涂、喷雾干燥、喷雾法、相变分离、流塑、热喷涂、拉伸、滴注、形成液滴(对于液体和液体-表面活性剂体系)、漆涂、模制、成型、挤出、旋转、超声波、振动、溶液聚合、乳液聚合、微电子聚合、注射、注塑或以其它方式操纵成基本上任何体积形状。所述体积形状可以例如包括以下形状:球体、丸状、薄片、片状、片屑、环状、透镜状、圆盘状、面板状、锥形体、截头圆锥形、正方形、矩形、桁架状、角状、通道、中空密封腔室、中空球体、块状、片状、涂层、膜、表层、颗粒、棒状、杆状、角状、厚板状、柱状、纤维状、短纤维状、管状、杯状、管道以及这些和其它更复杂的工程和建筑形状的组合和变型。
成型步骤、固化步骤和热解步骤可以以间歇过程、序列性地、连续地进行,其中有时间延迟(例如,在步骤之间储存或保持材料),以及这些和其它类型的加工顺序的组合和变型。此外,前体可以被部分固化,或者可以在前体形成体积形状之前开始和进行固化过程。这些步骤及其各种组合可以(在一些实施例中,优选地)在受控和预定的条件下进行(例如,材料暴露于预定的气氛,其整个处理过程中的温度分布,例如氧气减少,固化预成型体的温度在热解之前保持在约140℃)。还应当理解,用于成型、固化和热解的系统、设备或加工步骤可以是相同的设备、连续设备、间歇联结设备以及这些和其它类型的工业过程的组合和变型。因此,例如,喷雾干燥技术可以形成固化的颗粒,固化颗粒被直接进料到流化床反应器中用于热解。
聚硅氧碳前体制剂可以制成净的、非增强的、非填充的、复合的、增强的和填充的结构、中间体、最终产品以及这些和其它成分类型的材料的组合和变型。此外,这些结构、中间体和最终产品可以被固化(例如,绿色固化、最终固化或硬固化),可不被固化,可被热解为陶瓷,以及它们的组合和变型(例如,固化材料可以用衍生自与固化材料相同的聚硅氧碳的热解材料填充)。
前体制剂可用于形成“净”材料(“净”材料指的是所有和基本上所有的结构由前体材料或未填充的制剂制成;因此,不存在填料或增强剂)。
聚硅氧碳前体制剂可用于涂覆或浸渍由例如碳纤维、玻璃纤维或由聚硅氧碳前体制剂(相同或不同的制剂)制成的纤维制成的机织物或非机织物。因此,聚硅氧碳前体制剂可用于形成复合材料,例如增强产品。例如,所述制剂可以流入、浸渍到增强材料中,被增强材料吸收或以其它方式与增强材料结合,所述增强材料如碳纤维、玻璃纤维、机织物、石墨(grapheme)、碳纳米管、薄膜、沉淀物、砂、非机织物、毛纤维(coppedfibers)、绳索、编织结构、陶瓷粉末、玻璃粉末、碳粉末、石墨粉末、陶瓷纤维、金属粉末、碳化物球粒或碳化物组分、短纤维、丝束、上述材料的纳米结构、聚合物衍生的陶瓷,符合过程和最终产品的温度要求的任何其它材料以及它们的组合和变型。
聚硅氧碳前体制剂可用于形成填充材料。填充材料将是具有加入到聚硅氧碳前体制剂中的其它固体或半固体材料的任何材料。可以对填充材料进行选择,以提供固化产品、陶瓷产品或两者的某些特征。这些特征可能涉及或者是例如美学、触觉、热、密度、辐射、化学、成本、磁、电特性以及这些和其它特征的组合和变型。这些特征可以是除强度之外的特征。因此,填充材料可能不影响固化材料或陶瓷材料的强度,而可能增加强度,或者甚至可以在一些情况下降低强度。填充材料可以赋予颜色、磁性能、耐火性、阻燃性、耐热性、导电性、抗静电性、光学性能(例如反射率、折射率和虹彩)、美学性能(如建筑产品中的石材外观)、化学电阻率、耐蚀性、耐磨性、成本降低、耐磨损性、热绝缘、uv稳定性、uv保护性以及在最终产品或材料中可能是期望或必要或者既期望又必要的其它特征。因此,填充材料可以包括炭黑、铜导线、导热填料、导电填料、铅、光纤、陶瓷着色剂、颜料、氧化物、砂、染料、粉末、陶瓷细粉、聚合物衍生的陶瓷颗粒、造孔剂、碳硅烷、硅烷、硅氮烷、碳硅氮烷、硅氧烷粉末、陶瓷粉末、金属、金属络合物、碳、丝束、纤维、短纤维、含硼材料、磨碎纤维、玻璃、玻璃纤维、纤维玻璃和纳米结构(包括前述的纳米结构)等。
衍生自所述制剂或由该制剂制成的聚硅氧碳制剂和产品可以具有金属和金属络合物。填充材料将包括增强材料。在许多情况下,固化以及热解的聚硅氧碳填充材料可以视为复合材料。通常,在这种观点下,聚硅氧碳将构成本体相或基体相(例如,连续的或基本上连续的相),而填料将构成分散相(例如非连续相)。根据具体的应用、产品或最终用途,填料可以均匀地分布在前体制剂中,不均匀地分布,以预定和受控的分布梯度(如基于预定的沉降速率)分布,并且可以在不同的制剂中具有不同的量,然后可以在预定区域(例如,具有不同填料浓度的横纹层)内形成具有预定量的填料的产品。然而,应当注意的是,将材料称为“填充的”或“增强的”并不意味着该材料的大部分是聚硅氧碳(以重量、体积或以两者计)。因此,通常,聚硅氧碳与填料的比率(重量或体积)可以为约0.1:99.9至99.9:0.1。
可以使用聚硅氧碳前体制剂形成非增强材料,所述非增强材料是主要由、基本上由且优选地仅由前体材料制成的材料;但也可以包括具有不赋予强度的填料或附加剂的制剂。
固化可以在以下条件下完成:在标准环境温度和压力(“satp”,1个大气压,25℃)下,在高于或低于该温度的温度下,在高于或低于该压力的压力下以及在不同的时间段内。固化可以通过各种加热、加热速率和温度分布(例如,保持时间和温度,连续温度变化,周期性温度变化(例如加热、之后保持、冷却、再加热等))进行。固化的时间可以是从几秒(例如小于约1秒,小于5秒)至小于一分钟,至数分钟,至数小时,至数天(或可能更长)。固化也可以在任何类型的周围环境中进行,包括:例如气体,液体,空气,水,含表面活性剂的液体,惰性气体,n2,氩气,流动气体(例如吹扫气体),静态气体,o2减少,减压,加压,环境压力,受控分压以及这些和其它加工条件的组合和变型。对于高纯度材料,固化装置和固化过程的炉子、容器、处理设备、气氛以其它部件是洁净的,基本上没有且不给固化材料带去任何被认为是杂质或污染物的元素或材料。在实施例中,固化环境(例如炉子、气氛、容器以及它们的组合和变型)可以具有有助于或影响以下的材料:例如,预成型体、陶瓷和最终应用或产品中的组成、催化、化学计量、特征、性能以及它们的组合和变型。
优选地,在固化过程的实施例中,在以下范围的温度下进行固化:约5℃或更高,约20℃至约250℃,约20℃至约150℃,约75℃至约125℃以及约80℃至90℃。但也可以使用更高和更低的温度及各种加热分布,例如随时间的温度变化率(“斜率”,例如δ度/时间)、保持时间和温度。
固化条件(例如温度、时间、斜率)可以取决于例如预成型体的尺寸,预成型体的形状或保持预成型体的模具,在一些实施例中,可以全部或部分地由制剂预先确定固化条件以匹配以上所述,以防止应力断裂、脱气(offgassing)或与固化过程相关的其它现象。此外,所述固化条件可以使得优选地以受控的方式利用先前被认为是与固化过程相关的问题。因此,例如,可以利用脱气来产生具有开放或封闭结构的泡沫材料。类似地,固化条件可用于产生或控制材料的微结构和纳米结构。通常,固化条件可用于影响、控制或改变过程的动力学和热力学,这可以影响形态、性能,特征和功能等。
在固化聚硅氧碳前体制剂后进行交联反应,该交联反应在一些实施例中提供了尤其具有-r1-si-c-c-si-o-si-c-c-si-r2-的交联结构,其中r1和r2根据制剂中所使用的前体而变化且基于该前体。在固化材料的一个实施例中,它们的交联结构可以具有相对于另一硅原子的3-配位硅中心,硅与硅之间被少于5个原子分隔开。
在固化过程中,一些制剂可以表现出放热(即自热反应),这可以产生少量的热量来辅助或推动固化反应,或者可能产生可能需要管理和移除的大量热量,以避免诸如应力断裂等问题。在固化过程中,通常发生脱气并导致材料损失,该损失通常由剩余材料的量来界定,例如固化产率。本发明实施例的制剂、固化条件和聚硅氧碳前体制剂的实施例可以具有至少约90%、约92%、约100%的固化产率。事实上,在使用空气固化的情况下,由于从空气中吸收氧气,材料的固化产率可能高于100%,例如约101-105%。此外,在固化过程中,所述材料通常收缩,根据制剂、固化条件和预成型体形状的性质以及预成型体是否是增强的、填充的、净的或非增强的,这种收缩为约20%,小于20%,小于约15%,小于约5%,小于约1%,小于约0.5%,小于约0.25%或更小。
预成型体的固化可以通过具有必需的温度和环境控制水平的任何类型的加热装置或机械装置、技术或形态来实现,例如加热水浴、电炉、微波炉、燃气炉、炉子、强制加热空气、塔、喷雾干燥、降膜反应器、流化床反应器、激光器、间接加热元件、直接加热、红外加热、uv照射、rf炉,通过高剪切混合原位乳化,通过超声破碎原位乳化。
此外,在固化之后或在初始固化之后,固化材料(例如绿色材料、初始固化、最终固化或硬固化)可以保持在受控的环境下,例如固化材料可以保持在控制温度下,保持在预定的环境(例如水浴、低o2环境、高o2环境、液浴、具有附加剂的液浴、惰性气氛、雾化环境、具有附加剂的气体气氛等)中。这种控制保持步骤可用于影响最终产品(固化、热解或两者)的性质,例如强度,并且可用于向最终材料或产品提供附加剂或附加特征。
在热解预成型体或固化结构或固化材料时,将其加热至约600℃至约2300℃,约650℃至约1200℃,约800℃至约1300℃,约900℃至约1200℃以及约950℃至1150℃。在这些温度下,通常将所有的有机结构去除或者与无机成分组合形成陶瓷。一般在约650℃至1200℃的温度范围内,所得材料是无定形玻璃状陶瓷。当加热超过约1200℃时,所述材料通常可以基于纳米晶体结构或微结晶结构,例如sic、si3n4、sicn、βsic;当超过1900℃时,可以形成αsic结构,当超过2200℃时,通常形成cαsic。例如,热解的陶瓷材料可以是单晶、多晶、无定形的以及这些和其它类型形态的组合、变型和亚组。
热解可以在不同的加热和环境条件下进行,优选地包括热控制、动力学控制以及它们的组合和变型等。例如,热解可以具有各种加热斜率、加热周期和环境条件。在一些实施例中,可以升高温度并保持预定温度以有助于已知转变(例如放气、挥发、分子重排等),然后升高到对应于下一已知转变的下一保持温度。热解可以在以下条件下进行:还原气氛,氧化气氛,低o2,气体富集(例如火焰内或紧邻火焰),惰性气体,氮气,氩气,空气,减压,环境压力,加压,流动气体(例如吹扫气体,例如流速为约15.0ghsv至约0.1ghsv、约6.3ghsv至约3.1ghsv、约3.9ghsv),静态气体以及它们的组合和变型。
热解在一段时间内进行,优选地使预成型体完全热解。对于高纯度材料,热解装置的炉子、容器、处理设备和其它部件是洁净的,基本上不含或不含或者不给热解材料带去将被认为是杂质或污染物的任何元素或材料。恒定流速的“吹扫”气体可有助于在挥发性发生期间净化炉子。在一个实施例中,热解环境(例如炉子、气氛、容器以及它们的组合和变型)可以具有有助于或影响以下的材料:例如,陶瓷和最终应用或产品中的组成、催化、化学计量、特征、性能以及它们的组合和变型。
在热解过程中,材料可能通过脱气而损失。在热解步骤或周期结束时剩余的材料的量被称为成炭率(或热解产率)。该制剂实施例的制剂和聚硅氧碳前体制剂的sioc形成的成炭率为至少约60%、约70%、约80%和至少约90%、至少约91%及更高。事实上,利用空气热解,材料的成炭率可远高于91%,可能接近100%。为了避免材料在空气热解中的降解(注意,通常在以下气氛中进行热解:惰性气氛,氧气减少气氛,基本上为惰性气氛,最小氧含量气氛以及这些的组合和变化),可以使用专门定制的制剂。例如,可以使用高苯基含量(至少约11%、优选为至少约20重量%的苯基)的制剂、高烯丙基含量(至少约15%至约60%)的制剂用于空气热解以减少材料的降解。
热解可以在保持所需温度和环境控制的任何加热装置中进行。因此,例如,可以使用以下完成热解:燃气炉、电炉、直接加热、间接加热、流化床、窑炉、隧道窑、箱式窑、梭式窑、焦化型装置、激光器、微波炉以及它们的组合和变型以及可获得热解所需温度的其它加热装置和系统。
当在从原料到最终产品的各个阶段发生化学反应、分子排列和分子重排时,定制和预定的控制可以提供以下优点:降低成本,增加过程控制,提高可靠性,提高效率,增强产品功能,增加纯度以及这些和其它优点的组合和变型。这些转变何时发生的顺序可以基于前体的加工或制备以及前体制剂的加工或制备,并且还可以基于固化和热解条件。此外,这些步骤、制剂和条件的定制和预定选择可以通过各种转变(例如化学反应)提供增强的产品和加工特征,分子排列和重排以及微结构排列和重排。
以下是用于制备小体积形状的pdc预成型体和pdc的过程、装置、系统和方法的具体说明性实施例。
塔式系统
通常,塔式系统一般涉及在具有内腔的塔的顶部形成前体的珠粒、液滴或颗粒。内腔的内部环境可以对外部环境开放,与外部环境部分隔离或完全隔离。塔(特别是内腔)具有受控环境,可以是真空或者容纳液体或气体,并且优选地是气体。塔具有热源,该热源加热该腔(特别是珠粒在形成后滴入其中的腔)内的环境。珠粒在从加热的腔中落下时被固化。也可以就与预成型体的固化有关的其它因素对内腔环境进行控制。虽然通常优选的是珠粒沿着该腔落下,但是珠粒可以通过流动介质(例如,气流)携带或者可以通过密度效应携带。因此,尽管自上而下的运动是优选的,但也可以考虑自下而上的运动、半静态和水平的运动。可以使用声波装置、激光器、细丝、喷雾器、滴管、喷嘴、喷射器、振动装置以及其它机械、非机械装置及两种类型的装置组合,以在塔的腔的顶部形成珠粒。
可以与塔式系统一起使用的珠粒、液滴和颗粒成型装置、系统和设备的实施例以及其它实施例包括以下通用类型的液滴形成装置:例如压力驱动式液滴喷射装置,流驱动式液滴喷射装置,声式液滴喷射装置;基于二次流动介质的剪切诱导液滴产生(即剪切流液滴产生)装置以及它们的组合和变型。在压力驱动式系统中,一般使用阀来启动和停止液滴形成。这些阀可以是例如螺线管和压电类型。可以与这些成型装置和用于产生珠粒、液滴和颗粒的其它装置一起使用的致动器包括:例如压电致动、蓄压器致动、注射/容积式致动、振动致动、电磁(e&m)致动、相变致动以及它们的组合和变型。
