电解电极及其制造方法和在水净化系统中的使用方法与流程
本申请要求于2016年8月26日提交的美国临时申请序列号62/380,150的权益,该美国临时专利申请通过引用以其整体并入本文。
本发明总体上涉及电解,并且更具体地,涉及用于水处理电解和氯-碱工业(chlor-alkaliindustry)的电极。
背景
系统正被提出用于电解质中的污染物的电化学氧化。这些系统的实例包括采用电解来清洁废水的废水处理系统。这些系统将电压电位施加在各自与废水接触的阳极和阴极之间,以实现有机物质的电化学氧化。
这些系统中的阳极通常具有接触废水的半导体的层。半导体通常包括一种或更多种在系统的操作期间溶解到废水中的组分。半导体组件的溶解降低了阳极的使用寿命。
另外,某些阳极净化水的能力取决于阳极在水中产生反应性氯物质(reactivechlorinespecies)(rcs)的能力。然而,已知的阳极典型地以太低以致于对于某些废水处理应用不是合意的电流效率(currentefficiency)产生反应性氯物质。高效地产生rcs的阳极在氯碱工艺和工业中也是有用的。
因此,增加电解阳极(electrolysisanode)的使用寿命及其rcs产生的电流效率的电解阳极的改进是合意的。
概述
电解阳极具有第一导电金属氧化物层、接触第一导电金属氧化物层的第二半导体层以及一个或更多个第三半导体的岛(island),所述一个或更多个第三半导体的岛接触第二半导体层。根据阳极的示例性实施方案,第一导电金属氧化物层包括铱,第二半导体层包括二氧化钛,并且第三半导体包括二氧化锡。
根据另一个示例性实施方案,电解阳极可以包括钛基底(titaniumbase)、在钛基底上形成的ir0.7ta0.3o2层、在ir0.7ta0.3o2层上形成的钴掺杂的tio2层、以及在co-tio2层上形成的多个sno2的岛。
可以使用喷雾热解以施加每种半导体材料来制造阳极。
阳极可以被用于净化具有有机污染物和氨的水的系统中。阳极可以被配置成使得当被放置在水中时,第二半导体层的至少一部分和岛与水直接物理接触。
操作用于水的电解的水处理系统的方法包括使阳极与包含氯离子(chloride)的水接触,以及向阳极施加足以在阳极处产生反应性氯的阳极电位。
本公开内容还描述了水净化系统,所述水净化系统包括阳极,所述阳极具有第一导电金属氧化物层和第二半导体层,所述第二半导体层接触第一导电金属氧化物层。第二层被配置成至少部分地与包含氯离子的水直接接触。阳极还包括和一个或更多个第三半导体的岛,所述一个或更多个第三半导体的岛接触第二半导体层。第三半导体岛还被配置成与水直接接触。
本公开内容提供了电解阳极,所述电解阳极包括第一导电金属氧化物层;第二半导体层,所述第二半导体层接触第一导电金属氧化物层;以及一个或更多个第三半导体的岛,所述一个或更多个第三半导体的岛接触第二半导体层。在一个实施方案中,第一导电金属氧化物层包括铱。在另外的实施方案中,第一导电金属氧化物层包括ir0.7ta0.3o2。在又另外的实施方案中,第一导电金属氧化物层中的ir0.7ta0.3o2的质量负载量(massloading)是选自由约0.3mg/cm2和约0.05mg/cm2组成的组的值。在另一个实施方案中,第二半导体层包括tio2。在另外的实施方案中,第二半导体层包括钴掺杂的二氧化钛。在又一实施方案中,第三半导体是锑掺杂的二氧化锡、或二氧化锡。在另一个实施方案中,阳极还包括金属导体,所述金属导体接触第一导电金属氧化物层。在另外的实施方案中,金属导体是钛。在又另外的实施方案中,第一导电金属氧化物层被配置成克服第二半导体层和金属导体之间的肖特基势垒(schottkybarrier)。
本公开内容还提供了电解阳极,所述电解阳极包括钛基底;ir0.7ta0.3o2层,所述ir0.7ta0.3o2层在钛基底上形成;co-tio2层,所述co-tio2层在ir0.7ta0.3o2层上形成;以及多个sno2的岛,所述多个sno2的岛在co-tio2层上形成。在一个实施方案中,co-tio2层中的tio2的质量负载量是约0.5mg/cm2;其中sno2的岛中的sno2的质量负载量是约1.0mg/cm2;并且其中ir0.7ta0.3o2层的质量负载量是选自由约0.3mg/cm2和约0.05mg/cm2组成的组的值。在另一个实施方案中,sno2的岛中的每个具有在co-tio2层上形成的离散表面区域。
本公开内容还提供了水净化系统,所述水净化系统包括:阳极,所述阳极包括第一导电金属氧化物层;第二半导体层,所述第二半导体层接触第一导电金属氧化物层并且被配置成至少部分地与包含氯离子的水直接接触;以及一个或更多个第三半导体的岛,所述一个或更多个第三半导体的岛接触第二半导体层并且被配置成与水直接接触。在一个实施方案中,该系统还包括金属导体,所述金属导体接触第一导电金属氧化物层。在另外的实施方案中,金属导体是钛。在又一个实施方案中,该系统还包括阴极。在另外的实施方案中,阴极是不锈钢。在另一个实施方案中,该系统还包括电流源(currentsource),所述电流源被连接至阴极和阳极。在又一实施方案中,第一导电金属氧化物层包括铱。在另外的实施方案中,第一导电金属氧化物层包括ir0.7ta0.3o2。在又一实施方案中,第二半导体层包括tio2。在另外的实施方案中,第二半导体层包括钴掺杂的二氧化钛。在还另一个实施方案中,第三半导体是二氧化锡或锑掺杂的二氧化锡。在又一实施方案中,该系统还包括用于容纳水、阳极和阴极的电解槽(electrolysistank)。
本公开内容还提供了操作水净化系统的方法,所述方法包括使阳极与包含氯离子的水接触;以及向阳极施加足以在阳极处生成反应性氯的阳极电位,所述阳极包括:第一导电金属氧化物层;第二半导体层,所述第二半导体层接触第一导电金属氧化物层并且被配置成至少部分地与水直接接触;以及一个或更多个第三半导体的岛,所述一个或更多个第三半导体的岛接触第二半导体层并且被配置成与水直接接触。在一个实施方案中,第一导电金属氧化物层包括ir0.7ta0.3o2。在另一个实施方案中,第二半导体层包括钴掺杂的二氧化钛。在还另一个实施方案中,第三半导体是锑掺杂的二氧化锡、或二氧化锡。
本公开内容还提供了制造用于水处理的电解阳极的方法,所述方法包括将金属电极加热至第一预先确定的温度;通过使用喷雾热解将第一含水金属氧化物前体施加到加热的金属电极上,将第一导电金属氧化物层沉积在加热的金属电极上;通过使用喷雾热解将第二含水金属氧化物前体施加到第一导电金属氧化物层上,将第二半导体层沉积在第一导电金属氧化物层上;以及通过使用喷雾热解将第三含水金属氧化物前体施加到第二半导体层上,将第三半导体沉积在第二半导体层上。在一个实施方案中,第一含水金属氧化物前体包括溶解在异丙醇中的3.5mmircl3和1.5mmtacl5的溶液。在另一个或另外的实施方案中,第二含水金属氧化物前体包括通过羟基-过氧化方法(hydroxo-peroxomethod)制备的25mm钛-乙醇酸盐络合物和0.1的摩尔分数的掺杂剂前体co(no3)2的溶液。在还另一个或另外的实施方案中,第三含水金属氧化物前体包括溶解在异丙醇中的25mmsncl4的溶液。在又一实施方案中,金属电极是钛。在另一个实施方案中,该方法还包括在加热前,在10%hf溶液中蚀刻金属电极。在还另一个实施方案中,该方法还包括在第二预先确定的温度,将第一导电金属氧化物层、第二半导体层和第三半导体中的每个退火。在又一实施方案中,该方法还包括在沉积第一导电金属氧化物层、第二半导体层和第三半导体后,将电解阳极退火。在一个实施方案中,第三半导体在第二半导体层上形成为离散的岛。
本公开内容还提供了氯-碱工艺,所述氯-碱工艺包括使阳极与nacl盐水接触;以及向阳极施加足以在阳极处产生反应性氯的阳极电位,所述阳极包括:第一导电金属氧化物层;第二半导体层,所述第二半导体层接触第一导电金属氧化物层并且被配置成至少部分地与盐水直接接触;以及一个或更多个第三半导体的岛,所述一个或更多个第三半导体的岛接触第二半导体层并且被配置成与盐水直接接触。
前述的概述没有限定所附权利要求的限制。对于本领域技术人员而言,在查看以下附图和详细描述后,其他方面、实施方案、特征和优点将是或将变得明显。意图的是所有这样的另外的特征、实施方案、方面和优点都被包括在本说明书内并且受所附权利要求保护。
附图简述
应理解的是,附图仅为了说明的目的并且不限定所附权利要求的限制。此外,附图中的部件不一定按比例绘制。在附图中,相似的参考数字在整个不同的视图中表示相应的部分。
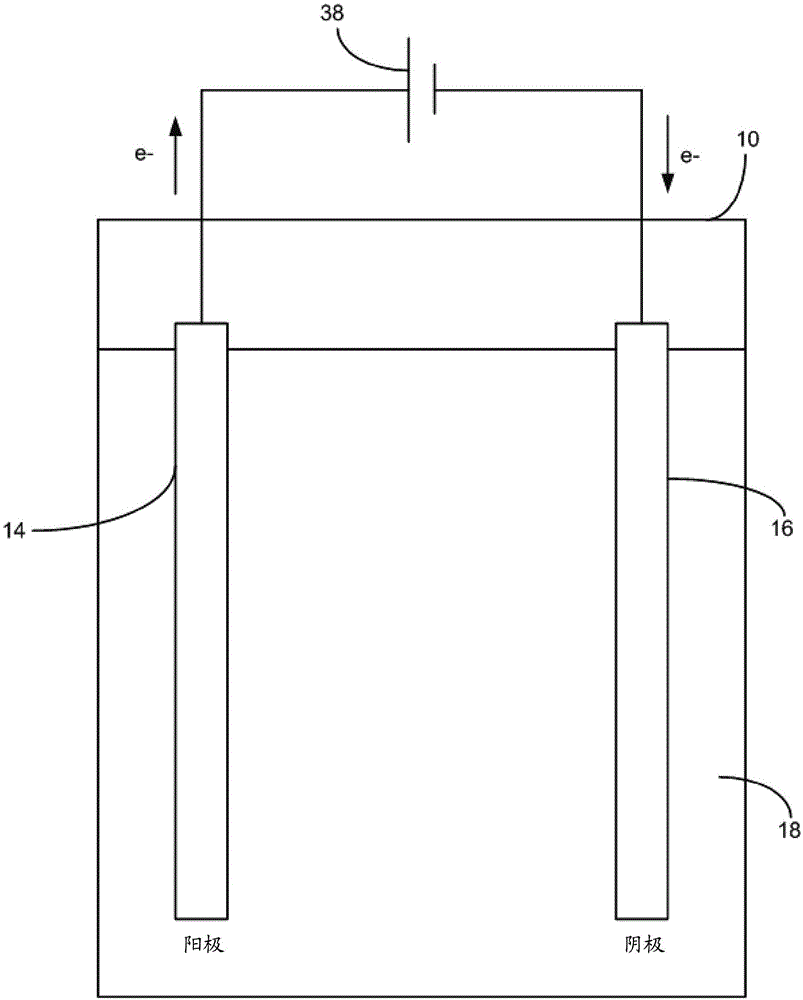
图1图示出了采用阳极的示例性电解系统。
图2图示出了第二示例性电解系统,例如连续水净化系统(continuouswaterpurificationsystem)。
图3是示出了图1和图2的阳极的概念性透视图。
图4是示出了岛和半导体中间层(semiconductorinterlayer)的示例性阳极的表面的场发射扫描电子显微镜(fesem)图像。
图5是阳极的示例性构造的概念性横截面图。
图6是将公开的阳极的氯析出速率(chlorineevolutionrate)和电流效率与现有阳极比较的图。
图7a-图7e是示出了使用所公开的阳极的电解的示例性实验结果的图。
图8a-图8b是示出了使用所公开的阳极的ba电解的示例性实验结果的图。
图9是阳极的反应性物质生成机制的示意图。
图10a-图10c是比较使用所公开的阳极和现有阳极的废水处理的实验结果的图。
图11a-图11e是示出了在不同电解质条件下的公开的阳极的氯析出速率并且示出了在处理废水的实验期间由所公开的阳极产生的反应性氯的消耗的图。
图12a-图12b是示出了在使用公开的阳极的废水电解期间,no3-形成和总氮去除(totalnitrogenremoval)的示例性实验结果的图。
图13a-图13c是示出了在废水电解期间,通过公开的阳极的总氯(tc)生成和游离氯(fc)生成的示例性实验结果的图。
详述
参考附图和并入附图的以下详述描述并说明了电解电极、水处理系统以及使用电解电极和水处理系统的方法和制造电解电极的方法的一个或更多个实例。足够详细地示出和描述了这些实例,提供这些实例不是用于限制而是仅用于例证和教导本发明的电极、方法和系统的实施方案,以使本领域技术人员能够实践所要求要求保护的内容。因此,在适当的情况下为了避免模糊本发明,描述可能省略了本领域技术人员已知的某些信息。本文中的公开内容是不应当被理解为过度地限制可能基于本申请最终被授权的任何专利权利要求的范围的实例。
在整个本申请中使用词语“示例性”意指“用作实例、情况(instance)或说明”。本文被描述为“示例性”的任何系统、方法、装置、技术、特征或类似物不一定被解释为相对于其他特征优选的或有利的。
除非内容清楚地另外指示,否则如在本说明书和所附的权利要求中使用的,单数形式“一(a)”、“一(an)”和“该(the)”包括复数指示物。
虽然与本文描述的那些类似或等效的任何方法和材料可以用于本发明的实践或测试中,但本文描述了适当的方法和材料的具体实例。
另外,除非另外陈述,否则“或”的使用意指“和/或”。类似地,“包含(comprise)”、“包含(comprises)”、“包含(comprising)”、“包括(include)”、“包括(includes)”和“包括(including)”是可互换的并且不意图是限制性的。
还应理解,在各个实施方案的描述使用术语“包含(comprising)”的情况下,本领域技术人员将理解,在某些特定情况下,实施方案可以使用语言“基本上由......组成(consistingessentiallyof)”或“由......组成(consistingof)”来可选择地描述。
水短缺已经被认为是正在出现的全球危机。为了有助于水的再循环(recycling)和再利用,分散化废水处理(decentralizedwastewatertreatment)已经被提议作为常规的城市废水系统的补充。电化学氧化(eo)通常比生物处理更有效,并且通常比均相高级氧化工艺(homogeneousadvancedoxidationprocess)更便宜。此外,紧凑的设计、易于自动化和小的碳足迹(carbonfootprint)使电化学氧化成为分散化废水处理和再利用的理想候选者。