使用这些形成和致动设备的珠粒、液滴和颗粒产生装置的实施例包括,例如喷墨装置(例如:离散流动,使得快速致动的力产生器引发的大压力梯度按需生成单颗液滴)、振动raleigh连续流解体(例如:连续流与声波波前相叠加引起raleigh解体)和气溶胶(例如:液体储库经历声能后引起表面调节使得液滴从储库表面喷射而出)型装置。
这些珠粒、液滴和颗粒产生装置可在如下频率下运转:约150hz及以上、约300hz及以上、约1000hz及以上、约2000hz及以上、约3000hz及以上、约150hz至约3000hz、约3000hz至约6000hz、约50hz至约7000hz,虽然在更高和更低的频率下也是可能的。这些珠粒、液滴和颗粒产生装置可以控制所形成颗粒的粒度分布(例如:在生产运行期间的粒度变化)小于约10%、小于约5%,优选地小于约3%,对于需要均匀性的应用而言,更优选地小于约1%。在优选的实施例中,珠粒产生装置使用对pdc前体制剂的粘度低敏感或者几乎没有敏感性的形成和致动设备。因此,在所述优选实施例中,制剂的粘度变化(例如由不同制剂的使用所带来的)对液滴的产生影响很小,甚至没有影响。
可用于珠粒、颗粒和液滴生成的其他装置和方法包括,例如水雾化、气体喷流雾化、超声雾化器、空气辅助超声波、喷雾器、烟雾机、超声波挤出系统、叶轮-乳化系统、悬浮聚合、涂覆和剥落系统、微孔泡沫系统、化学泡沫系统、气体辅助雾化、充气发泡系统、起泡系统,喷雾冷却、喷雾热解、离心雾化、火花侵蚀、直接喷雾雾化,以及这些装置、方法的组合和变化。
图3至图7、图9至图10提供了珠粒、液滴和颗粒产生装置及过程的实施例的例子。参见图3,显示的是高压喷射系统300。所述系统300具有第一喷射流302和第二喷射流303,在pdc前体入流301上形成两翼。喷射流302和303在冲击区(见圆圈304)撞击,导致pdc前体形成颗粒306,其被固化305并由移除装置307(可以是阀、闸门、输送机等)收集和除去。喷射流302、303可以是高压水、蒸汽、高压气体以及它们的组合和变化。参见图4,显示的是雾化系统400。所述系统400有一条气流管线401进料到喷嘴402。喷嘴402在进料管线403中具有pdc前体。pdc颗粒305在区域306中形成并固化。固化颗粒305通过移除装置407(可以是阀、闸门、输送机等)被除去。排气孔和过滤器408允许过量气体逸出并管理内部压力。参见图5,显示的是超声系统500。所述系统500具有超声波发生器501,其刺激pdc前体502使得pdc前体进入并喷出喷嘴503,在此处pdc颗粒505在载流气体506中形成,所述载流气体506可继续前往固化所述颗粒505。参见图6,显示的是压电系统600。所述系统600具有一个包围着喇叭603的压电发生器601和一个容纳pdc前体的通道602。喇叭与压电协同作用产生pdc颗粒605。参见图7,显示的是反射共振系统。所述系统具有喷嘴701,其上有pdc前体进料702a和702b。pdc前体通过进料进入气流703离开喷嘴702,如箭头703a所示,撞击反射器,产生向后的或反向的气流704,并在区域706中形成pdc颗粒705(颗粒也可在其中被固化)。参见图9,显示的是泡沫系统900。所述系统具有一个泡沫发生器901,其上的进料管线902中含有pdc前体;有一个泡沫破碎机903,pdc颗粒在其中成形并固化;还有一个移除装置907。参见图10,显示的是喷雾热解系统1000。所述系统1000具有气体/燃料入流1002、pdc前体进料1001、第二进料1003(可以是第二种pdc前体制剂、o2源、燃料或者其他作为附加剂的气体或材料)。pdc前体在区域1004形成颗粒并热解。
现参见图1,图中提供了塔式珠粒预成型和固化系统100的实施例。系统100具有一个固化塔101,用于保持聚硅氧烷前体批料的罐119;一个计量装置118,用于将批料沿着进料管线117传送到分配总管103。混合、搅拌、混装、泵送、流控制、反应器和调节装置也可用于前体批料的转移、处理和计量。分配头103具有喷嘴组件104、105、106、107、108和109,分别配有喷嘴104a、105a、106a、107a、108a和109a。隔热罩110、111、112保护喷嘴组件和分配头不被塔架101的热量或喷嘴组件和分配头过热所损坏,否者其反过来会对喷嘴组件和分配头的温度产生不利影响。例如,它们防止温度上升到使批料在分批头或喷嘴组件中固化进而引发堵塞的点。所述隔热罩可使用空气(例如运用气刀)、金属的、陶瓷的、气体、油、流体、化学品热交换器、反射器和水等等。
塔架101具有包含加热单元的壁102,以及用于加热单元的日照和控制的装置。在图1的实施例中,塔构造出两个区域:第一或成形区113、第二或固化区114。根据正在形成的珠粒、丸、颗粒或球体的大小(例如体积、质量),成型区113应具有足够的高度,并根据该高度选择合适的温度,使得前体材料的液滴离开喷嘴转移到固化区114之前或之时(例如从区域113到区域114),形成预定形状,例如,尽可能完美的球体。固化区114应具有足够的高度,和与高度匹配的温度,将粗加工的支撑剂固化成足够坚硬的结构,使得它们在撞击分流器115并被收集和储存在盘116时形状不会被影响。额外的固化,例如硬固化可发生在盘116、另一熔炉或在塔的第三区中。
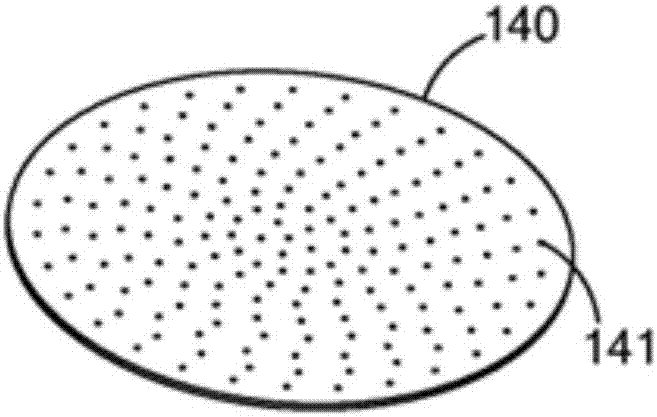
尽管在图1的实施例中使用了两个温度区和六个喷嘴,使用更多或更少的区域和喷嘴亦可行。因此,可以存在单个区域或喷嘴、两个区域或喷嘴、数十个区域或喷嘴,或更多的区域或喷嘴,以及它们的组合和变化。可使用如图1a所示的分布成形板。所述分布成形板140具有10个、100个或更多开口(例如141),通过这些开口形成液滴。还应当理解的是,除了喷嘴之外,还可以在塔顶部使用其他类型的装置来帮助即将形成珠粒、丸和球体的前体材料液滴初步形成及塑形。因此,可以使用细丝、控制前体以可控的速率和条件滴下的振动丝,以及说明说中讨论的各种喷涂、分配和成形的技术和装置。也可使用其它装置将前体批料形塑造成球形结构,然后固化所述结构,对预成型体形状的不利影响很小或者没有。
通常,例如,为了制造珠粒、丸或支撑剂,可以在塔式实施例以及其他实施例中使用表1列出的一个或多个工艺参数和设备。
表1
参见图2,图中提供了一种用于制造球形和其它类型珠粒的塔式珠粒生产系统的实施例,用于,例如水力压裂支撑剂、研磨剂和添加剂等等。所述实施例的制造方法包括若干操作,例如混合、搅拌、分配、固化、冷却和储存。应当理解的是,不是所有列出的操作都是必需的,并可使用额外的操作和这些操作的其他组合方式来制造预期珠粒。
在所述实施例中,进料和催化剂分别储存在独立储罐201和206中。进料通常包括制备具有所需性质的珠粒所需的pdc前体材料。
所述进料可含单一成分或多个成分的混合物。此外,可以使用一个或多个储存罐来容纳一种或多种单一成分或多成分混合物。
进料通过齿轮泵203从管线202泵送到混合器210。在制造过程中使用的泵可为任何种类的泵,能充分地将进料顺着管线移动,在这个例子中是将其输送到混合器210。管线202具有泵203、控制器204和阀211、216b。例如,在所述过程中使用的泵可以是离心式又或者是正位移式设计,带有组件(例如叶轮和活塞)和操作参数(例如压力和流速),它们被选择从而充分处理进料。在这种情况下,是齿轮泵203。
所述泵203由控制器204控制,控制器204可包括适合于操作的任意类型的反馈回路。例如,控制器204包括了能感应管线202中的进料流并根据预设的流量值来调整泵203的fic回路。
系统中可使用额外的控制器来控制如流量、压力、温度这些过程变量,通过完全或部分地打开或关闭阀、泵、加热器或其他操作来响应从控制器接收到的信号从而控制进料、液体或催化剂的水平,控制器将设定值与由传感器检测并提供的过程变量测量值进行比较。可以在整个系统中通常用于控制、采样、安全效率操作及类似的位置部署泵208、阀211抽头管线或喷口(未示出)。
催化剂罐206中的催化剂通过管线207经由计量泵208进料到静态混合器210。在那里,来自管线202的催化剂和进料结合在一起以形成所需混合产物,这些混合物通常是均匀的。与齿轮泵203一样,计量泵208不必具有任何特定的设计,并且可以以任何通常理解的方式进行控制。
可以使用各种混合器。例如,混合器210可以是静态的,例如板式混合器。混合器也可以是动态的。混合器可以定位在水平、垂直或倾斜的位置,其可以包括桨叶、搅拌器、叶轮和其它部件,并且其尺寸、形状和操作要足以提供上游材料的均匀混合物。优选地,混合器是连续的或半连续的,并且可以使用一个或多个混合器。
进料和催化剂的混合物通过管线212中的阀211进入到混合罐213。混合罐213可包括任何类型的混合器,并包括已述的用于进料和催化剂混合物的合适混合组件。混合罐213还可包括合适的出口,例如排水通道217和217a。
混合罐储存混合物240,还可在速度、温度和压力下运行以产生清洁并符合期望的混合物(blendedmixture)。搅拌机可以通过普遍接受的方式进行控制,并对如水平、温度、压力、流量和粘度这些过程变量进行了描述。
用于冷却或加热的温控介质可以被供给到整个所述混合罐中或者部分所述混合罐中,或者被供给到临近整个或部分所述混合罐的位置。在这种情况下,冷却介质通过管线205循环到围绕着罐213下部的外表面的护套241。
在混合罐内部,混合物240可由通过管线214加入的清洗溶液洗涤。虽然优选在混合罐中洗涤,但催化剂和进料也可在混合之前分开清洗,并在下游的其它位置混合。
所述混合物(blendedmixture)通过齿轮泵218与控制器219按预定的方式协作经管线217给进。通常,进料和混合系统的压力由本领域已知的背压或压力控制系统设定,并通过一个或多个压力指示器(例如压力指示器220)进行监测。
所述混合物(blendedmixture)经管线221进料到柱222,柱222通常包括进料区223、冷却区224和固化区225。
进料区223包括喷嘴头226,喷嘴头又包括设置在喷嘴歧管227中的一个或多个喷嘴(未示出)。进料区223的温度要确保流动性和适当的粘度,同时降低或控制管线229中的反应温度,所述管线是前体进料到支管型喷嘴227的循环回路。
根据本实施例的方法,进料区223中的一个或多个喷嘴会因分配混合物而振动。振动可以通过振动控制器228来实现,所述振动控制器可以连接到进料区中的一个或多个部件,包括管线221、喷嘴歧管227或喷嘴。喷嘴振动器控制器可以是已知用于提供和控制喷嘴振动的任意控制器。
喷嘴的形状和尺寸应能获得可用的、一般为球形的预期支撑剂。优选地,喷嘴大小范围约在20微米至800微米之间,最优选地在约50微米至400微米之间。
预期在生产过程中使用的喷嘴包括由buchi和brace等制造的喷嘴。
当通过一个或多个喷嘴进料时产生珠粒。优选地,所选择的喷嘴产生尺寸大约是孔口两倍的珠粒。
珠粒向下流经借助温度控制介质230维持的冷却区224。珠粒流出冷却区224进入固化固化区225。
固化区225包括固化柱231和加热器232。所述固化柱可以由任何典型材料构成,以允许滴下的和固化的颗粒形成预期形状的支撑剂。所述固化柱可自制或从各个供应商处购买。
喷嘴可以5hz至300hz的频率震动,优选频率15hz至200hz。据了解,振动频率与液滴尺寸、射流速度、流体粘度和表面张力有关。使用聚硅氧碳前体制剂制备直径约200微米至1000微米的球形珠粒,例如美国专利公开号2014/0326453中公开的那样(其全部内容通过引用并入本文),喷嘴振动频率,例如,可达约700hz至1500hz。
固化柱保持在一个温度。加热器232与固化柱相关联以保持柱的温度。加热器可以布置在整个柱的一个或多个区域中。优选地,加热器布置在区域中。
珠粒通过落水管233离开固化区225到斜坡234。斜坡234(可以是任何类型的输送系统,例如传送带或螺旋输送机)可以保持在恒定的预定温度。如虚线符号235所示,存在几个进料到斜坡234、容器240和这两者的柱组件。
这些珠粒离开斜坡234并在容器240或类似物中收集。此时,这些珠粒可称为绿色支撑剂,可被储存或进一步加工。容器240可以具有通风口或过流242,或者可被密封,具有受控环境,或者作为贮存罐在受控的保存环境中储存珠粒。
或者,珠粒可以通过额外的手段进一步处理以实现固化。例如,将珠粒进一步暴露于高温以硬化。硬固化可以在惰性环境中进行,例如氩气和低氧水平。珠粒可以暴露于一个或多个额外的热处理步骤,包括暴露于1000-1100℃的温度。
也可以使用像是实施例图1a中的板。板的直径可以大于1英尺、大于2英尺、大于4英尺和大于6英尺。当使用较大的直径(例如大于2英尺)时,解决了当珠粒落入柱时沿柱宽均匀加热所有珠粒的技术问题。优选地,沿喷嘴宽度施加到珠粒的辐射热量是均匀和扁平的,例如,珠粒所暴露的能量在柱长上的任意一点和在任意长度的柱宽上是相同。在考虑柱的气流和温度分布时,诸如珠粒形成动力学、催化剂、材料性质(例如组成、相、粘度、内应力等)这些因素应予以考虑。柱中的湍流也是考虑的因素,且其通常应该被控制以防止珠粒彼此间或与侧壁间不必要的接触,直到至少它们已经充分地成形,使得这样的碰撞不会影响它们的形状。潜在的湍流源是在柱中由前体蒸发可能引起的增压。例如,如果加入柱中的300g前体发生10%蒸发,则柱中压力可增至约190psig。为了解决由于柱中增加的压力可能引起的问题,优选地,柱可以在减压下运行,其中过量气体可通过控制压力的方式从柱中除去,降低、减缓或控制柱中任何压力的增加,并优选地解决、减少、消除或减轻任何柱压引起的湍流。
例如,减压区可保持在靠近喷嘴的柱顶部。以这种方式,珠粒可在它们落入柱子中具有更大压力、流量和更多湍流的区域之前成形并最初固化形成壳,优选地形成硬壳。此外,可以使用,特别是在形成区和高压区中使用层流。这将进一步帮助珠粒成型和并固化,避免产生不必要的或有害的碰撞。