eo的性能由反应性物质的电化学生成来确定,反应性物质的电化学生成很大程度上取决于阳极材料的性质。在过去的几十年中,已经研究了对于氧析出反应(oxygenevolutionreaction)(oer)具有高过电位的非活性阳极(non-activeanode),例如基于sno2、pbo2和硼掺杂的金刚石(boron-dopeddiamond)(bdd)的非活性阳极。尽管sno2阳极和pbo2阳极对于羟基自由基(·oh)生成的优异的电流效率,但sno2阳极和pbo2阳极具有差的电导率和稳定性。bdd阳极的应用受其高成本和复杂制造的阻碍。相反地,pt族金属氧化物(例如,ruo2和iro2)是用于oer的高效且稳定的催化剂,在氯化物的存在下呈现出高的氯析出反应(cer)活性,尽管它们典型地对于羟基自由基生成是较低效率的。因此,对于cer和自由基生成两者均具有高活性的耐用阳极的开发是持续的挑战。
电解质组成是eo性能中的另一种因素。先前,·oh被认为是在eo期间对有机物质去除的主要贡献者。最近的研究已经指出,碳酸根、硫酸根和磷酸根基团也是强有力的氧化剂。与这些阴离子相比,废水中的氯离子(cl-)可以更容易地被氧化成反应性氯物质。在cl-的存在下观察到的有机化合物的增强的电化学氧化已经归因于与游离氯(cl2、hocl和ocl-)的反应。最近的研究表明,cl·和cl2·-可能主要负责有机化合物降解。然而,缺乏验证在电化学期间这些自由基的存在或形成机制的直接实验证据。在有机污染物的电化学氧化期间cl-溶液中的反应性物质形成和反应性的定量描述未曾被充分地阐明。
本文公开了对氯和自由基生成具有高活性的多功能sbsn/coti/ir异质结阳极和sn/coti/ir异质结阳极。还公开了实验方法和动力学建模方法的组合,其帮助解释阳极反应性物质生成机制并建模其在电解质中的稳态浓度。公开的阳极表示相对于用于电解的已知异质结金属氧化物阳极的改进,并且该建模提供对废水电解机制的新见解。
图1是水净化系统8的简化图示,所述水净化系统8包括容器10、阳极14和阴极16以及电压源38,所述容器10用于容纳电解介质18例如废水,所述阳极14和阴极16用于在废水电解中使用,所述电压源38用于向阳极14和阴极16提供电流。系统8可以通过利用高级氧化工艺(advancedoxidationprocess)(aop)以将有机物质分解成小且稳定的分子例如水和co2,来净化具有有机物质的水。为了简化的目的,图示出了单个阳极14和阴极16,尽管可以采用另外的阳极14和/或阴极16。
水净化系统8可以被用于净化废水。废水包括通常与废产物相关的有机物质和在尿液中天然存在的氯离子。因此,废水可以天然地作为电解介质18操作,或者电解质例如nacl可以任选地被添加至废水中。
本文参考其他附图来描述阳极14的详细构造的实例。通常,阳极14具有第一导电金属氧化物层、接触第一导电金属氧化物层的第二半导体层以及一个或更多个第三半导体的岛,所述一个或更多个第三半导体的岛接触第二半导体层。根据阳极14的示例性实施方案,第一导电金属氧化物层包括铱,第二半导体层包括二氧化钛,并且第三半导体包括二氧化锡。根据另一个示例性实施方案,水净化阳极14可以包括钛基底、在钛基底上形成的ir0.7ta0.3o2层、在ir0.7ta0.3o2层上形成的co-tio2层以及多个sno2的岛,所述多个sno2的岛在co-tio2钛层上形成。可选择地,岛可以由掺杂有sb的sno2制成。
阴极16可以由合适的金属导体例如不锈钢制成。
在水净化系统8的操作期间,阳极电位38以足以在阳极14处产生活性氯的水平被施加在阳极14和阴极16之间。
阳极14具有相对高的反应性氯物质(rcs)生成的速率。由于许多废水电解系统使用rcs来降解有机物质,所以阳极14高度地适合用于在这些系统中使用。
图2图示出了包括多个阳极14和阴极16的另一个合适的电解系统15例如水净化系统的实例。该系统包括具有储器(reservoir)的容器10。阳极14和阴极16被定位在储器中,使得阳极14和阴极16彼此交替。阳极14和阴极16彼此平行或大体上平行。电解介质18被定位在储器中,使得阳极14和阴极16与电解介质18接触。电解介质18包含一种或更多种电解质,并且可以是液体、溶液、悬浮液或液体和固体的混合物。在一个实例中,电解介质18是包含有机物质、氨和氯离子(cl-)的废水。氯离子可以作为将盐添加至电解介质18的结果存在于电解介质18中,或者电解介质18可以包括为氯离子的天然来源的尿。电解系统还包括电压源(未示出),所述电压源被配置成驱动电流通过阳极14和阴极16,以便驱动电解介质18中的化学反应。
图2中图示出的电解系统包括入口20和出口22。电解系统可以作为连续反应器操作,因为电解介质18通过入口20流入到储器中,并且通过出口22流出储器。可选择地,电解系统还可以作为间歇反应器来操作。当电解系统作为间歇式反应器操作时,电解介质18可以是固体、液体或组合。
图3是阳极14的示例性构造的概念性透视图。阳极包括:集电器30,例如金属基底;第一导电金属氧化物层32;第二半导体层34;以及第三半导体的岛36。
集电器30可以是片材或箔,或可选择地,集电器30可以是多孔材料,例如网(mesh)或织物。用于集电器30的合适的材料包括阀金属,例如ti。
第一导电金属氧化物层32位于集电器30和第二半导体层34之间。第一半导体32接触第二半导体34,形成异质结。第一半导体32还接触集电器30。第一半导体32可以起选自由以下组成的组的一种或更多种功能的作用:钝化集电器30、用作第二半导体34和集电器30之间的电子穿梭器(electronshuttle)以及减少第二半导体34和集电器30之间的电子转移(electrontransfer)的欧姆电阻。在某些情况下,集电器30和第二半导体34之间的直接接触将导致电子从第二半导体34转移至集电器30的肖特基势垒。可以选择第一半导体32,使得用于通过第一半导体32从第二半导体34至集电器30的电子转移的能垒(energybarrier)小于将由从第二半导体34直接至集电器30的电子转移造成的能垒。