液-液系统
通常,在液-液系统中,液体前体形成珠粒、液滴或颗粒,然后优选地在所述液体中固化。前体珠粒、液滴或颗粒可在被置入液体内之前首先暴露于气体中;然而,对于这些类型的方法,优选珠粒、液滴或颗粒直接在液体中形成,其中前体在形成和初始固化期间不暴露于任何气体。聚合物乳化、溶液聚合、溶液珠形成、纳米乳化和类似类型的体系可将前体制剂制备成小体积形状。在这些系统中,液-液处理使前体制剂形成小的、非常小的,即几英寸至微米、亚微米级的珠粒、液滴或颗粒;然后优选地在第二液相中固化它们。因此,这些类型的系统通常在另一种液体中,例如散装液体相、第二液体、连续相或连续体中形成液体前体材料的珠粒。形成后的珠粒在第二液相(例如连续相)中被充分固化,使得它们的形状和其它预定特性被锁定到后续加工或使用(例如储存、热解和加工等)所需的程度。也可制造更大和更小的珠粒。
珠粒、液滴和颗粒产生和成型装置、与液-液系统一同使用的包括上述塔式系统所讨论到的系统及设备的实施例。
此外,液-液技术可以与其它成型和固化技术组合。因此,例如液-液前体悬浮液可被喷雾干燥用以引发固化同时驱除过量的h2o。类似地,液-液前体悬浮液可用于本说明书中提供的其它成型和固化技术。因此,在液-液实施例以及其他实施例中,珠粒或颗粒可以在液体中成型,在液体中固化,在非液体环境中成型,在非液体环境中固化,并包括这些的组合及变化。
在第一和第二液相中形成的珠粒可加入到第三或第四液相中,用另一种聚合物、表面活性剂以及这些和其它材料的组合材料涂覆颗粒或珠粒,以固化颗粒。
本系统的实施例可以控制与液-液体系中粒度分布有关的参数,其中包括表面张力、粘度、温度、分散相(例如,前体)的体积分数、聚合(例如固化)、方法(process)、表面活性剂、吸附表面活性剂的作用和系统的流体动力学流动条件。
在一些实施例中,由于与乳剂相关的表面积(和相应的表面能)增加,散装物料在分散体上的分离可能在热力学上更有利。所以需要能量输入来产生乳剂。因此,理论上“稳定”的乳剂的概念存在热力学缺陷,但通过向系统中加入表面活性剂/乳化剂而具有动力学可能,这阻止了形成的液滴聚结成体相或结块。
优选地,在本系统的实施例中,理论上最终粒度分布是在液滴分裂和液滴聚结现象之间形成的稳态的结果。通过增加液滴分解的容易性和增加液滴聚结的阻挡作用,乳剂可形成平均粒度越来越小的颗粒。
通常更高的温度、分散相体积分数和粘度倾向于增加平均粒度。更高的表面活性剂浓度和更高能量的流体动力学流动条件倾向于降低平均粒度。
通常,在聚合(例如固化)时,聚合物表现出与初始物相不同的固体胶体性质,例如单体前体,或者对于聚硅氧碳前体来说是液体聚合物。相对于液体前体,优先考虑固化前体的吸附及附聚特性,以更好地确保固化的颗粒不聚集。对于一些实施例,聚集或结块可能并非不可取。此外,对于一些应用,例如如果附聚物可以在稍后的时间容易地分离,则这种聚集可以是非常有利的,无论是从安全方面(例如避免处理非常小的颗粒和除尘问题)还是性能方面(例如需要很少的能量将聚集体分解成一些应用中偏好或需要的尺寸较小的颗粒)考量。
虽然预期过许多尺寸的颗粒。优选通过液-液系统(例如乳液和溶液体系)获得大小约在0.1微米(μm)至4mm(5目,美制)范围内的粒度,虽然还可以获得更大或更小的颗粒。多种粒度系统的实施例将包括例如微系统(例:分散粒度约10-100nm)、微型系统(例:分散粒度约100-1000nm)、宏系统(例:分散粒度约0.5-200μm;约400目(37μm)至10目(2000μm))。应该指出,这个命名法是定义一个粒度范围,并不一定限制用于获得所述粒度的过程。
悬浮聚合过程(例如聚合引发剂可溶于分散的单体相)和乳液聚合过程(例如引发剂可溶于连续相)可用于形成小体积前体形状,这些前体可以被固化,或者稍后被固化并热解。
以下的讨论和理论通过pdc前体体积形状、pdc预成型体,优选聚硅氧碳体积形状的制备进一步解释说明液-液系统的过程。值得注意的是,提供这些解释或理论,来强调主题为用于制备体积形状的pdc预成型体的液-液体系统或与之相关的实施例的新颖的、创造性的特征和性能,并不是必须的。然而,各种理论的提出进一步推动了这一重要领域技术。这些理论除非另有明确说明,绝不限制、限制或缩小要求发明的保护范围。
表面张力通常为系统贡献积极能量,因此当表面能最小化时自由能最小化。这在确定系统的吉布斯自由能进行建模方面可能被证明是有利的,并被视为作出许多贡献。因此,考虑体积和表面能量贡献(在恒定温度和压力环境中):
系统的体积能量取决于体积。在乳液中,没有出现体积变化——仅将给定体积的液体重新排列成不同的几何取向。
dg=debulk+desurface
然而,吉布斯能量的表面能含量根据以下几个因素会改变:
debulk=0
γ是界面的表面张力,a是面积。
因此可理论化为,通常情况下最小自由能状态需要使恒定体积系统的表面积最小化。然而,如果在吉布斯自由能表面中存在全局最小值而这个全局最小值条件(即体积分离,bulkseperation)并非动力学可得,则不能排除亚稳态乳液的形成。
通常存在两种类型的准稳定态的乳液,即亚稳态乳液和稳态乳剂;虽然其他特征和类型也可能存在。在这种情况下,稳定是指相对恒定的粒度分布。
第一种一般类型是亚稳态乳液。通过以下将讨论的机理,连续介质中颗粒的自由能可以具有局部最小值。如果这个最小值足够小,建立全局最小值条件(或体积条件,bulkcondition)的动力学路径是无法进行的,因此可以产生一种不需要额外的能量输入或亚稳态乳液的局部平衡条件。然而,在实际系统中真正的亚稳态是罕见的。
第二种类型准稳定乳液——稳态乳液更为常见。因此,大多数“稳定”乳液根本不处于热力学平衡,仅仅是液滴“分解”速率和液滴“聚结”速率相等的稳态条件下的系统。在一些实施例中,这通常就足够了,优选地,乳液保持完好以便有足够的时间固化珠粒,或者在随后的使用或处理前(例如硬固化、进一步固化、研磨或热解)防止珠粒结块。
在典型的液-液体乳化过程中,以体积(bulk)或大于所需粒度的形式将待分散相(例如单体或聚硅氧碳前体聚合物)引入到连续相(例如,水相)。为了实现乳化,由于与较小液滴相关的表面积增加,通常应添加能量到体系中以诱导待分散相分解成越来越小的液滴。实现所需粒度的最小能量输入可以用以下公式来量化:
desurface=γda
恒定的表面张力(γ)可合理地假定,直到典型的微尺度。因此,由于随着直径减小,表面积快速增加,获得非常小的粒径需要高能量输入。
由于表面积的局部最小化,液滴的最小能量形状是球体。通常,为了分解液滴,一般应将其先制成非球形或细长形状。拉普拉斯压力:
其中d是粒径,抵抗被称为维持力的伸长率。施加到细长液滴上的力被称为破坏力,并且通常以剪切力来描述。
无量纲韦伯数:
其中ρ是密度,v是速度,l是系统的长度尺度,是用于理解当提供某种能量输入时液滴是否会分解的有用工具。韦伯数给出了破坏力和维持力之间的比例,高于某个韦伯数(达到足够的时间),引起液滴分解。
存在可以向液滴提供破坏力的几种机制。仅举几例,剪切力可以在剪切层流、湍流漩涡或收敛/发散流系统中提供。不同的均化/乳化技术利用不同的机制来诱导剪切分解。可以利用各种系统和装置在更大的系统中提供这些力,例如100加仑、200加仑、500加仑、1000加仑和更大的系统。
通常,如果流动条件的长度尺度与颗粒的直径相当,颗粒将经受显着的剪切力。因此理论上,不具备短长度尺度部件(shortlengthscalecomponents)的高能量整体流不太可能分解液滴。因此提出使用按尺度依赖的能量参数来关联由各种技术提供的剪切力的大小。这是比能:
其中p是系统的功率,v是流量。
经验证明平均液滴尺寸与ev成反比。然而,在不同的流动条件(湍流、收敛等)下,几何的特定方面超过ev区分粒度。下图说明了在形成聚硅氧碳预成型液滴方法的多个实施例中ev对粒度的影响。
应当注意的是,足够的能量密度对促使液滴分解是必要的,但是一些实施例中的能量密度可能不足以使液滴分解。液滴分解的动力学可能是复杂的,但是经验证明存在分解发生的特征时间尺度,这取决于许多材料和机制的参数。因此,在一些实施例中较长的时程(例如大于30分钟、大于1小时、大于2小时等)对于达到稳定状态可能是必要的。
在缺乏体积条件(bulkcondition)的热力驱动力的理论系统中,乳化将通过液滴分解进行,直到达到超过最小韦伯数的最小长度尺度。然而,实际系统被驱使“聚结”或聚集,以便最小化表面积从而最小化表面能。
没有吸引力或排斥力的系统将具有一个作为两个颗粒分离的函数的平带电位,除了重叠点之外,相对于非附聚颗粒其电势基本上是负的(表示聚结)参见图18,其中分离1801对自由能1803绘制曲线1802。
在这种系统中,通常颗粒之间运动的驱动力是热力学和流体动力学(介质的流动状态)。如果两个颗粒接触,除非提供足够的破坏力,否者它们会在较低能量状态下聚结并保持接触。
然而,实际系统具有的吸引力和排斥力将自由能修正为分离的函数。
范德华力(或感应偶极子振荡力)作用在非常短的距离上,并用于非极性颗粒的相互吸引。所得到的曲线与图19性质类似,其中分离1901对自由能绘制出曲线1902。
在非常短的距离处,原子电子云开始重叠并发生born排斥。所产生的电势,包括范德华力,与经典的lennard-jones电势相似。然而在一些实施例中上述效果不是理解较大粒度聚结的一个重要因素,例如微型系统(如数百纳米)和更大的粒度。
因此理论上,如果不修正系统以囊括额外的排斥力,自由能曲线随着颗粒聚集而单调递减;因此通常不可避免地导致聚结。为了得到所需粒度的亚稳态或稳态平衡粒子,通常应采用动力学屏障阻挡聚结。液-液系统中存在几个动力学屏障的来源,例如乳化体系。
如果颗粒本身或更常见地颗粒表面带电,则库仑力存在于系统中并可起到排斥颗粒的作用。这种库仑排斥比范德华力在更远的距离便可发生。作为分离曲线函数的所得自由能对具有带电表面的系统而言具有以下特征,如图20的曲线图所示,其中自由能2003对分离2001作曲线2002,且存在动力学屏障δg2004。因此,为了使两个颗粒达到对于聚结能量有利的分离点,通常应该给予系统足够的能量以克服δg。这种能量可来自于例如流体动力或热运动。
系统中带电表面的来源可以是多种多样的。表面物质可以被直接电离产生残余电荷。这可通过许多机制实现,但通常是依赖于ph。因此,改变连续相的ph可产生电荷排斥影响。
如果系统中存在较低价的离子被交换为原始状态,则所述系统中存在的所述较低价的离子可形成负电荷。
带电吸附物质的存在可导致带电的表面相互作用和排斥。这是乳化剂起作用以促进液滴分解和破坏液滴聚结的一般机制之一。
理论上,颗粒间介质(连续相)的介电常数以标准方式改变了库仑排斥的影响。然而,连续相的存在具有产生带电双层的附加影响。所述效应用于通过近端定位反相带电物质(proximouslylocatinginverselychargedspecies)来屏蔽表面电荷。因此,如果分散相的表面带负电,则连续相中的正电荷将形成双层结构以抵消负电荷。层的厚度取决于溶液的离子浓度——连续相中存在的离子越多,由于屏蔽离子的可用性越大,层就越薄。双层越厚,因系统带电引起的排斥效应越大。因此,连续相较低的离子浓度导致更大的电斥力并因此提高乳液的稳定性。所得双电层的电势称为电动电势。
通常,空间排斥力是指通过将物质吸附到用于阻止颗粒重叠的颗粒表面上施加的屏障。通常有两种空间排斥机制:修正范德华力和熵排斥。如果在分散颗粒的表面上存在具有类似于连续介质性质的一层厚的吸附物质,则通常范德华力被屏蔽。此外,通过与相邻颗粒接触而吸附在分散颗粒的表面上的聚合物的压缩将使系统的熵从平衡状态降低,从而诱发熵排斥。因此,吸附的物质可以作为聚结的屏障。空间排斥机制通常依赖于表面活性剂/乳化剂的浓度,并且在一些实施例中对表面活性剂/乳化剂浓度可能高度依赖。
当两个颗粒被驱动以聚结时,它们通常通过连续介质移动。它们移动时会置换液体,从而导致能量耗散。因此,诱导聚结所需的能量通常大于流变活性连续体缺失时所需的能量。此外,对于液-液乳化液,分散相在颗粒碰撞期间将经历几何变形,导致进一步的流变耗散。
有人建议用薄膜的物理学解释聚结现象,因为表面被破坏而聚集的动力学途径是由薄膜现象决定的。通常有一个临界膜厚度,低于这个厚度膜会自发瓦解。
前面的讨论描述了减少或增强碰撞引发的聚结的理论和机制。然而,其他的聚结机理在乳化体系中也可能是有效的。例如,其他机制之一是ostwaldripening或扩散物质转移。由于表面积最小化导致的能量最小化的根本驱动力仍然是质量传递的积极动力。通过认识到存在较大直径颗粒的体系所占的体积百分比越大,总体表面积越小,就可以直观地了解这一点。因此,牺牲较小颗粒促进较大颗粒生成通常是热力学有利的,且允许扩散通过颗粒曲率引起的浓度梯度进行。扩散转移现象将趋于在更长的时间尺度上发生,然后碰撞聚结,直到系统中出现非常小的直径。质量传递速率大致恒定,因此直径变化率随着直径的变化而急剧变化。
通常,在微型系统和较小的系统中通过两个主要过程来实现珠粒、液滴或颗粒成型、聚合\交联或固化。这些过程是是乳液聚合和悬浮聚合。
在制备pdc珠粒、液滴和颗粒的乳液聚合方法中,分散相通常是前体制剂,连续相通常是水相的。为了引发固化,引发剂(例如催化剂、热或这两者)被加入到所有系统上或被活化。引发剂优选溶于连续相。因此理论上前体制剂聚合物的表面用作成核位点,但在连续相中发生颗粒聚合或生长。前体制剂聚合物的初始乳液同时用作成核位点和颗粒生长的储库。通常这会影响pdc的生长动力学和粒度分布,例如聚硅氧碳、预成型颗粒。
由于存在一组可控的成核事件,因此理论上可以同时开始成核,使几乎所有的颗粒都能产生相对单分散的最终颗粒。然而,颗粒的生长速率被扩散地限制,并且根据系统和前体的具体实施例,乳液的完全形成可能需要很长时间(例如大于30分钟、大于1小时、大于12小时、超过24小时)。