第一半导体32可以包括第一金属氧化物,由第一金属氧化物组成或基本上由第一金属氧化物组成,所述第一金属氧化物包括氧、一种或更多种电活性金属元素和任选地一种或更多种稳定元素(stabilizingelement),由氧、一种或更多种电活性金属元素和任选地一种或更多种稳定元素组成,或基本上由氧、一种或更多种电活性金属元素和任选地一种或更多种稳定元素组成。电活性元素的实例包括,但不限于ir。稳定元素的实例包括,但不限于ta。因此,第一半导体32可以包括第一金属氧化物,由第一金属氧化物组成或基本上由第一金属氧化物组成,所述第一金属氧化物包括氧、一种或更多种电活性金属元素例如ir和一种或更多种稳定元素例如ta,由氧、一种或更多种电活性金属元素例如ir和一种或更多种稳定元素例如ta组成,或基本上由氧、一种或更多种电活性金属元素例如ir和一种或更多种稳定元素例如ta组成。作为实例,第一金属氧化物可以包括氧、铱和一种或更多种稳定元素例如ta,由氧、铱和一种或更多种稳定元素例如ta组成,或基本上由氧、铱和一种或更多种稳定元素例如ta组成。在一个实例中,第一金属氧化物包括氧、铱和钽,由氧、铱和钽组成,或基本上由氧、铱和钽组成。例如,第一金属氧化物可以是ir0.7ta0.3o2。
第二半导体层34具有暴露的表面区域35。在某些情况下,表面区域37的至少某些与电解介质18直接物理接触。选择第二半导体层34,使得与半导体岛36组合的第二半导体层34增强反应性氯的析出。
第二半导体层34可以包括第二金属氧化物,由第二金属氧化物组成,或基本上由第二金属氧化物组成,所述第二金属氧化物包括氧和一种或更多种选自第iv族的元素、由氧和一种或更多种选自第iv族的元素组成,或基本上由氧和一种或更多种选自第iv族的元素组成。在某些情况下,第二半导体层34包括氧和钛,由氧和钛组成,或基本上由氧和钛组成。例如,第二半导体层34可以包括钴掺杂的二氧化钛(co-tio2),由钴掺杂的二氧化钛(co-tio2)组成,或基本上由钴掺杂的二氧化钛(co-tio2)组成。
第二半导体层34被覆涂(overcoat)有第三半导体的离散的岛36。岛36与第二半导体层34接触,并且当阳极14被放置在电解系统8或15中时,岛36还可以接触电解介质18。可以使用喷雾热解在第二半导体层34上形成岛36。
岛36可以各自包括第三金属氧化物,由第三金属氧化物组成,或基本上由第三金属氧化物组成,所述第三金属氧化物包括氧和一种或更多种元素、由氧和一种或更多种元素组成,或基本上由氧和一种或更多种元素组成。在某些情况下,第三半导体岛36包括氧和锡,由氧和锡组成,或基本上由氧和锡组成。例如,岛36可以各自包括二氧化锡(sno2),由二氧化锡(sno2)组成,或基本上由二氧化锡(sno2)组成。可选择地,作为另一个实例,岛36可以各自包括掺杂有锑的二氧化锡(sb-sno2),由掺杂有锑的二氧化锡(sb-sno2)组成,或基本上由掺杂有锑的二氧化锡(sb-sno2)组成。
岛可以是在第二半导体层的顶部上的材料的离散表面沉积物,如图3-图5中示出的。它们可以在物理上彼此分开。在某些情况下,每个岛可以具有约8μm至1μm或更小的一般直径(generaldiameter)。虽然它们在第二半导体层上的覆盖率可以是任何合适的量,但在某些情况下,它们可以覆盖第二半导体层的表面积的约50%。岛36为rcs和自由基生成提供活性位点。尽管在图3的实例中以规则的间距间隔和相同的尺寸示出,但岛36不一定必须被放置在此布置中,并且可以具有相对于彼此不均匀的间距和不同的不规则的尺寸(例如,图4)。岛36的数目和它们在第二半导体层34上的密度可以是任何合适的值。
第二半导体层34和覆涂的第三半导体岛36增强活性氯的析出。表面岛36还用作用于游离的自由基生成的反应位点。
在阳极14的操作期间,使阳极14的暴露的面35与电解介质18接触,该暴露的面35包括第二半导体层34的表面37的至少某些和表面的第三半导体岛36。可以使岛36的侧面和顶部与电解介质接触。
图5是阳极14的示例性构造的概念性横截面图。在此实例中,集电器30是ti箔。第一导电金属氧化物层32是使用本文以下描述的喷雾热解方法在ti箔上形成的ir0.7ta0.3o2的层。第二半导体层34是使用喷雾热解在ir0.7ta0.3o2层上形成的钴掺杂的二氧化钛(co-tio2)的层,如以下描述的。并且,第三半导体岛36是在co-tio2层34上形成的锑掺杂的二氧化锡(sb-sno2)的岛。还描绘了通过层26-30的电流流动e-。
图5示出了阳极上的活性位点,在活性位点中形成活性物质。例如,图5图示出了氯循环,其中氯离子在阳极表面处被氧化,以便生成反应性氯物质(rcs)。反应性氯物质是游离的氯加上氯自由基。游离的氯的实例包括但不限于cl2、hocl和clo-。氯自由基的实例包括但不限于cl·和自由基离子cl2·-。
如图5中示出的,在rcs生成的一个实例中,sb-sno2岛36促进cl-的单电子氧化,以在电解介质18中产生游离的cl·。原子氯自由基(cl·)可以与介质18中的氯离子相互作用,以形成自由基离子cl2·-。自由基离子cl2·-可以在阴极表面处被还原,以在电解介质18中生成两个氯离子。co-tio2层34的暴露的表面生成cl·和cl2。这又在电解介质18中生成hocl。hocl是酸(次氯酸),其因此在电解介质18中部分地解离成质子(h+)和次氯酸根(clo-)。次氯酸根和水可以在阴极16的表面处反应,以生成氯离子和羟基离子,氯离子和羟基离子各自从阴极16接收电子。次氯酸、次氯酸根、原子氯自由基和自由基离子cl2·-用作反应性氯物质(rcs)。
本发明还可以通过以下实施例来例证,这些实施例通过例证的方式提供并且不意图是限制性的。
实施例
实施例1
制备第一阳极。清洁的ti金属箔充当基底,该基底用作集电器(例如1cm×1.5cm)。ti基底用砂纸抛光,并且在10%hf溶液中蚀刻持续一分钟。然后,通过喷雾热解,将金属氧化物层沉积在清洁的ti表面上。