在悬浮聚合中,引发剂(例如催化剂)在乳化之前可溶于pdc前体(例如聚硅氧碳前体),而引发剂通常在乳化前就被加入到(例如早已存在于)分散相中。因此对于分散相,固化发生在原位。引发剂可以是催化剂、热,优选这两者。因此理论上,粒度分布直接由乳液中的分散相分布控制——这些通常应该成为最终的颗粒。进一步推论,聚合/固化可以比乳液聚合发生得更快是因为在这个过程中没有扩散速率限制动力学。
预期不同系统和前体剂型的各种实施例提供特定的最终(例如固化至可以使用或进行进一步加工的程度,优选没有变形和附聚的风险)粒度和分布。进一步解决了可能影响粒度的几个因素,以用于属于本系统范围内的各种附加实施例的过程决策。
对于乳液聚合,最终粒度通常主要由成核和生长动力学决定,而不是通过初始的乳液液滴分布来确定。一方面,一些乳液聚合的实施例中,生长是颗粒的同时成核。此外,在一些实施例中,获得乳液聚合所需的长时间稳定的稳态乳液是具有挑战性的。通常,优选确保例如在聚合期间反应器保持均匀的热力学和动力学条件,以促使单分散颗粒的生长。
由于乳液聚合体系中颗粒生长的动力学,理论上,与较大的粒度相比更小的粒度(几百纳米)倾向于需要较低的产能。这可以通过注意到在颗粒生长的乳液中存在有限数量的成核位点来理解。当达到所需的粒径时,反应通常停止,但是除非成核位置的数量足够多以致于占用了初始前体制剂聚合物的总体积,系统中可存在过量未固化的前体乳化。对于液-液前体制剂,其固化反应在珠粒或颗粒内完全发生或基本上完全发生(解释了集肤效应和表面效应),通常不是考虑的因素。
在优选的实施例中,粒度分布可大致呈正态,例如高斯分布。
对于悬浮聚合,通常乳液液滴粒度和分布将直接对应于系统的颗粒粒度。
理论上,液滴大小和分布很大程度上是液滴分离和液滴聚结之间稳定状态的结果。以下是各种材料性质、热力学条件和乳液机理对分解、聚结和粒度分布的影响的其他描述。液滴粒度分布通常取决于过程,并且与流体系统中的长度尺度上的能量分配统计有关。系统中表面活性剂/乳化剂的存在可以对液滴分解和液滴聚结现象产生影响,并且在一些实施例中可以改变液滴分解和液滴聚结现象。
理论上,一些实施例中表面活性剂对液滴分解的主要贡献之一是降低表面张力。理论上,吸附的熵影响降低了系统的自由能。因此具有吸附物质的颗粒的表面张力减小。
对于所述减少的发生,最常见的机理是具有亲油性和亲水性末端的表面活性剂。亲水端与前体聚合物和具有连续体或连续相(例如水相)的亲水末端缔合。通常以表面活性剂覆盖的颗粒的表面百分比越大,表面张力的降低越大。
通常,吸附有表面活性剂的颗粒表面的百分比取决于连续体中表面活性剂(其又取决于浓度)的活性。表面活性剂浓度越大,颗粒表面被覆盖得越多,表面张力越小。通常这是一个非线性过程,活性的热力学使收益减少。此外,如下所述添加表面活性剂可以改变连续相的粘度,进一步影响分解现象。
特定体系中的一些表面活性剂已经显示出促进自发乳化。这通常带来微乳液特性,其理论上是热力学可能的。
如上所述,表面活性剂的吸附也可以产生另外的聚结动力学屏障。这对令人沮丧的聚结现象和稳定乳液都有影响,从而实现更小的液滴尺寸。这些障碍可以是空间或静电。同样,高表面活性剂浓度可以影响连续相的粘度,而这种影响在一些实施例中强烈地影响聚结现象,如下所述。
通常,固体纳米颗粒可用于代替化学表面活性剂,尽管它们在降低表面张力方面可能不太有效,但它们具有破坏聚结的能力。
用于给定方法,合适的表面活性剂高度依赖于使用的材料体系、方法的温度范围(如下所述)、所需的粒度分布,以及该方法中进一步的步骤(即干燥/粉末形成)和其他因素。通常,优选地,选择具有最大表面张力降低的表面活性剂,因为这是一个因素,并且在一些实施例中可以是影响液滴大小和所需能量输入的关键参数。
下面的表2是跟踪平均粒度作为表面活性剂浓度相对于分散相浓度的函数的典型经验结果的曲线。
表2
通常存在一种表面活性剂浓度,高于所述浓度时观察不到液滴直径附加下降,或者可忽略不计。这通常是由于高浓度对液滴分解的不利影响。基于实验证明,所述浓度与分散相体积分数无关,因此与吸附的热力学有关。
对于其中表面活性剂是离子的(诱导电荷效应)的系统,不认为存在这样的阈值。
通常,正常情况是能量输入和乳化剂浓度可以用作独立的杠杆来调节颗粒直径。可以通过增加乳化剂浓度来节省能源,但可能会带来下面阐述的后果。
高浓度的乳化剂可以改变连续体对前体制剂的扩散性。这在一些实施例中影响,且在一些实施例中强烈影响乳液聚合方法的动力学,但在一些实施例中通常不会影响悬浮聚合的动力学。
在用于扩散的热力学驱动力太大而不能使颗粒保持亚稳态之前,也可能存在最小稳定的粒径直径。这受到,并在一些实施例中受到,乳化剂浓度的强烈影响,因为聚集态和分散态之间的自由能差异减小。
由于引入大量的乳化剂对于获得微乳液可能是重要的,所以通常应考虑这些化学品对聚合后的工艺步骤的影响。具体来说,某些类型的表面活性剂可使干燥颗粒更困难,从而使得粉末形成过程更加复杂。选择乳化剂是考虑后续工艺和最终用途要求的一个因素。
粘度是确定最终粒度分布的一个因素,并且在一些实施例中是关键参数。例如,要考虑到两相的分散相粘度、连续相粘度和相对粘度。分散相的粘度可与粒度直接相关。应注意的是,韦伯数不考虑分散相的粘度。当分散相的粘度高时,会发生流变消散。因此,需要额外的能量以获得与较低粘度分散相相同的液滴直径。然而,同样的效果也可以影响聚结:由于碰撞聚结可能引发流变学耗散,因此理论上可以降低聚结事件的可能性。这种效应通常不如分解效应那么强,并且由于存在另外的聚结动力学途径(例如奥斯特瓦尔德成熟)而进一步抵消,如图5的曲线所示,其中平均下降直径2103对能量密度2101绘制曲线2102。
几个因素可超过均匀介质的特性粘度影响系统的粘度。
颗粒相互作用——对于高分散相浓度,颗粒往往相互作用。空间排斥和库仑相互作用引起粘度修正。具体来说,将其视为液体接触中的两个球体,可以将其理论化为彼此碰撞,导致流变消散和动量传递增加系统粘度。如果两个带电粒子经过彼此,附加的力就会产生动量传递,并增加系统的粘度。
乳化剂浓度——加入乳化剂可以影响,并在一些实施例中直接影响连续介质的均匀粘度。然而,由于电粘性和空间排斥性,粘度可以不同的方式依赖于乳化剂浓度。
在高浓度分散相下,颗粒相互作用可以引起系统的非牛顿粘性行为。这可能使系统的流体动力学变得复杂,并且倾向于产生更大的平均粒度。
通常,对连续相粘度主要的独立影响是能量输入的粘性耗散。连续相的粘度可以对系统的流体动力学条件产生很大的影响,并且可能导致湍流和涡流长度尺度的改变。然而,这种影响通常具有高度的器件特异性,因为会合流相对湍流混合技术具有不同的几何形状和流动条件。
对于一些实施例而言,重要的是分散到连续相的相对粘度。当大不相同时,倾向于产生较大的粒径。这可能是由于剪切力的低效通信或跨越相位边界耗散。图22显示了平均液滴直径2203对粘度2201的典型曲线2202(表明液滴大小作为粘度的函数)。
温度可影响聚结和分解现象的动力学,并在一些实施例中存在显着影响,因此粒度分布可以取决于温度,而且理论上可以多种方式依赖于温度。
还应当注意的是,温度影响聚硅氧碳前体制剂的固化速率和固化程度。
粘度通常是温度的函数,且不同的材料可以具有与给定温度变化相关的不同的粘度变化大小。因此,分散相和连续相的独立粘度和相对粘度可以通过温度和冲击液滴的尺寸来改变。
温度反映系统的平均动能。尤其是在一些实施例中(例如对于小颗粒),这可直接影响颗粒的布朗运动,因为更高的平均动能克服由表面活性剂建立的动力学障碍,导致聚结速率增加。具体来说,如上图所示,如果能量势垒δg大于几kt(其中k是玻尔兹曼常数,t是温度),则在动力学上抑制聚结。然而,随着温度(t)的增加,所需的屏障的大小也额外增加。
表面活性剂对颗粒表面的吸附通常是温度依赖的(由于活性的温度依赖性)。通常,吸附更高的温度增加吸附,产生较厚的空间壳,尽管这可以是系统依赖的。温度对静电活跃(electrostaticallyactive)的表面活性剂的负面影响往往比对依赖于立体机制的系统小。
扩散也是温度依赖的(对于热活化扩散机制),因此较高的温度通常增强奥斯特瓦尔德成熟。
成核速率随温度而变化。对于乳液聚合的过程,较高的温度可大大提高颗粒的成核度,从而降低平均粒径。
由于温度影响等温系统中的粒度,因此应考虑温度变化对颗粒分布的影响。图23中曲线追踪了在等温和非等温过程的非固化系统中平均液滴尺寸对时间的函数。在非等温过程中,温度随时间稳定上升。
通常,在实施例中温度的升高导致更大的平均粒度。因此,在一些实施例中,作为时间的函数的所述变化可导致较少的单分散粒子。因此,图23提供了粒度2303对温度2301的等温和非等温行为曲线2302。
此外,经验证明,与等温系统相比非等温乳化趋向于具有更陡峭的瞬变。较高的温度降低分散相的粘度,从而降低了界面张力并增强分解。然而,一般在实施例中温度对聚结现象的影响可以以更长的时间间隔压倒最初的优势。
温度的影响是高度依赖于系统的,因此应当认识到,在乳化体系的各种实施例中,这些温度效应以及其它温度效应可以更大或更小的程度存在。例如,温度效应应该与温度对固化速率的影响平衡,特别是聚硅氧碳前体制剂的固化速率。
各种因素都可以影响系统的温度。例如,一些典型的因素是来自均化器的能量耗散和放热聚合反应。应当注意,随着分散相的浓度增加,放热反应的作用通常变得更大。
分散相的体积分数可以影响系统,以下变化可导致反向乳化,在这种情况下分散相和连续相反转。通常,大体积分数可以诱发非牛顿粘性效应,增强粘度,导致放热系统的温度变化,增加聚结概率,降低表面活性剂的最终功效,以及这些的组合和变化。在大多数实施例中,体积分数对粒度的影响趋向于为负——体积分数越大,平均粒度越大。典型的结果显示在图24中,对两种不同的表面活性剂浓度2402、2401,用液滴尺寸2402b、2401b对体积分数2402a,2401a作曲线。
通常,这些理论和解释适用于液体连续相内的固体和液体分散体系。因此,适用于广泛的pdc前体。对于聚硅氧碳前体,液体分散体优选在液体连续相中固化成固体。因此,在从液体到固体的这种过渡中,在一些实施例中可能出现区别,并且相对于其他因素主要涉及在碰撞聚结和颗粒分解期间颗粒的流变学考量。
当液滴形成薄层(skin)并且通过机械乳化混合器重新评估,它们可分解并形成较小的非均匀颗粒。
在确定最终粒度时,应考虑粘度演变的几个方面。随着聚合或固化的进行,液滴的粘度增加。此外理论上在一些实施例中首先形成固化材料的初始表层。在固化期间发生的这些和其它变化(例如液态转变为半固体或固体状态)导致上述原理下的分解和聚结现象的改变。可能出现的一个效应的例子是,如果固化时间很长导致粘性逐渐演化,则稳态颗粒分布尺寸可能会蠕变。乳液可能失去稳定性并且聚结。
分散相表面上的表面活性剂的吸附特性可在聚合的固体和单体液体之间变化。因此,表面活性剂的空间或静电排斥性能可以根据固化程度而变化。这可能会导致固化后聚集。因此优选同时适用于单体液体和聚合物固体的表面活性剂。
这些影响的另一个例子涉及固体通过不同机理聚集的因素而不是液体凝聚。具体地说,它们不需要减少表面积来粘附。这意味着聚集动力学可以在液相中变化,并且在一些实施例中变化很大。液-液乳液可以是稳定的。但固-液乳液可天然聚集。或者可以根据速率和固化程度聚集,例如初始固化的固体的内聚力,或固体的表层。
也应考虑固体絮凝的一般倾向。通常当所有其他因素相同时,由于系统中缺乏流动,较高密度的固相将因重力而自然地聚集在反应器的底部。一旦聚集,范德华力可以用足够的力粘附絮凝物,需要高剪切以获得所需的粉末尺寸分布。此外应注意的是,随着悬浮液体的尺寸减小到亚微米范围,伴随着着表面积/克增加,液滴或固化液滴(固体)相的表面能变得越来越重要。系统能量的最小化是通过优选使颗粒/溶剂表面能最小化的机制来减少表面积进而发生的。
液滴分解和凝聚的许多实施例和方法本质上是动力学的,固具有与它们相关的速率。因此,时间依赖对乳化体系的影响是通常应该进行评估、利用和考虑的因素。
通常,系统具有与达到稳定状态相关的特征时间(对于没有聚合的静态热力学条件)。其影响因素包括扩散转移、涡旋形成、分解动力学及其组合和变化。图25显示了粒子分布2503和rpm2504(例如叶轮驱动系统)作时间2501的函数曲线2502。在此,叶轮rpm直接对应于韦伯数计算中的流体速度,因此可见rpm增加导致平均粒度减小应归于增加的剪切力和比能。
在许多实施例中,当所有其他因素通常恒定,粒度向稳态的演变可以是或表现为时间常数。
表面活性剂吸附也可以是时间依赖的。由于例如热力学或动力学的原因,“快速吸附”表面活性剂容易吸附。快速或缓慢的吸附物质的影响是确定最终粒度分布的一个因素。当认识到一旦被吸附,表面活性剂分子通常不会永久地附着在液滴的表面上就可以理解。相反,它们不断地与连续体交换。这通常发生在几何结构改变例如碰撞或分裂时。如果表面活性剂不容易被再吸收则可能发生聚结。
图26定性地显示出实施例中物质吸附速度的影响,其中曲线2602显示了物质吸附的不同速率下,能量2601与平均液滴直径2603之比。更快的吸附物质通常倾向于降低平均粒度。
对于直径小于约1微米的分散体,可考虑额外的物理学,这可以一步影响粒度分布和聚合动力学。在这些较小的粒度下,影响系统自由能的曲率是显着的。这可影响表面活性剂的吸附概率以及固化反应的聚合动力学等。两种效应通常取决于不同的前体和体系,并且可能随不同的前体和系统而变化。
通常,从实验室到大型工业规模生产(例如每天数百、数千和数十万磅)的各种尺寸和形状的支撑剂、珠粒和颗粒可用溶液形成系统完成,但优选为球形。这种类型的系统可以用于制造固化的体积形状,例如珠粒和颗粒,其可用作例如颜料,用作水力压裂中的支撑剂,在烃生产中提供高纯和超纯(例如:大于99.999%,大于99.9999%纯度)(>5ninespure,>6ninespure)的sioc,例如作为制造高纯度sic和相关电子应用的源材料。
通常,这些系统仅需要最小的组分提供制备大量颗粒、珠粒和支撑剂的能力,并且如果需要具有非常高的均匀性,例如粒度分布(例如至少约70%或更多的粒度分布在5%范围内,至少约80%或更多的粒度分布在5%范围内,至少约90%或更多的粒度分布在5%范围内)。这些窄分布以非过滤或非筛分形式获得,即不需要过滤、筛分或后处理。
通常,这些系统具有优选为温度控制的容器。容器可以是例如罐、槽、槽、通道和其它形状与类型的结构,其容量为大约1加仑或更小,至100加仑或更大,至1000加仑或更大。容器内有液体,液体内前体批料形成体积形状然后固化。容器中的液体可以是任何液体(这种液体被称为连续体、连续相,也可以称为成形液体,除非另有说明,否则所有这些术语都是可互换的),允许前体在成形液体中形成离散的的体积形状。对于pdc前体制剂,成形液体优选是水、蒸馏水、去离子水,但也可以是极性液体、非极性液体、醇以及它们的组合和变化。优选地,对于聚硅氧碳前体制剂,成形液体在一个实施例中是具有降低的氧含量(例如,溶解的o2)的去离子水,例如优选少于约o2、少于约10ppm的o2、少于约5ppm的o2,少于约1ppm的o2和少于约0.1ppm的o2。在一些实施例中,对于一些前体制剂,可以使用更高的o2含量,例如20ppm、50ppm、100ppm和300ppm等。成形液体也可以具有并优选具有表面活性剂。此外,可以利用其它技术或方法,例如水的ph、离子强度和/或电导率的变化结合或独立于表面活性剂。还可以使用例如连续相的添加剂,例如流变改性剂、稀释剂、消泡剂、抑制剂、其它催化剂、反应性稀释剂等。表面活性剂可以是具有极性和非极性官能团的任何类型的表面活性剂。例如在聚硅氧碳-水体系中,表面活性剂可以是单甘酯和甘油二酯(例如atmos300、arlacel165)、脱水山梨糖醇脂肪酸酯(例如span20、span80)、聚氧乙烯脱水山梨醇脂肪酸酯(例如tween80、tween61)、聚氧乙烯山梨糖醇酯(arlatonet、atlasgn-1441)、聚氧乙烯酸(myrj52、myrj45),聚氧乙烯醇(brij72、brij58、renex36)和离子表面活性剂(十二烷基硫酸钠、atlasgn-263)。一般来说,考虑到成本和纯度,最好使用需要的表面活性剂。通常,表面活性剂可以以约0.1%表面活性剂的浓度使用形成液体(例如水)或更浓,0.5%或更浓,1%或更浓,5%或更浓,7.5%或更浓。表面活性剂也可以加入到前体制剂、成形液体中和两者中。在前体和成形液体中可以使用相同或不同的表面活性剂。
表面活性剂可以以连续或间歇的方式加入到连续相中。表面活性剂-连续相混合物可以再循环,且随着颗粒或珠粒固化被除去,额外的表面活性剂被加入到连续相中以替代在固化颗粒上损失的表面活性剂。
优选地,成形液体温度通过例如与容器相关联的加热和冷却装置来控制。应控制液体的温度以解决问题,尤其是任何放热,来增强珠形成和均匀性。优选地,成形液体在大气压下(更优选地,表面处在惰性气体中),其温度保持在约80℃至约90℃至小于100℃。通常,在形成液体中形成并固化体积形状,例如珠粒,使其在较大或较小程度上固化。作为说明,将使用珠粒作为体积形状,应当理解,这不限于形成其它类型的体积形状,并且以下适用于这种其它形状。珠粒可能仅在成形液体中部分固化,并且随后或后续的固化可能发生在热裂解之前,或作为热解的预备步骤。基于前体制剂,部分固化的和固化的珠粒的储存应优选在少氧及低温条件下进行
充分混合以搅拌成形液体,为前体提供剪切,并控制粒度、聚结、温度均匀性和尺寸分布等等。可以以各种方式将前体添加到液体中,从简单地通过空气注入到位于大气表面下方的喷嘴、喷雾、筛网、分配头、气体(例如大气)中产生的液滴,然后落入连续相的表面以及这些和其他装置和方法的组合和变化来将前体引入到成形液体中。可以有单个、两个、三个、四个、数十个、数百个以上的前体引入装置,并且每个前体引入装置可以具有多个引入端口或开口,这取决于系统的尺寸、生产率,固化率,预期的粒度,预期的粒度分布等因素。
图14是溶液形成系统和过程的实施例的示意图。所述系统可用于批次和连续生产固化珠粒。溶液成形系统1401具有容纳成形液体1402的容器1403。成形液体1402存在表层1407,即其上方的大气。成形液体可以是例如去离子水、含约1%的表面活性剂、含优选少于约15ppm的o2、少于约10ppm的o2,少于约5ppm的o2,少于约1ppm的o2和少于约0.1ppmo2。
1403具有温度控制装置1404,其具有控制和电力电缆1405。应当理解,温度控制装置可以位于容器内,如图所示可以在容器内的任何位置,在容器壁、容器外、流体入口线、储罐上,以及这些的组合和变化。容器具有位于容器1403内的搅拌器1406,搅拌器具有驱动构件1412,例如驱动轴和叶轮1413。搅拌器叶轮1413位于液体1402表面1407的下方并且靠近开口1411用于进料管线1408的喷嘴1410,例如前体引入装置1415。
前体引入装置1415可以具有例如供给管线1408、喷嘴1410,形成用于将前体引入到表面1407下方的成形液体1402的开口1411。引入装置可以具有多个进料管线,这些管线可以在容器的壁内,因此本质上在容器壁中形成用于引入液体的开口。容器壁导入开口可以在容器的侧壁、底部和两侧。单个进料管线可以具有与其相关联的一个、两个、三个或更多个开口。进料管线连接到聚合物的补充、储存或其他处理设施。应当注意的是,通常当前体在进料管线中时其已被催化。尽管在一些实施例中,催化可以发生在其它位置,例如位于进料管线中的内联混合器。
应当理解的是,搅拌器可以位于相对于成形液体的表面、容器和前体引入装置的其它位置。还可以有一个、两个、三个或更多个与容器相关联的搅拌器。搅拌器可以完全位于液体的表面下方,并且优选地位于所述位置,以最小化在驱动轴表面界面处的气体夹带。搅拌器可以位于不同的水平,当使用多个搅拌器时,例如,可以将所述搅拌器定位在与前体引入装置相邻,可从所述装置移除其它搅拌器。搅拌器可以在成形过程中不同的时间段以及它们的组合和变化下以不同的速度运行,来促进颗粒形成、粒度分布和固化,命名几个参数及效应。
图15显示了溶液形成系统和过程的实施例的原理图。溶液形成系统1500具有容纳成形液体1502的容器1501。成形液体1502具有作为其上方介质的表面1508。(注意虽然介质优选为气体,但它可以是液相、减压气体、高压气体、环境压力下的气体和真空)。成形液体1502可以是例如含表面活性剂的去离子水。
容器1501具有包裹容器1501壁1516的温度控制装置1503。应当理解,温度控制装置可以是外部的也可以在容器上,如图所示,位于容器内任何地方,在容器壁、成形液体入口管线、储罐中,及它们的组合和变化。容器具有位于容器1501内的搅拌器组件1504,搅拌器组件1504具有驱动构件1512,例如驱动轴和叶轮1513。整个搅拌器组件1504位于成形液体1502表面1508的下方。应当理解,搅拌器可以位于相对于成形液体表面、容器和前体引入装置的其它位置。
前体引入装置1515具有进料管线1505和分配组件1530。分配组件1530具有筛网1506,筛网1506具有大量开口,例如1507,通过它前体得以进料,例如泵送、流动、重力进料等。开口的直径可用于调整粒度,并有助于和确定颗粒的形状。例如,圆形开口等可以用于球体。优选地,筛网1506和筛孔例如1507位于成形液体1502的表面1508下方。
引入装置可以具有多个进料管线,这些管线可以在容器壁上,并且因此进入容器壁上的筛网以便引入成形液体。容器壁导入开口可以在容器的侧壁、底部和两侧。容器壁上可以有一个、两个、三个、四个或更多这些筛孔。
图16显示了溶液形成系统和过程的实施例的透视图。所述实施例优选地提供连续操作。形成系统1600具有容器1601,在该例中为矩形槽。容器1601具有四个区域1601a、1601b、1601c、1601d。四个区域可以具有相同的或不同的条件。因此,四个区域可以具有不同的温度、表面活性剂水平、搅拌程度、温度、流速以及它们的组合和变化。例如,可以将第一区域1601a为颗粒形成及其预定尺寸设置最佳条件。第二区1601b可具有用于初始固化的最佳条件。第三区域1601c可具有防止最终固化期间附聚的最佳条件,并且第四区域1601d可以是固化颗粒的去除或收集区域。容器1601包含沿箭头1604的方向流动的成形流体1603。根据区域以及所述区域的预定目的或功能以及其它因素,将流体或多或少地搅动。通过分配头1602将前体添加到成形流体1603中,分配头部1602可以是筛网、几个筛网、喷嘴、狭缝,以及这些和其他的引入前体装置的组合和变化。通过颗粒去除装置1605将固化的颗粒从系统中除去,其可以是细网状物收集系统或筛网。返回管线1606使形成的流体返回并通过入口管线1607进入容器。
如此,颗粒例如支撑剂在分配集管1602处或附近形成,并且由流动的成形液体1603沿箭头1604的方向承载。系统1600的各个区域提供必要的条件产生固化的颗粒,例如固化的支撑剂。
如果固化的材料被热解并转变成陶瓷,优选去除多余的水;并且更优选地,在发生热解之前所述材料是干的。在一定程度上,固化材料应在低于150℃、低于140℃、至少低于100℃的温度下储存,最好还应储存在还原氧中。在热解之前、固化的颗粒从成形液体中除去后也可以进行附加的固化。这种最终的或进一步的固化和热解可以一起发生(例如串联在同一炉中),或者可以是分离的步骤(例如步骤之间不同的炉子和储存时间)。
可以使用诸如图14、图15和图16实施例中的系统来实施的方法的实施例,所述实施例可用于从前体中产生球形或非球形颗粒。对于水性体系,应注意的是温度通常为或低于水的沸点,例如100℃(在标准温度和压力下)。对于较高沸点温度的成形流体,或增加水性流体的压力,可以使用更高的温度。通常,这些系统可以与液体形式的任何类型的前体一起使用,只要前体在加工温度下不与成形流体反应,例如不在100℃或低于100℃与水反应。通常,使用聚硅氧烷前体的水性体系的实施例可以产生约5毫米直径至约1微米的颗粒,尽管可以考虑更小和更大的尺寸。由这些实施例制成的颗粒可以具有不同的形状,并且将包括例如硬质球、球形、十二面体、小面以及其它体积形状。
可用于这些系统,特别是水相系统的聚硅氧碳前体制剂的实例是:100%mhf;95%mhf-5%tv;46%mhf-34%tv-20%vt;70%mhf-20%tv-10%vt;75%mhf-15%tv-10%vt;85/15mhf/dcpd原位反应共混;70/30mhf/dcpd原位反应共混;和65/35mhf/dcpd原位反应共混;60/40mhf/dcpd原位反应共混;和82/18mhf/dcpd原位反应共混。另外,为了提高固化珠粒的硬度,减少固化时间,可以加入1%至50%的tv,1%至20%的低分子量(mw<1000)vt。催化剂可以包括例如1-20ppm的铂、稀碱、稀酸、胺催化剂以及其它类型的催化剂。
例如,以下工艺参数可用于制备500微米的珠粒,该方法可以放大应用于更大的体积,然后可将珠粒固化以形成支撑剂
—将192克65/35hhf/dcpd+8克tv混合至少5分钟
—加入1克p01催化剂,并彻底混合至少10分钟
—单独地,将400克去离子水与4克hlb9.1表面活性剂混合,并用机械搅拌器剧烈搅拌以溶解表面活性剂
—将催化的聚合物倒入水/表面活性剂混合物中并用机械搅拌器以100-500rpm搅拌,以将聚合物分解成所需尺寸的液滴(通常2-3分钟搅拌)
—将聚合物/水/表面活性剂混合物从上面倒入1升已加热到63-68℃的去离子水中,并使用低剪切搅拌方法搅拌45分钟至1小时,同时保持整批材料温度范围在45℃至50℃
—珠粒变硬而不粘稠后,将液体倒入板中或通过一组筛子从水/表面活性剂混合物中分离珠粒和凝固的硅氧烷
—然后将所述珠粒干燥并进一步固化,加热1-2小时至50℃-60℃,然后加热1-2小时至80℃-85℃,随后1-2小时在115℃下完成固化
—所述珠粒随后被热解成陶瓷
hlb是亲水亲油平衡。一般来说,可将hlb数值分配给要用于形成乳液的成分组,然后选择表面活性剂或表面活性剂的混合物以匹配所述数值。通常,乳化剂的hlb表现的是其亲水亲油平衡,例如乳化剂亲水性(亲水性或极性)和亲脂性(油性或非极性)基团的尺寸和强度的平衡。通常,乳化剂由同时结合亲水基团和亲脂基团的分子组成。
具有亲脂性的乳化剂被指定为低hlb数(低于9.0),而具有亲水性的乳化剂被指定为高hlb数(高于11.0)。在9-11范围内的是中间体。
当混合两种或更多种乳化剂时,计算所得到的共混物的hlb。例如,包含70%的tween80(hlb=15)和30%的span80(hlb=4.3)的共混物的hlb值。计算如下:
tween80span80
70%x15.0=10.5
30%x4.3=1.3
混合hlb=11.8
通常,对于非离子表面活性剂,hlb值通常为0至20,对于离子表面活性剂,hlb值可超过50。例如在本液体体系的实施例中,表面活性剂和表面活性剂组合或共混物的hlb值可以为约2至约18,约5至约15,优选约8至约12。在这些范围之外的其它hlb值也可以被使用,并且对于某些类型的前体可能是优选的。
参见图17,显示了溶液形成系统和过程的实施例的工艺流程图1701。因此,催化的前体1701形成珠粒1702,珠粒固化1703(缓慢混合并且温度约为45-55℃)。将珠粒、水和表面活性剂转移1705,把固化的珠粒1708除去1707。将去除的水转移1709,从水中除去硅氧烷1719,返回到1716表面活性剂和水的混合站1717。在约50-115℃下干燥并进一步固化1712,之后从有用的珠粒1714中把非珠废料1715(可用作或进一步加工成颜料,研磨剂等)除去1713。站1717混合水和表面活性剂,并返回去离子水1704作为水源。将表面活性剂(例如hld9.1)1718加入到站1717处的水中。水和表面活性剂用于珠粒成型1702。
喷射成形系统