使用喷雾热解,将含水金属氧化物前体用5psi空气雾化,并且喷雾到加热的(例如,300℃)ti金属基底上。然后,将产生的氧化物膜在500℃退火持续十分钟。重复此程序以达到对于每个半导体层的期望的质量负载量。在对于层达到期望的质量负载量后,最终退火在500℃进行持续一小时。
关于用于每个半导体层的前体,ir0.7ta0.3o2层前体包含在异丙醇中的3.5mmircl3和1.5mmtacl5。tio2前体包含通过羟基-过氧化方法制备的25mm钛-乙醇酸盐络合物。将掺杂剂前体co(no3)2以0.1的摩尔分数添加至tio2前体中。sb-sno2前体包含溶解在异丙醇中的25mmsncl4和1.24mmsbcl3。
如本文提到的,为了简单,仅具有ir0.7ta0.3o2层的阳极被表示为ir。具有sb-sno2岛、co掺杂的tio2层和ir0.7ta0.3o2层的多层阳极被表示为sbsn/coti/ir。没有sb-sno2掺杂的阳极被表示为coti/ir(具有co掺杂)或ti/ir(没有co掺杂)。
ir0.7ta0.3o2、tio2和sno2的质量负载量分别是0.3mg/cm2、0.5mg/cm2和1.0mg/cm2。可选择地,可以使用其他质量负载量,包括在前述值周围(例如±25%)的质量负载量。
使用surfacesciencem-probeesca/xps,对第一阳极进行x射线光电子能谱(xps)。形态和元素组成用装配有oxfordx-maxsddx射线能量色散光谱仪(eds)的zeiss1550vp场发射扫描电子显微镜(fesem)获得。
通过喷雾热解制备的阳极的形态比刷涂阳极的典型的“裂纹泥(cracked-mud)”纹理更致密且更平滑。元素绘图还指示,通过喷雾热解制备的阳极的子层ir、顶层ti和co掺杂剂的较好的分散。
图4是示出了岛36和半导体中间层34的示例性第一阳极14的表面35的场发射扫描电子显微镜(fesem)图像。sb-sno2的沉积在co-tio2层34的顶部上产生孤立的岛36而不是薄膜(图4)。这与先前报告的通过喷雾热解制备的sb-sno2阳极的形态一致。sb-sno2岛被确定为在2μm-4μm高的范围内,并且位于0.5μmco-tio2层的顶部,co-tio2层覆盖0.5μmir0.7ta0.3o2层。来自第一层32的iro2可以热扩散到tio2层34中。
coti/ir阳极表面34主要包括tio2,如通过在tio2或co-tio2涂层中的xps光谱中的独特的氧化铱峰(62ev和65ev)的损失所证明的。与没有iro2底层(under-layer)的coti相比,coti/ir的ti2p峰移动至轻微较低的结合能。此移动归因于从iro2至tio2的电荷转移,因为iro2具有比tio2更高的功函数。此相互作用指示iro2层充当电子穿梭器,以克服co-tio2层和ti基底之间的肖特基势垒。因此,基于观察到的含ir的阳极的电荷转移电阻(rct)的减小,促进了电子转移(即,rct从用于coti的122kω降低至用于coti/ir的4kω)。
tio2层的性质可以通过金属离子掺杂来改性。钴(co)掺杂显著地增加氧空位(oxygenvacancy)(531ev-533ev)相对于晶格氧(latticeoxygen)(529ev-531ev)的分数。此移动反映了coti/ir相对于ti/ir的氧结合能的弱化。
在30mmnacl中的线性扫描伏安法期间,ir阳极在ma/cm2呈现出1.32v的起始电位(onsetpotential),对应于0.5v的氧析出的过电位(在ph7,为0.82v)。这相当于先前报告的关于纳米晶体iro2催化剂的过电位。虽然tio2涂层或co-tio2涂层几乎不影响起始电位,但sb-sno2的沉积将起始电位升高至1.38v,这紧密地匹配氯析出反应(cer)电位(1.36v)。观察到的起始电位的移动可能是由于氧析出反应(oer)的抑制,如通过sbsn/coti/ir阳极相对于ir阳极的oer的电化学活性表面积的减小所证明的。尽管co-tio2中间层仅轻微影响oer起始电位,但对于oer活性的抑制是至关重要的。在没有co-tio2涂层的情况下,ir0.7ta0.3o2层通过sb-sno2岛中的裂缝接近电解质,这增加用于oer的电化学活性表面积并且减小起始电位。
实施例2
使用实施例1中描述的喷雾热解方法来制备第二阳极。第二阳极在本文中被表示为sbsn/coti/ir*。第一示例性阳极和第二示例性阳极之间的差别在于,在第二阳极的情况下,ir0.7ta0.3o2质量负载量被减小至约0.05mg/cm2。
基于其高的起始电位(1.56v)和用于oer的低电化学活性表面积,ir0.7ta0.3o2(sbsn/coti/ir*)的质量负载量的83%降低导致相对无活性的第二阳极。在30mmnacl中的线性扫描伏安法期间,第二阳极在ma/cm2呈现出1.56v的起始电位。
此外,ir0.7ta0.3o2的质量负载量影响总阳极稳定性。加速寿命测试(acceleratedlifetimetest)示出,sbsn/coti/ir*阳极在25ma/cm2的寿命是720小时,而sbsn/coti/ir的寿命可以多达4年。
加速寿命测试(alt)在1mnaclo4中在1.2a/cm2的电流密度进行。在alt测试中,使具有0.25cm2的表面积的阳极样品经历300ma电流,产生1.2a/cm2的高电流密度。alt的目的是在严苛的恒电流条件下测试电极,以加速电活性层的溶解或分离。失活将通过电池电压(e电池)的急剧增加来反映。当电池电压达到9v时,电极被认为是失活的,因为高于此电压,ti金属基底将被腐蚀。基于在高电流(iacc=1.2a/cm2)从alt观察到的寿命(tacc),在操作电流(i)的实际寿命(t)可以通过经验等式来评估:
t=(iacc1.7*tacc)/i1.7(等式1)
sbsn/coti/ir*和sbsn/coti/ir的tacc分别是0.5小时和52小时,其在25ma/cm2将寿命分别作为360小时和37,507小时(约4.3年)给出。
实施例3