喷射成形技术可以用于形成固化的和热解的小体积形状。例如,喷雾干燥方法和系统可用于形成小的、均匀的或这两者兼具的聚合物衍生的陶瓷形状。特别地,喷雾固化技术可以用于使用聚硅氧烷形成固化的体积形状。喷雾干燥技术可用于净前体制剂、填充的前体制剂、部分催化的前体制剂、前体制剂的乳液、前体制剂的悬浮液等。
使用喷雾干燥技术,固化的,基本上是圆形,球形和其它形状的颗粒可以由聚合物衍生的陶瓷,特别是聚硅氧碳产生。通过喷雾干燥(例如固化方法)将聚硅氧碳前体加工成固化的,基本是圆形,球形和热解的颗粒,平均粒度大于约200微米。也可产生较大和较小粒度的所述颗粒,例如从亚微米到1000微米。
喷雾干燥包括将前体流体原料雾化成液滴喷雾,其与热气接触时固化成单颗的pdc预成型体颗粒。在本发明之前,喷雾干燥pdc,喷雾固化材料,特别是喷雾固化聚硅氧碳前体,被认为是未知的。
图11是使用本文所述的喷雾固化方法制备基本为圆形和球形的固化的pdc颗粒的流程图。图11a显示了用于如本文所述的喷雾固化方法的固化室,所述固化室提供并流和逆流的组合。图11b显示了用于如本文所述的喷雾固化方法的固化室,所述固化室提供并流。
特别地,预期喷雾固化方法用于制备平均粒度大于或小于约200微米,基本为圆形和球形的固体pdc颗粒。在某些实施例中,颗粒的平均粒度大于约300微米,或大于约400微米。如本文所用,除非另有说明,短语“平均粒度”描述了由一批颗粒的筛分分布计算出的粒度。
如本文所用,除非另有说明,短语“固体陶瓷颗粒”描述了具有小于约10%颗粒体积的内部空隙的陶瓷颗粒。在某些实施例中,固体陶瓷颗粒具有小于约5%颗粒体积的内部空隙。
现参考图11,使用喷雾固化方法制备基本为圆形和球形的固体陶瓷颗粒的方法包括前体制备1100、雾化1102、接触1104、固化1106、排出1108和热解1110。
在前体制备1100中,制备包含pdc制剂,优选为聚硅氧碳前体。pdc制剂的粘度限于那些使制剂适合于在雾化过程1102中通过压力喷嘴或旋转轮泵送的粘度,并可用于生产能被热解的绿色颗粒以形成固体陶瓷颗粒,其基本为圆形和球形。
在雾化过程1102中,将前体制剂进料至雾化设备。合适的雾化设备包括但不限于旋转轮雾化器、压力喷嘴雾化器、双盘式雾化器和双流体喷嘴雾化器。旋转轮、压力喷嘴和双流体喷嘴雾化器为喷雾干燥领域技术人员熟知,且包括可以从各种来源(例如niro,inc)购买的喷雾干燥器。
是否使用旋转轮、压力喷嘴或双流体喷嘴雾化器取决于最终干燥的pdc颗粒中所需的性质,例如粒度、分布和形状以及期望的生产能力。通常,旋转轮雾化器产生细颗粒,而在压力下操作的压力喷嘴和双流体喷嘴可以产生相对较大的颗粒。
当使用旋转轮雾化器时,pdc前体被供给到雾化器的旋转轮中心,并且通过离心力移动到轮的周边。雾化发生在轮缘。液滴粒度和所得喷雾中液滴的粒度分布取决于赋予前体的能量大小和新形成的液滴与转轮附近的湍流气流之间的摩擦效应。液滴喷雾从转轮水平地喷射,但快速地尾随由气体分散器产生的气流模式,将热气引导到空气室中,例如以可控方式固化、热解和这两者。用带有旋转轮雾化器的喷雾干燥器生产的颗粒,其粒径随着雾化器轮速的减小而增大。通常,在给定的雾化器轮的最佳工作范围内,进料速率的影响不大,并且在操作过程中进料速率的波动不会改变所生产的陶瓷粉末的粒度分布。与旋转轮雾化器一起使用的腔室直径通常应足够大,以防止在雾化器水平面的室壁处形成半湿沉积物。相比之下,直径较小但圆柱高的腔室可与压力喷嘴和双流体喷嘴雾化器一起使用。
当使用压力喷嘴雾化器时,在压力下前体被供应给喷嘴。在双流体喷嘴的情况下,前体和固化气体通过单独的喷嘴进料。气体的进料被加压,而前体的进料可以被加压或虹吸/重力进料。在本文所述的使用双流体喷嘴的实施例中,前体进料被加压。
压力能被转化为动能,前体作为易分解成液滴的高速薄膜从喷嘴孔流出。从压力喷嘴雾化器或加压双流体喷嘴产生的液滴尺寸与压力成反比,与进料速率和进料粘度成正比。压力喷嘴或加压双流体喷嘴的容量随压力的平方根而变化。在需要高进料速率和/或高容量喷雾固化的某些实施例中,可以使用多喷嘴系统。
现在转到接触1104步骤,离开雾化设备的前体的液滴喷雾与进入固化室的热固化气体相遇。液滴和固化气体如何初始接触,以及液滴/颗粒如何在整个固化室中移动,通常可以描述为并流、逆流或其组合。
在某些实施例中,例如图11a所示的实施例,显示了与压力喷嘴雾化器一起使用的提供并流和逆流的组合的固化室。
图11a是包括固化室1124(例如固化、热解或这两者)和压力喷嘴1122的喷雾固化装置的简图。喷雾干燥器通常包括本文不需要详细描述的附加部件,因为喷雾干燥器及其部件对于喷雾干燥领域的普通技术人员是已知的。在图11a中,前体通过压力喷嘴1122从进料源1120进料。尽管图11a中仅示出了一个压力喷嘴,但可以使用多个喷嘴。适用于将前体进料的各种类型的设备是本领域普通技术人员是已知的,并且可以包括,例如,具有或不具有过滤器的进料泵。压力喷嘴1122将前体雾化成液滴并将液滴向上喷射到干燥器室1124中,其由箭头a所示。热气通过入口1128从气体源1126送入固化室1124,并进入固化室1124,在固化室中接触所述前体液滴。因此,热气体从前体喷射到固化室的点上方的点进入,并且在室中以大致向下的方向流动。最初,前体液滴在固化室中大致向上流动,从而形成逆流。然而,在某个时刻,液滴会耗尽它们的垂直轨迹,并开始在室中以大致向下的方向流动,从而形成并流。在如图11a所示的固化室中的液滴具有延伸的垂直轨迹,这允许它们有更长的空中固化时间。尽管图11a显示了与组合并流和逆流固化室一起使用的压力喷嘴雾化器,这种固化室也可以与旋转轮雾化器和双流体喷嘴雾化器一起使用。
在某些实施例中,例如图11b所示的实施例,并流固化室与压力喷嘴雾化器一起使用。图11b是包括固化室1134和压力喷嘴1132的喷射固化装置的简化图。前体通过压力喷嘴1132从进料源1130进料。压力喷嘴1132将前体雾化成液滴并以大致向下的方向(以“a”表示)将液滴喷入干燥器,例如,固化室1134。热气从气体源1136注入固化室1134,并沿大致向下的方向(以“b”表示)流入固化室1134。因此,热气和前体液滴在室中沿大体向下的方向流动,从而形成并流。
虽然图11b显示了与并流固化室一起使用的压力喷嘴雾化器,但是并流固化室也可以与旋转轮雾化器和双流体喷嘴雾化器一起使用。
适用于将热气进料到固化室以用于液滴固化的各种类型的设备是本领域普通技术人员已知的,并且可以包括,例如,具有或不具有空气过滤器的加热器。
通常对于喷雾干燥技术,液滴一般不旋转,因为它们通过固化室投射出来,因此,这样的可能性是存在的:液滴的一侧暴露在来自入口的气体中,该气体比液滴的另一侧暴露的气体热(在此分别称为“热侧”和“冷侧”)。在这种情况下,在热侧上固化可能更快,并且在液滴表面上形成的膜变厚。例如,固化在热侧比在凉侧更快。液体和在液滴中的固体迁移到热侧。此时,可以预计到凉侧将收缩,这将导致具有凹坑的中空绿色颗粒(greenparticle),而不是本文所述的固体绿色颗粒(greenparticle)。然而,根据本文所述的方法,颗粒可以是固体、均匀的和球形的,而不是中空的或不规则的形状,这是因为调整了一个或多个固化和配制参数,减轻了不均匀固化的可能性。
关于气体入口温度,对进入固化室的气体的温度进行控制并且优选进行预定。因此,在某些实施例中,气体入口温度的范围在约100℃至约200℃,或约200℃至约300℃,或约300℃至400℃,或约400℃至约500℃。在其它实施例中,气体入口温度的范围为约150℃至约200℃,或约200℃至约250℃。优选地,使用此类范围的较低端的温度以减慢颗粒的固化速率,这反过来又有助于生产绿色陶瓷颗粒(greenceramicparticles),所述绿色陶瓷颗粒可以被热解而生产基本上为圆形和球形的固体陶瓷颗粒。
再次参考图11,排出1108步骤包括从干燥器室分离和排出绿色陶瓷颗粒。在某些实施例中,使用两点排出系统。在两点排出系统中,从室的底部完成绿色陶瓷颗粒的粗粒部分的初次排出,从旋风和袋滤室系统的底部完成较细部分的排出。在某些其它实施例中,使用单点排出系统。在单点排出系统中,从干燥器室完成绿色陶瓷颗粒的回收。例如,在图11a和11b所示的原理图中,在重力的影响下,从固化室将绿色陶瓷颗粒至少部分排放到排出部件1130和1131中。
除了图11a和11b所示的部件之外,合适的固化装置还可进一步包括风扇和管道、废气清洁设备(旋风分离器,袋滤室,洗涤器)和控制仪器。这些进一步的部件和设备是本领域技术人员所熟知的。
在排出1108步骤之后,使用常规的热解设备随后将绿色陶瓷颗粒热解1110,形成大体上为圆形和球形的固体陶瓷颗粒。
随后可以将绿色珠粒(greenbeads)进行热解,形成pdc,优选聚碳氧硅衍生的陶瓷支撑剂、颜料和转化为sic的高纯度原料。
美国专利no.7387752中提出的系统方法和装置可以与聚合物衍生的陶瓷前体一起使用,以制造小体积形状的固化材料,用于制造支撑剂、颜料和超纯sioc和sic。特别地,这些装置、系统和方法可用于制备聚硅氧碳预成型体和陶瓷。美国专利no.7387752的全部公开内容在此通过引用的方式并入本文。
注塑成型和反应成型系统
注塑成型工艺是众所周知的,并且已经被开发用于对塑料部件进行模塑。这些工艺通常涉及使用旋转螺杆通过加热的螺杆筒将颗粒进料来熔化塑料颗粒或树脂颗粒。加热的螺杆桶与通过塑料颗粒的剪切供应的热量将树脂颗粒加热到其熔点以上。螺杆用负载轴向支撑,并且当材料移动到螺杆的前部时,压力的增强迫使螺杆向后,直到在螺杆前方产生了所需体积的塑料。此时,旋转的螺杆停止,通过向前移动螺杆迫使塑料通过喷嘴而将熔融的塑料注入冷却的模具型腔,以提供所需的模塑部件。模具型腔被冷却,注入的塑料被固定为所需部件的形状中。这些已知的技术和操作需要:螺杆的向前运动通常应该填充模具型腔以获得质量好的、致密模塑部件。
对于pdc制剂的注塑成型以形成注塑成型的pdc预成型体,根据本发明的教导,改进和使用典型的注塑成形机。可以作为pdc预注塑成形机使用的这种注塑成形机的实例见于美国专利nos.8328548,6267580和专利公开no.2009/0011064和2002/0030295。本发明的pdc注塑成形机和方法的实施例通常被设计成将液体输送到模具型腔,并且将模具型腔保持升高的温度以固化pdc制剂,以形成固化的pdc预成型体。通常,在机器的液体pdc制剂部分中催化液体制剂,或者在加入到机器的该部分之前催化液体制剂。可以在液体pdc制剂部分加热液体以控制模具中的粘度和最终固化时间,或者可以不是,并且可以保持在环境温度或低于环境温度,例如60f。将液体pdc制剂注入模具中,将模具保持在pdc制剂的固化温度,并且pdc制剂在模具中固化成预成型体。预成型体可以是硬固化,最终固化,绿色固化(greencure),以及这些和其它固化水平的变化和组合,例如,仅固化外皮即可。
例如,聚硅氧碳制剂可以由注模设备(包括反应注塑成型设备)制成,以及其它。例如,模具可以由钢、铝、环氧树脂和聚氨酯制成。
在一个实施例中,提供了一种用于对微固化pdc预成型体进行模塑的注塑成形机,所述微固化pdc预成型体包含约0.001至3.5立方厘米之间的pdc喷射体积每制成的体积形状。具体地说,微型注塑成形机利用气动缸或气缸将pdc制剂输送到注塑成形机的注射部分。线性电动机驱动注射部分以将pdc制剂通过喷嘴注入模具型腔中以完成微固化pdc预成型体的注塑成型。
塑料成型技术、机器、方法和工艺,特别是这些用于成型的技术,优选注塑成型技术,小、非常小的部件(例如,微型模塑部件)可用于制造pdc小体积部件,例如固化的pdc预成型体,尤其是聚硅氧碳体积形状和预成型体。
在实施例中,用于对pdc微型预成型体(例如,微固化pdc预成型体)进行模塑的注塑成形机(包含,例如,液体pdc注射体积为小于约0.1立方厘米(cc),小于约0.01cc,小于约0.001cc,小于约0.0001cc,约5cc至约0.0001cc,约4cc至约0.0015cc,约3.5cc至约0.001cc)包括液体pdc制剂部分,其操作性地连接到注射部分和模塑部分。设置有阀部件以打开和关闭液体pdc制剂部分和注射部分之间的连接。在将pdc制剂注入模塑部分时,在高达约100000psi的压力下(尽管这些压力通常是塑料注射所需的压力,而本发明的液体pdc制剂以及本发明控制和预选这些液体的粘度的能力可能需要较小的压力,例如,小于100000psi,小于50000psi,小于25000psi),线性电动机装置件与注射部分相关联,以允许约0.01秒,约0.0015秒的模塑时间(例如,根据液体pdc制剂的粘度,可以使用更多和更少的时间)。
用于注塑成型的现有技术方法通常足以使用超过3.5至5.0立方厘米的注射尺寸将正常尺寸的部件进行模塑;然而,当微固化pdc预成型体需要非常小的注射体积(小于约5cc,小于约4cc,小于约3.5cc或更小)时,现有的较大规模的工艺和技术存在明显的问题。例如,用于运输pdc制剂的螺杆或螺旋钻装置通常应该在直径上小型化以接受和加工pdc制剂。(应当注意的是,在一些实施例中,pdc制剂可以是固体或半固体,并且可以以类似于模塑机中的树脂颗粒的方式进行加工,然而,目前优选的pdc制剂是液体制剂,更优选为被催化的液体制剂)。如果螺杆太大,则相对于被模塑的部件,其将含有太大的pdc制剂体积。在这种情况下,pdc制剂(被催化且在每个成型周期之后可能在螺杆筒中保持加热)在保存时将随着时间推移而固化。然而,如果螺杆或螺旋钻是小型化的并且螺杆飞行深度小于颗粒尺寸,如果使用颗粒,则存在以下问题:接收颗粒并将前体或颗粒进料到螺旋钻中以允许前体压缩和熔化的问题。此外,因为pdc前体通常是液体,螺杆组件可以被替换为柱塞或活塞式组件。通常,催化的pdc制剂剪切相对而言非常迟钝,对小型化螺杆的较小尺寸的考虑不应成为问题。
此外,对于对微固化pdc预成型体进行模塑通常需要范围为约0.025至0.30mm的薄壁厚度。期望的是用力将液体注入到这些薄壁微固化pdc预成型体中,而不过早固化、凝固。在一些模具和装置中,这可能需要更高的压力和更短的注射时间。在这些情况下,还可以控制配方、催化剂量和温度,以控制固化速率,包括增稠。因此,这些要求中的许多可以通过pdc的能力(预定和控制其粘度,以及预先确定和控制其固化速率)来解决。
因此,对于对微固化pdc预成型体进行注塑成形,注塑成形机优选产生足够高的注射压力并且具有基本上小于0.5秒的受控喷射速度曲线。而且,现有技术和工艺利用液压来产生注射压力和注射速度曲线。然而,液压油不易与洁净室设施兼容。因此,医疗级装置、超纯材料和相关的微固化pdc预成型体的注塑成形分别受到现有技术的限制。
在注塑成型机的一个实施例中,为克服这些已知的注塑成形机以及制造微型模塑预成型体的的问题,建议注塑成形机包括其中前体被引入注射柱塞前部的系统。本发明的系统被改进以用于制造pdc预成型体。这种机器利用或需要气缸驱动注射柱塞、结构和机构。因为模具型腔被填满,因此这种注塑成形机不能停止注射过程,除非在注塑过程中增加压力。通过测量压力产生量的增加来控制注塑过程是较不优选的方法,因为模塑部件的高度变化性,这对大多数模塑操作来说是不能令人满意的结果。
因此,除了其它,实施例给注射成型机提供了用于对微固化pdc预成型体进行模塑的方法,使用0.001至3.5立方厘米的pdc制剂注射体积对微固化pdc预成型体进行塑模的方法,以及对由液体pdc制剂部分和注射部分(其允许利用约0.00015至4.5立方厘米的前体注射体积)组成的微固化pdc预成型体进行塑模的方法。
注塑成形机的注射部分的一个实施例包括注射缸,所述注射缸位于并固定在气缸体内,与树脂流动通道轴向对齐,其与喷嘴配合以允许将pdc制剂注入模具中。注射部分保持在模具的中心线上。精密配合的注射销部件被安装在注射缸的孔内,并且相对于孔保持着非常紧密的公差,在约0.012mm或更小的范围内。在注射缸的孔内注射销的精密配合,以及与注射销啮合的线性电动机的使用,允许在熔融树脂注入模具部分的阶段通过树脂流动通道和喷嘴以非常高的速度施加高压。此外,精密配合阻止了在模塑过程中喷射销和气缸孔之间的回流。位于注射部分和液体pdc制剂部分之间的阀部件在注射过程期间被关闭,以防止pdc制剂回流入低压容量液体pdc制剂缸。阀部件是锥形阀,其优选由气缸驱动。阀部件位于液体pdc制剂缸体内部并保持在适当的均匀温度。
当pdc制剂被液体pdc制剂缸压入树脂流动通道和注射缸中时,阀部件被关闭,并且注射销向前驱动,以通过喷嘴和浇口将前体流压入闭合的模具型腔。
注射销由电动机装置驱动。术语电动机装置可用于描述耦合到滚珠螺杆装置的旋转电动机,其将旋转运动转换成线性运动。然而,本发明的优选实施例是:电动机装置是线性电动机,直接向注射销提供线性运动。术语“线性电动机”用于描述以直线运动电驱动而不是以旋转运动电驱动的电动机。在本发明中有用的一种线性电动机是由trilogylinearmotor,webster,tex制造和销售的线性伺服电动机或步进电动机。线性电动机提供线性运动,其啮合并控制与注射销啮合的速度和压力。
线性电动机的电子控制提供了非常高速的注射销的运动,同时保持了注射销的精确控制和定位。注射销的位置被连续监测,并通过线性测量装置(例如lvdt)反馈给电子控制系统。注射销由线性电动机啮合并推动,但不一定直接耦合到线性电动机。如果需要,消除注射销和线性电动机之间的直接耦合,避免相对于注射销和线性电动机精确对准的必要性。根据需要,树脂流动通道内的注射销的向前轴向运动将约0.00015至4.5立方厘米的注射体积注入模具中。
在模具循环完成之后,注射销在负载下向后轴向移动,这是因为阀部件打开并且来自液体pdc制剂缸的pdc制剂进入树脂流动通道以迫使注射销从模具部分向后移动。制剂流入树脂流动通道,在预定的注射体积的pdc制剂从液体pdc制剂部分进入注射部分的重新加载循环期间返回注射销。
在液体pdc制剂流入树脂通道后,称为制备预定的注射体积的pdc制剂,模具部分轴向移动离开喷嘴,模具被打开以允许模塑的微固化-pdc预成型体从模具型腔排出。此后,阀部件关闭,模具部分轴向移动以啮合喷嘴,以重复用于预定注射体积的模塑和固化循环。
如上所述,注射喷嘴与注射销配合,以便加热的树脂或前体材料通过浇口开口进入模具型腔。模具型腔被设计成这样:模塑的微固化pdc预成型体在每个操作周期之后通过顶出销或抽吸被轻易地从模具型腔中移除。通过利用前体或直径约0.5至6.0mm的树脂流动通道,易于实现介于约0.00015至4.5立方厘米之间的前体注射体积。此外,由于流动通道的尺寸减小,因此,可以模塑的部件的数量减少,利用包含在液体pdc制剂室中的pdc制剂,从而确保模塑效率最高,而不会在装载之间pdc制剂意外固化。
转到图12,为注塑成形机的实施例的横截面图,其显示了液体或颗粒装入注塑成形机的液体pdc制剂部分。图12a是图12的注塑成形机的横截面图,显示了液体pdc制剂部分中的温度控制和用预定的注射体积的pdc制剂填充注射部分。图12b是图12的注塑成形机的横截面图,显示了通过线性电动机的移动,将pdc制剂通过树脂流动通道和喷嘴注入模具中。图12c是图12的注塑成形机的横截面图,显示了来自注射部分的模具部分和模具开口的轴向运动,以排出模塑的微固化pdc预成型体。图12d是局部放大图,显示了图12的液体pdc制剂部分和注塑成形机的注射部分之间关闭的阀部件。图12e是局部放大图,显示了在液体pdc制剂部分和注射部分之间打开的阀部件以允许在图12的注塑成形机中预定的注射体积的pdc制剂流入注射部分。图12f是局部放大图,显示了来自图12的注塑成形机中的液体pdc制剂部分的pdc制剂填充树脂流动通道的过程中注射销的位置。图12g是局部放大图,显示了图12的注塑成形机中的液体pdc制剂部分与注射部分之间的阀部件的位置。
现在转到图12a至12g,其中,使用相同的数字来表示几个视图中相同或相似的部分。图12、12a至12g的实施例涉及用于对微固化pdc预成型体进行模塑的注塑成形机和用于制造这些预成型体的方法。微固化的pdc预成型体通常是固体,并且可以是支撑剂、颜料、添加剂和其它固体体积形状,它们也可以具有范围为约0.025至0.3mm之间的壁厚。如附图的图12-12c所示,微型注塑成形机1210由液体pdc制剂部分1212,注射部分1214和模塑部分1211组成。液体pdc制剂部分1212适于将进料pdc制剂限制在注塑成形机的注射部分。注塑成形机1210包括由彼此成一体的上部1217和下部1218组成的加热气缸体1216。气缸体1216的上部和下部优选包括其中的加热器孔1220,参见例如图12d和图12e、12d和12e。加热孔位于整个气缸体1216中,并且适于在其中容纳电筒式加热器1221,从而为气缸体提供均匀温度。
液体pdc制剂部分1212包括螺旋螺杆或螺旋进料部件1222,其由步进电动机(未显示)驱动以顺时针旋转的。螺旋螺杆部件的上端1223适于从包含pdc制剂供给的料斗1225接收pdc制剂1224。液体pdc制剂部分1212进一步包括液体pdc制剂气缸1226,所述制剂气缸驱动液体pdc制剂室或孔1213(位于温度控制的气缸体1216内并包含pdc制剂)内的液体pdc制剂柱塞1227。所述孔1213适于从螺旋螺杆部件1222接收pdc制剂1224,位置如图1所示。液体pdc制剂柱塞1227与气缸体1216中的孔1213配合至如图12a所示的位置。液体pdc制剂柱塞1227的尺寸相对于所述孔1213设定,以允许在前体的压缩和潜在加热期间被捕获的空气逸出柱塞和孔壁。
另外,参见例如图12和12f,导管1229离开所述孔1213并与注塑成形机1210的注射部分1214的树脂流动通道1232连通。位于管道1229内的是高压阀部件1231,其在开口和关闭位置之间是可操作的,如图12d和12e所示。所述导管1229适于与树脂流动通道1232相交,以便将pdc制剂材料输送并填充注射通道,如下文所述。
注塑成形机1210的注射部分1214包括树脂流动通道1232、注射缸1233和注射销1234,所注射销与耦合到线性驱动装置或电动机装置1236的推销1235啮合,例如参见图12~12c和12f。注射缸1233可移除地安装到位于气缸体1216的上部1217和下部1218之间的孔1237。注射缸1233包括延伸其长度的孔1238(图12f),孔1238定义了其中的树脂流动通道1232,并适于接收在其中前后运动的注射销1234。树脂流动通道1232与喷嘴1240轴向对齐,喷嘴1240与模具部件1244中的浇口1241啮合,以允许pdc制剂的注入或pdc制剂通过浇口注入到由模具部件1244和1245所定义的模具中,如图12c清楚所示。如果需要,可以围绕气缸体设置温度控制器1242,其中树脂流动通道与喷嘴1240啮合以促进和保持制剂,最优选在该过程中的该点被催化。温度控制如图12~图12c所示。
注射销部件1234适于容纳于注射缸1233的孔1238内,并适于相对于孔保持非常紧密的公差,在约0.012mm或更小的范围内。注射缸内的注射销的这种精密配合允许在注射阶段以非常高的速度施加高压,同时防止在注射操作期间注射销和注射缸1233之间的pdc制剂回流。如图12d所示,位于液体pdc制剂部分1212的导管1229中的阀部件1231在注射步骤(图12b)期间被关闭,以防止pdc制剂回流到低压容量液体pdc制剂缸中。如图12d和12e所示,阀部件1231是由气缸1239提供动力的锥形阀。阀部件1231位于加热缸体内部并保持在适当的均匀温度。
在本发明的另一个实施例中,阀部件1231与液体pdc制剂缸1226和柱塞1227同心定位,以预定方式控制pdc制剂通过导管1229从液体pdc配制部分流到注射部分。在图12g中,阀部件1231的锥形端1230在结构上被设置为啮合管道1229的入口,以在注射步骤(图12b)期间阻止pdc制剂流入注射部分,并且防止pdc制剂回流到压力容量液体pdc制剂缸。
填充注射部分的过程如图12a所示。pdc制剂1213被液体pdc制剂柱塞1227压缩,阀部件1231打开,如图12e和12g所示,液体pdc制剂柱塞1227迫使pdc制剂进入树脂流动通道1232和注射部分1214的注射缸1233。这填充了树脂流动通道,位置如图12a所示,并由图12f所示。
液体pdc制剂柱塞1227通过气缸1226移动到上部1217中的室或孔1213中。围绕液体pdc制剂柱塞和室的气缸体1216被温度控制到特定催化的pdc制剂被模塑的适当温度。通过液体pdc配制气缸1226和温度控制施加在液体pdc制剂柱塞1227上的力有助于室或孔1213内的稳定性。
阀部件1213位于导管1229(图12~12e)中或与导管1229(图12g)相关联,并且位于树脂流动通道和注射缸以及液体pdc配制室孔1213之间,当喷嘴保持抵靠模具部件1244和浇口1241时,阀部件1231被打开。阀部件1231通过气缸1239或通过同心安装的气缸在打开和关闭的位置之间移动(图12g中未示出)。在一段时间内,阀部件1231打开,注射部分接收并填充有液体前体,并且喷嘴1244抵靠模具放置,而预先模塑的pdc预成型体是冷却的。这防止了在填充步骤期间前体材料离开喷嘴40进入模具。
线性电动机1236控制注射销1234的运动。在用前体填充注射部分期间,通过线性电动机1236保持对注射销的小的负载或压力。因为在填充期间通过液体pdc制剂柱塞对液体pdc制剂室中的前体施加更大的压力,进入注射部分1214的pdc制剂将注射销1234推回远离喷嘴1240,图12f中的流动通道箭头的位置。迫使喷射销和线性电动机远离喷嘴有助于防止在包含于pdc制剂中的液体pdc配制室或孔1213中形成空隙。此外,注射销与线性电动机的啮合提供了部件模塑所需的注射体积的预定控制。当注射销在注射缸内被迫轴向向后,反馈到线性电机控制器的线性位置编码器传感器将注射销停止在预定位置。随着注射销从喷嘴向后轴向移动,制剂保持在压力下,所以对树脂流动通道内的前体注射体积的浓度(用于下一个微固化pdc-预成型体的随后的模塑)进行适当和预定的控制。当线性电动机1236达到通过树脂流动通道、喷嘴和浇口预注入模具的所需注射体积的合适位置,线性电动机停止,液体pdc制剂缸上的负载被去除。然后,关闭阀部件1231(图12d)以去除液体pdc制剂缸上的负载。此后,线性电动机36从注射缸向后轴向移动约1mm,以减轻注射销前面的制剂的压力。
如图12c所示,在将注射体积填充到注射部分并完成将前体注入模具(图12b)之后,模具部件1244和1245从喷嘴1240轴向移动并相对于彼此打开。在模具型腔打开期间,顶出或升降销1243或者抽吸软管(未显示)用于从模塑型腔移除模塑的微固化pdc预成型体1250。喷嘴1240在该时间段内与模具保持距离,其被加热到或保持在固化温度,以防止喷嘴的加热和随后的硬化,例如包含在喷嘴中的pdc制剂的固化。模具部件以轴向对齐的关系联接在一起,并通过模具气缸1247相对于喷嘴轴向移动。
当模具关闭并轴向移动以啮合喷嘴时,注射销处于向后位置。模具通过气缸1247与喷嘴的啮合防止了在喷嘴1240和浇口1241之间的前体泄漏。前体随后通过致动电动机装置1230将前体注入模具的空腔中,以将顶出销向前驱动。
术语“电动机装置”可以用于描述耦合到滚珠螺杆装置的旋转电动机,其将旋转运动转换成线性运动。然而,本发明的优选实施例是:电动机装置是直线式电动机1236,其直接向注射销1234提供线性运动。术语“线性电动机”用于描述以线性运动电驱动的电动机而不是以旋转运动电驱动的电动机。在本发明中有用的一种线性电动机是由trilogylinearmotor,webster,tex制造和销售的线性伺服电动机或步进电动机。线性电动机提供线性运动,接合和控制与注射销啮合的速度和压力。