使用上文实施例1中描述的相同的程序来制备第三阳极,但形成未掺杂的二氧化锡(sno2)的岛,而不是sb-sno2岛。sno2前体包含溶解在异丙醇中的25mmsncl4或sncl2,并且以与实施例1相同的质量负载量施加并以与实施例1相同的方式退火。
实施例4
通过使用sbsn/coti/ir阳极(第一阳极)和sbsn/coti/ir*阳极(第二阳极)以在受控的条件下使用不同的电解质进行电解并且还通过应用sbsn/coti/ir阳极和sbsn/coti/ir*阳极以电化学处理人类废水,来测试sbsn/coti/ir阳极和sbsn/coti/ir*阳极。在废水测试的情况下,测试结果示出,可以使用第一阳极,在两小时电解后以最小能量消耗(370kwh/kgcod和383kwh/kgnh4+)去除化学需氧量(cod)和nh4+。尽管游离的自由基物质有助于cod去除,但公开的阳极提高反应性氯产生,并且因此比那些设计成提高游离的自由基产生的阳极更有效。
与计算机动力学模拟结合的测试和实验示出,虽然·oh和cl·初始地在sbsn/coti/ir阳极(第一阳极)暴露的表面35上产生,但溶液中形成的占主导的自由基是二氯自由基阴离子cl2·-。反应性自由基(例如·oh)的稳态浓度(steady-stateconcentration)比反应性氯的稳态浓度小十个数量级。
为了进行测试,构建了类似于图1中示出的示例性电解电池(反应器)。该电池包括与不锈钢阴极(例如,2×1.5cm2)平行的阳极,其中在阳极和阴极之间具有5mm的间隔。电池使用不同的电解质用于不同的测试。对于使用nacl电解质的电池,电压相对于ag/agcl/饱和nacl参比电极(例如,可获自basi,inc.)来控制。对于使用na2so4电解质的电池,电压相对于hg/hg2so4参比电极(例如,可获自gamryinstruments)来控制。
电池的电化学双层电容(cdl)在静态30mmna2so4溶液中,以多个扫描速率(0.005v/s-0.8v/s),通过循环伏安法(以开路电位为中心的0.1v窗口(window))在非法拉第范围(non-faradaicrange)内测量。电化学阻抗光谱(eis)测量在静态30mmnacl电解质中进行。正弦波的振幅是10mv,其中频率在从0.1hz至100khz的范围内。eis光谱通过将阳极的helmholtz层视为包括溶液电阻、电荷转移电阻(rct)和电容的randles电路来拟合。
进行使用第一阳极和第二阳极以及比较阳极的电解。在反应器中使用前,第一阳极和第二阳极在30mmnacl中以25ma/cm2预处理持续一小时。电池的未补偿电阻(ru)通过具有200ma电流偏置的电流中断来测量。对于ru,调节所有阳极电位,并且相对于正常氢电极(normalhydrogenelectrode)(nhe)来报告。所有电解实验以恒电流模式,其中电流密度为25ma/cm2或50ma/cm2。cer测试通过30mmnacl溶液的恒电流电解来进行。在15分钟内,以两分钟间隔取出样品。
使用dpd(n,n-二乙基-对苯二胺)试剂(hach方法10101和10102)测量总氯(tc)浓度和游离氯(fc)浓度。计算氯析出速率和电流效率(ce)。恒定电流地进行苯甲酸(ba)的电解。使用具有作为洗脱剂的10%乙腈和90%0.1%甲酸的zorbaxxdb柱,色谱法地分离ba。
由于第一阳极和第二阳极的结构,在电解期间第一阳极和第二阳极的氯析出显著地改进。在30mmnacl溶液的电解期间,用tio2涂覆ir阳极显著地增加氯析出反应(cer)活性和电流效率,如通过图6的图示出的。图6示出了在30mmnacl中以25ma/cm2测量的某些已知的比较阳极以及第一阳极、第二阳极和第三阳极的氯析出速率和电流效率。用于每个阳极的阳极电位作为每个棒上的数字被示出。在棒的顶部的误差棒代表标准偏差。cer活性的增加由顶部tio2层和ir0.7ta0.3o2子层之间的相互作用引起,因为仅tio2位点被暴露,并且没有ir0.7ta0.3o2子层的tio2阳极不没有cer活性(数据未示出)。图6中指示的和本文讨论的商业阳极是购自韩国的nanopac的可商购的基于iro2的cer阳极,该商业阳极用于与公开的阳极比较。
通常接受的是,cer遵循volmer-heyrovsky(v-h)机制。volmer步骤包括cl-的吸附和电子的放电:
mox+cl-→mox(cl·)+e-(等式2)
在heyrovsky步骤中,吸附的cl·与来自本体电解质(bulkelectrolyte)的cl-组合,并且释放cl2:
mox(cl·)+cl-→mox+cl2+e-(等式3)
两个cl·经由volmer-tafel反应的重组还可以产生cl2:
2mox(cl·)→2mox+cl2(等式4)
对于oer具有最优的氧结合能的催化剂通常对于cer具有高的活性,这导致oer和cer之间的竞争。然而,其他人的密度泛函理论(dft)计算已经示出,朝向cer的选择性可以通过ruo2上的单层tio2涂层来提高,这轻微地增加cer的能垒,但剧烈地升高oer的能垒。与这些计算一致,具有与ruo2类似的氧结合能的被施加到iro2上的tio2涂层显著地增加用于氯产生的电流效率。因此,通过在ir0.7ta0.3o2层上的tio2覆涂提供的oer的减小的活性表面积和增加的oer起始电位显著地改进第一阳极和第二阳极的cer。
在分子水平,cl·的解吸(等式3和等式4)被认为是cer的速率限制步骤。考虑到氧结合能和氯结合能之间的正线性关系,通过tio2层的co掺杂降低氧结合能可以有助于cl·解吸,与ti/ir相比,提高coti/ir的cer活性。
基于oer被阳极中的tio2涂层和co-tio2涂层的抑制,ti/ir阳极和coti/ir阳极呈现出相比于ir阳极的增加的cer(图6)。然而,sb-sno2岛的较低的电导率导致较高的操作阳极电位。sbsn/ir阳极和sbsn/coti/ir*阳极的相对较差的性能指示,不存在coti层或ir质量负载量的减小对cer活性是有害的。通常,coti/ir和sbsn/coti/ir的cer活性比商业阳极的cer活性高(图6)。