为了得到高质量的模塑的微固化pdc-预成型体,实现对模具的填充和随着制剂固化成预成型体所保持的压力的控制。通常,在用前体填充模具型腔的第一部分期间,线性电动机1236以预设速度将柱塞向前移动,而与前体中产生的压力无关。对于小的薄壁的微固化pdc预成型体以及对于固体形状,优选以非常高的速度(高达125cm/秒的速度)。可以用伺服驱动器控制线性电机速度,以便在充填阶段以预定步骤改变电机的速度。当对复杂几何的微固化pdc预成型体进行模塑时,这是必需的,这是因为当模具被填充时,期望具有恒定的前体流动前沿(constantflowfrontofprecursor)。
当模具型腔几乎被填充时,填充量为95%,注射运动从速度控制切换到负载或压力控制。这通过用线性编码器感测注射销1234的位置来实现,并且当到达模具型腔几乎被填充的预定位置时,控制系统切换到压力控制。然后,施加到注入前体的压力由与不同值相关的时间步长控制。通常,最初需要更高的压力,然后是更低的压力。这使得来自注射缸的制剂随着固化流入微固化的pdc-预成型体。
理想地,耦合到滚珠螺杆装置的线性电动机或旋转电动机适用于对微固化的pdc预成型体进行模塑,这是因为它们可以控制来自单个伺服控制器的速度、位置和负载。对于模塑的微固化pdc预成型体,这些类型的电机能够施加高达100000psi的压力并实现0.01秒的注射时间。此外,根据本发明的实施例,这些类型的电动机提供了非常快速地启动和停止针对小注射体积的制剂所需要的能力。将制剂注入模具并保持压力时间完成后,加热模具以固化pdc制剂。虽然这种固化正在完成,如前所述,模塑过程重复了用pdc制剂填充注射部分并排出模塑部件的步骤。
注塑成形机1210的实施例利用气缸来驱动液体pdc制剂柱塞的运动并驱动模具部分相对于注射部分的轴向移动。注射阀运动通过利用线性电动机实现,以在注射期间提供高速和高压。这种气缸和电动机装置的使用有利于洁净室的气氛,如果这是需要的,但对于形状是不需要的,例如支撑剂和颜料,对于所有类型的使用和应用(包括医疗和电子领域)以容许对所有类型的微固化pdc-预成型体进行塑模,。
此外,对注射缸、注射销、树脂流动通道、喷嘴和在加热的气缸体16的中心线1252(图12d和12e)处的模具进行定位防止了随着部件温度的改变造成的各部件的不对准。这种中心线的定位将各部件之间的尺寸差减小到小于0.1mm。通过安装加热的气缸体16(含有注射缸、注射销、树脂流动通道和喷嘴作为注塑成形机架1252的中心线位置(图12d~12e))并确保与模具部分1211的轴向对准和配合,有利于提高位置精度。
其它成形技术
印刷技术、丝网印刷技术(screentechnologies)、基底印刷技术、轧印技术(niptechnologies)以及例如图8和13-13a所示的其它技术,可用于从聚合物衍生的陶瓷前体制备小体积形状结构,以制备小体积形状的预成形体,尤其从聚碳氧硅前体制剂制备这种结构。
因此,例如,可以使用以下技术、设备和方法来形成小体积预成型体:印刷技术(例如,凹版印刷);3-d印刷;激光诱导空化(例如,美国专利申请公开no.2012/0236299,其全部内容在此通过引用并入本文));基于轧印的技术;基于柔性基板的技术;喷雾冷却和离子凝胶化。
转到图13和图13a,为辊隙形成组件和系统1300的透视图,以及辊1302之一的详细透视图。辊隙形成系统1300具有第一辊1301和第二辊1302,如箭头1305、1304所示进行反转。所述辊表面的一个或两个具有特定表面,例如1309具有一系列成形槽,例如1307a,1307b,1307c,例如小凹口,杯状物,器皿,容器。在操作过程中,随着两个辊1301、1302相互啮合和反向转动时,形成辊隙1303。前体通过分配组件被进料到辊隙1303的顶部,如箭头1306所示。辊隙1303迫使前体进入成形槽,例如1307a,1307b,1307c。优选地,成形槽覆盖整个辊表面1309,并且可以包含在具有成形槽集合的插入组件或插入行1308中。以这种方式,槽会被堵塞或损坏,可以除去所述插入行,进行清洗,更换并进行后续的操作。
在所述辊1301、1302中,存在较高和较低压力的区域,使得前体可以在固化的同时保留在成形槽中,然后在固化时从成形槽中排出。
基底成型技术可用于制造小体积的形状。通常,在这些系统中,可以使用可以成脊状的、柔性的或连续的基底。例如,可以使用连续的丝网(例如金属,例如黄铜或合成长网线,这取决于温度、纯度和其它加工和最终用途要求),固体表面或柔性基材来容纳或携带前体材料,例如液体pdc前体的薄层或膜被铺展在或以其它方式放置在基底的表面上。这些基底(substrate)或基底(bases)可以是移动的,带状的,连续状的或批次的,例如,托盘。前体材料可以在线的开口或基底的表面上固化、硬固化,然后从基底上除去。在线的实施例中,固化材料可以从线中排出。在线和其它实施例中,固化材料可被机械地除去,超声波除去,振动脱离或以其它方式与基底分离。此外,根据固化的类型,固化材料可以剥落,因此通过剥离与基底分开。
对于这些方法,基底可以是一次性的,例如,其不需要从固化的pdc材料物理去除;相反,它被消耗,因此在固化期间被去除,更优选在热解过程中被去除。这些系统提供了一种制造薄且非常薄的pdc固化和热解材料的片材和片晶的方法。因此,这些系统可以制造薄于1mm,薄于100微米,薄于50微米,薄于10微米,薄于1微米和薄于0.1微米的片材和片晶,以及更厚和更薄的薄膜、圆盘和片晶以及其它基本上平面的体积形状。
液体pdc前体的薄膜或层可以通过任何成膜设备(例如,分配机,切片,气刀,辊,喷雾器等)形成在基底上。
基底上pdc前体的薄膜或层可以用本文所述的任何加热和固化技术和条件来固化。此外,由于pdc前体的薄的性质,优选使用电磁辐射作为能量来源来固化、热解pdc前体的薄膜,或这两者。因此,例如可以使用微波,相干光,单色光,广谱光(例如,高强度白光)和其它形式的电磁辐射。如果使用白光,则白光可以具有宽波段,例如从近紫外到近红外,从390nm到790nm,从约400nm到约760nm,具有至少约100nm的波段,至少约200nm的波段,至少约300nm的波段,以及它们的组合和变化。也可以使用uv波长(通常约10nm至400nm)的光,因此可以使用uv灯,uv激光,uv-led或uv辐射源。为了增强uv固化,可以向前体中加入uv活性催化剂(例如,在380至405nm之间吸收最佳的催化剂)。可以使用360~420(uv到紫色)的波长。高强度电磁辐射,例如光可以脉冲或连续。也可以选择电磁能量波长或波长,以通过pdc前体提供预定量的能量吸收。pdc前体还可以具有添加剂,例如着色剂,其增加或选择性地确定被吸收的能量(例如,电磁能量)的量。因此,pdc前体和电磁波长可以是预定的并且匹配的以优化固化、剥离和这两者,以及末端或中间pdc材料的其它特性。基底也可以对电磁辐射是透射的,因此,pdc前体的膜可以从两侧固化。此外,当使用pdc前体材料时,基底可以是水平的、垂直的或在水平和垂直之间的角度。基底也可以是圆形的,例如,沿着扬基干燥器造纸鼓(yankeedryerpapermakingdrum)的线条。
转到图8,显示了基底成形装置和工艺800的实施例。该系统800具有成形槽803,所述成形槽在移动的基底803(例如,在该实施例中为连续的带)上提供pdc前体802的薄层。能量源804(例如,热源,rf,光源等)被提供以固化pdc薄膜802,并且当pdc前体与基底分离时,导致形成片晶805。
应当理解的是,在本说明书中使用标题是为了清楚的目的,并不以任何方式进行限制。因此,标题下描述的过程和公开的内容应当结合本说明书的全部内容进行阅读,包括各种实施例。在本说明书中,标题的使用不应限制本发明提供的保护范围。
实施例
通过本发明目前的成形系统和方法的实施例,提供以下实施例以说明聚硅氧碳前体(可以形成固化的预成型体的聚硅氧碳前体)和热解材料的各种实施例。这些实施例仅用于说明目的,不应视为限制本发明的范围。应当理解的是,术语聚硅氧碳批料包括催化的批料和未催化的批料。除非另有说明,实施例中使用的百分比是总批量、预成型体或结构的重量百分比。
实施例1
一种聚硅氧碳批料,具有75%mh,15%tv,10%vt和1%催化剂(10ppm铂和0.5%luprox231过氧化物)。
实施例2
一种聚硅氧碳批料,具有70%mh,20%tv,10%vt和1%催化剂(10ppm铂和0.5%luprox231过氧化物)。
实施例3
将具有50%(体积)飞灰(flyash)的聚硅氧碳批料加入具有70%mh,20%tv,10%vt和1%催化剂(10ppm铂和0.5%luprox231过氧化物)的聚硅氧碳批料。
实施例4
将直径为0.5μm的40%(体积)的al2o3加入聚硅氧碳批料,所述聚硅氧碳批料具有70%mh,20%tv,10%vt和1%催化剂(10ppm铂和0.5%luprox231过氧化物)。
实施例5
一种聚硅氧碳批料,具有100%tv。
实施例6
一种聚硅氧碳批料,具有10%mh前体(约800的分子量),73%的甲基封端的甲基以及聚硅氧烷前体(约1000的分子量)和16%的tv前体,和1%的oh封端。
实施例7
一种聚硅氧碳批料,具有100%tv和低于约0.5%的过氧化物催化剂。
实施例8
一种聚硅氧碳反应混合批料,具有85/15mhf/pcpd。
实施例9
一种聚硅氧碳反应混合批料,具有85/15mhf/pcpd,以及1%p01催化剂和1%过氧化物催化剂。
实施例10
一种聚硅氧碳反应混合批料,具有85/15mhf/pcpd,以及1%p01催化剂和3%tv(作为固化速率加速剂)。
实施例11
一种聚硅氧碳反应混合批料,具有65/35mhf/pcpd。
实施例12
一种聚硅氧碳反应混合批料,具有70/30mhf/pcpd。
实施例13
一种聚硅氧碳批料,具有50~65%mhf,5~10%四乙烯基,和25~40%二烯(二烯=二环戊二烯或异戊二烯或丁二烯),优选用p01或其它铂催化剂催化。
实施例14
一种聚硅氧碳批料,具有60~80%mhf和20~40%异戊二烯,优选用p01或其它铂催化剂催化。
实施例15
一种聚硅氧碳批料,具有50~65%mhf和35~50%四乙烯基,优选用p01或其它铂催化剂催化。
实施例16
一种聚硅氧碳反应混合批料,具有85/15mhf/pcpd,优选用p01和
实施例17
一种聚硅氧碳反应混合批料,具有65/35mhf/pcpd,优选用p01和
实施例18
通过使用反应型工艺,使用以下配方制备前体制剂。反应温度在72℃保持21小时。
实施例19
一种聚硅氧碳制剂,具有0~20%mhf,0~30%tv,50~100%h62c和0~5%羟基封端的二甲基聚硅氧烷。
实施例20
一种聚硅氧碳制剂,具有40%mhf,40%tv,和20%vt,并且其氢化物基团与乙烯基的摩尔比为1.12:1,可以用于形成强陶瓷珠粒,例如,用于水力压裂烃生产地层(hydraulicallyfracturinghydrocarbonproducingformations)的支撑剂。
实施例21
一种聚硅氧碳制剂,具有42%mhf,38%tv,和20%vt,并且其氢化物基团与乙烯基的摩尔比为1.26:1,可以用于形成强陶瓷珠粒,例如,用于水力压裂烃生产地层(hydraulicallyfracturinghydrocarbonproducingformations)的支撑剂。
实施例22
一种聚硅氧碳制剂,具有55%mhf,25%tv,和20%vt,并且其氢化物基团与乙烯基的摩尔比为2.36:1,可以用于形成强陶瓷珠粒,例如,用于水力压裂烃生产地层(hydraulicallyfracturinghydrocarbonproducingformations)的支撑剂。
值得注意的是,没有要求提供或解决作为新颖的和开创性的工艺、材料、性能或其它有益的特征和性质(这些是本发明实施例的主题或与本发明实施例相关)的基础的理论。然而,在本说明书中提供了各种理论,以进一步推进该领域的技术。在本说明书中提出的这些理论,除非另有明确说明,绝对不限制、限定或缩小本发明所提供的保护范围。利用本发明可能不需要使用或实践这些理论。应当进一步理解的是,本发明可能导致新的和迄今未知的理论来解释本发明的方法、制品、材料、设备和系统的实施例的功能特征;这种后续发展的理论不应限制本发明所提供的保护范围。
还值得注意的是,尽管本说明书着重于小pdc体积形状,以解决长期以来对获得这种制品的方法和系统的需要,本说明书的系统、技术和方法可以应用于更大的形状和结构。因此,除非另有特别说明,本发明的保护范围不应被限制,并且延伸和涵盖更大的形状和体积。
在本说明书中阐述的系统、装置、技术、方法、活动和操作的各种实施例可用于各种其它活动以及用于本文中所阐述的领域以外的其它领域。此外,例如,这些实施例可以与以下一起使用:将来可能开发的其它设备或活动;基于本说明书的教导可能部分被改进的现有设备和活动。此外,本说明书中阐述的各种实施例彼此之间可以以不同和各种组合使用。因此,例如,本说明书的各种实施例中所提供的配置可以配合使用;并且本发明所提供的保护范围不应限于特定实施例,在特定实施例、例子中或在特定图中的实施例中所阐述的配置或排列。
在不脱离本发明的精神或基本特征的情况下,除了在本文中所明确公开的那些,本发明还可以其它形式体现。本文所描述的实施例在各方面仅被认为是说明性的而不是限制性的。
- 还没有人留言评论。精彩留言会获得点赞!
- 制造金属、陶瓷制品的金属、陶瓷粉末与聚合物混融物及聚合物从成型品中水解脱除方法
- 打印金属、陶瓷制品的金属、陶瓷粉末与聚合物混融材料及聚合物在成型品中的脱除方法
- 制造金属、陶瓷制品的金属、陶瓷粉末与聚合物混融物及聚合物从成型品中酸催化脱除方法
- 聚合物基陶瓷复合介电材料及其制备方法
- 一种Zr-C-Si聚合物陶瓷先驱体及其制备方法与应用的制作方法
- 一种耐高温陶瓷基地聚合物的制备方法
- 制造金属、陶瓷制品的金属、陶瓷粉末与聚合物混融物及聚合物从成型品中的脱除方法
- 用于石质或陶瓷基材的混杂聚合物涂层、石质或陶瓷基材及其获得方法
- 有机金属聚合物陶瓷先驱体及其制备方法与应用的制作方法
- 一种可陶瓷化高碳型聚合物基复合材料及其制备方法