图7a-图7e是示出了使用公开的阳极的电解的示例性实验结果的图。图7a和图7b分别示出了使用sbsn/coti/ir阳极(第一阳极)在ma/cm2的30mmnacl(图7a)和60mmnacl(图7b)的电解的结果。图7c示出了使用coti/ir阳极在25ma/cm2的60mmnacl的电解。图7d示出了使用sbsn/coti/ir阳极(第一阳极)在25ma/cm2的60mmnacl的电解。图7e示出了使用sbsn/coti/ir*阳极(第二阳极)在25ma/cm2的60mmnacl的电解。图表中的符号代表实验数据,并且虚线代表预测的模型结果,除了图7d,在图7d中虚线代表模型拟合结果。
用coti/ir阳极电解nacl溶液导致cl-的逐渐损失,其中对应的产生hocl/ocl-和clo3-(图7c)。在sbsn/coti/ir阳极的情况下观察到类似的结果(图7a、图7b、图7d),除了较高的clo3-的产生。sbsn/coti/ir*阳极的测试示出clo4-的形成(图7e),这与先前的研究一致,先前的研究证明非活性电极比活性电极更容易地产生clo4-。
公开的阳极还产生其他自由基,其他自由基在电解测试期间被实验地测量。除了游离氯之外,nacl水溶液的电解还生成自由基,例如·oh、cl·和cl2-。恒定电流地进行苯甲酸(ba)的电解以测量自由基生成。ba被选择作为自由基探针化合物(radicalprobecompound),因为它与·oh、cl·和cl2-反应(表1中给出的速率常数),但不与游离氯反应。
表1.na2so4电解和nacl电解中的关键反应。
表1.(续)
a活性位点mox和h2o的浓度被设定为1。
bh+和oh-的浓度分别被设定为3.16×10-9和3.16×10-6。
图8a和图8b是示出了涉及ba的电解的实验结果的图。这些图图示出了在可变电流密度(l:25ma/cm2,h:50ma/cm2)和初始cl-浓度(30mm和60mm)下的在sbsn/coti/ir*阳极(图8a)和sbsn/coti/ir阳极(图8b)的情况下ba降解。误差棒代表标准偏差。
仅在sbsn/coti/ir*阳极(图8a)的情况下观察到在25ma/cm2的30mmna2so4电解质溶液中的ba降解。当电流密度增加至50ma/cm2时,在sbsn/coti/ir*阳极(图8b)的情况下观察到ba降解,但在没有添加的sb-sno2岛的coti/ir阳极和ti/ir阳极的情况下,没有观察到ba降解(数据未示出)。ba可以经由在bdd电极上以高氧化电位(即2.4vnhe)直接氧化来降解。然而,此途径在本研究中被排除,因为在不存在或存在1mmba的情况下,在na2so4中观察到线性扫描伏安法中的相同的电流响应(数据未示出)。由于在30mmnano3电解质中观察到相同的ba衰变动力学(decaykinetic),因此排除硫酸根基团的贡献。因此,ba的降解归因于与·oh的反应。假定·oh的生成是零级反应,生成速率(rho·;m/s)可以通过用动力学模型拟合ba降解数据来评估(表2)。在·oh生成方面,sbsn/coti/ir*发现比sbsn/coti/ir更有效(图8a相对于图8b)。对于sbsn/coti/ir*阳极和sbsn/coti/ir阳极,稳态·oh自由基浓度分别被计算为2.6×10-15mol/l和1.4×10-15mol/l。
表2.通过动力学建模评估的速率常数
在30mmnacl的存在下,加速ba降解(图8a和图8b)。这意味着在cl-的存在下,生成更多的自由基。熟知的是,cl·以与·oh类似的速率与有机分子反应。然而,参与v-h步骤(代表图9中的volmer步骤和heyrovsky步骤的线)的cl·被假定为是表面结合的并且迅速地与局部的cl·或cl-结合,并且因此不可能有助于ba降解。此断言由在coti/ir阳极和ti/ir阳极的情况下观察到的缺乏ba降解来支持,尽管fc产生(数据未示出)。·oh自由基可以被cl-猝灭,以生成更小反应性氯自由基(图9)。其中·oh是唯一自由基物质的动力学模型未能模拟观察到的提高的ba降解速率。因此,必须存在另外的反应性自由基输入。一种可能性涉及sb-sno2的贡献,这促进cl-的单电子氧化以产生游离的cl·(图4和图9):
cl-sb-sno2>cl·+e-(等式5)
先前已经报告了在电解期间在sno2上生成游离的cl·,尽管没有证实实验证据。然而,动力学模型结果为多种氯自由基物质对总速率的贡献提供了自洽的动力学论证。动力学模型还被用于模拟在na2so4电解质和nacl电解质两者中的ba的降解,条件是包括等式5。
通过模型拟合获得的用于cl·形成的一级速率常数(kcl·;s-1)发现比用于·oh析出的高多于两个数量级(表2)。与sbsn/coti/ir*的kcl·相比,sbsn/coti/ir的较高的kcl·可以通过从co-tio2位点接收更多cl·的sb-sno2岛来解释(图5)。
尽管导致cl·和·oh的生成的电子转移反应是在阳极上的初始自由基形成步骤,但游离的自由基浓度(·oh、o·-、cl·、cl2-和cloh·-)的整个组的另外的建模示出在浓度上占主导的自由基物质是cl2-。模型模拟还指示cl·和cl-的组合是用于cl2-形成的主要途径。
cl-浓度从30mm增加至60mm导致观察到的ba降解速率的降低(图8a和图8b)。此明显的抑制可以归因于通过fc清除·oh和cl·所起的作用。在60mmnacl观察到的减小的ba降解速率和减小的自由基浓度成功地通过动力学建模结果来预测(图8a和图8b中的虚线)。
多种游离的自由基对于ba降解速率的相对贡献通过由特定的目标自由基模拟ba的氧化来评估,同时排除模型中的其他自由基反应。似乎cl·是对ba氧化的主要贡献者,随后是cl2·-和·oh,即使cl2·-具有最高的浓度。这是由于,与cl2·-相比,ba与cl·的高得多的反应速率(1.9×1010相对于2.0×106m-1s-1)。
基于校准的速率常数,动力学模型能够预测反应性物质形成和稳态浓度。通常,据发现,fc浓度比自由基浓度高十个数量级。
实施例5
使用装配有bader-deuflhard积分仪的kintecus5.75化学动力学建模软件进行nacl电解质电解期间的cer和自由基产生的动力学建模。使用的模型包括37种反应,这些反应在上文表1中示出。ph保持在8.5恒定,这是在nacl电解(ph在电解的一分钟内迅速升高至8.5)和废水电解期间的典型条件。通过将实验数据与动力学模型拟合,获得模型的未知的速率常数。
进行动力学建模以评估在cl2的析出中涉及的关键反应的速率常数(以下等式6)和cl2、hocl和ocl-的ph依赖性平衡(表1)。
hocl/ocl-直接氧化成clo3-,并且clo3-直接氧化成clo4-也被考虑:
总动力学可以被认为是系列中的一级反应:
d[cl-]/dt=-k1[cl-](等式10)
d[fc]/dt=k1[cl-]-k’2[mox+1][fc]
=k1[cl-]–k2[fc](等式11)
d[clo3-]/dt=k2[mox+1][fc]-k’3[mox+1][clo3-]=k2[fc]-k3[clo3-](等式12)
d[clo4-]/dt=k’3[mox+1][clo3-]=k3[clo3-](等式13)
用于coti/ir阳极和sbsn/coti/ir阳极的fc形成速率(k1)被发现比clo3-形成速率(k2)高多于两个数量级(参见表2)。
仅对于sbsn/coti/ir*阳极计算clo4-形成速率(k3),并且低于fc形成速率和clo3-形成速率(k1和k2),这与先前示出clo3-氧化成clo4-缓慢的研究一致。在sbsn/coti/ir阳极的情况下,增加的电流密度(50ma/cm2相对于25ma/cm2)没有显著地增加fc浓度,而是导致更大的clo3-产生(图7a-图7e)。
模型拟合示出,电流密度的增加导致表观速率常数的增加(参见表2),这可以通过butler-volmer公式来解释。也就是,fc产生速率(k1)和fc氧化速率(k2)的同时增加导致等式11中的d[fc]/dt的不太明显的增加,这解释了在50ma/cm2的无效率的氯累积(图7a-图7e)。
建模示出,cl-浓度的增加比增加的电流密度更有效地增加fc浓度。在coti/ir阳极、sbsn/coti/ir阳极和sbsn/coti/ir*阳极的情况下在25ma/cm2电解期间,cl-浓度加倍(即60mm相对于30mm)导致约双倍峰值fc浓度(图7a-图7e)。
在60mmnacl中电解过程的建模给出与实验数据一致的结果(图7a-图7e)。然而,在sbsn/coti/ir*阳极的情况下clo4-产生的实际增加小于预测的。这可能是因为高cl-浓度通过阻断用于将clo3-氧化成clo4-的活性位点来抑制clo4-形成。然而,如呈现的动力学模型为cer的优化提供了简单但强有力的工具。
实施例6
还测试了活性sbsn/coti/ir阳极(第一阳极)和非活性sbsn/coti/ir*阳极(第二阳极)在其用于家庭(例如人类废物)废水处理的潜力方面。
对于废水测试,人类废水从位于californiainstituteoftechnologycampus(pasadena,ca)的公共太阳能厕所原型收集。废水的化学需氧量(cod)通过重铬酸盐消化(dichromatedigestion)(hach方法8000)来确定。总有机碳(toc)通过auroratoc分析仪来分析。阴离子(cl-、clo3-、clo4-、no3-和po43-)和阳离子(nh4+、na+、ca2+和mg2+)通过离子色谱法(ics2000,dionex;ionpacas19柱和ionpaccs16柱)同时检测。
在cod去除方面,sbsn/coti/ir阳极(第一阳极)优于商业阳极,但在30mmcl-中在25ma/cm2,比sbsn/coti/ir*阳极(第二阳极)低效(图10a)。
图10a-图10c是比较使用公开的阳极和现有阳极的废水处理的实验结果的图。图10a是示出了由sbsn/coti/ir阳极和sbsn/coti/ir*阳极(第一阳极和第二阳极)和商业阳极在多种电流密度(l=25ma/cm2,h=50ma/cm2)和初始cl-浓度(30mm、60mm)下电解的废水中的化学需氧量(cod)的浓度相对于时间概况的图。图10b是示出了由sbsn/coti/ir阳极和sbsn/coti/ir*阳极(第一阳极和第二阳极)和商业阳极在多种电流密度(l=25ma/cm2,h=50ma/cm2)和初始cl-浓度(30mm、60mm)下电解的废水中的nh4+的浓度相对于时间概况的图。图10c是示出了在通过不同阳极的4小时电解后的总有机碳(toc)的去除的图。图中的误差棒代表标准偏差。实验数据从sbsn/coti/ir阳极收集,除了分别从第二阳极和商业阳极收集的标记有sbsn/coti/ir*或商业的数据。
假定cod的直接氧化不显著,那么用sbsn/coti/ir阳极在25ma/cm2获得的cod去除应当排它地经由fc介导的氧化进行。相反地,计算示出,自由基介导的氧化途径在sbsn/coti/ir*阳极上贡献多达cod去除的80%(图11a-图11e)。此结果表明,通过“非活性”sbsn/coti/ir*阳极产生的自由基比单独的fc对于cod去除更有效。然而,toc分析示出,尽管完全的cod去除,但在sbsn/coti/ir*阳极的情况下,没有观察到人类废水中的有机碳的完全矿化(图10c)。这可能部分地是由于作为占主导的自由基物质的cl2·-的贡献。cl2·-经由夺氢(hydrogenabstraction)、亲电加成和电子转移与有机物(organic)反应。残余的toc可能是由于氯化副产物的形成,或者是由于甲酸盐和草酸盐的累积。例如,使用可商购的基于ir的阳极的最近研究发现,经由氯介导的eo的四小时人类废水处理将形成三卤甲烷和卤代乙酸。考虑到浓度通常在关于在消毒处理后的二级流出物和游泳池水报告的那些的范围内,处理过的水应当对于非饮用的再利用是安全的。
对于sbsn/coti/ir阳极系统,将cl-浓度增加至60mm显著地提高有机物质去除。这可能是由于提高的fc析出,这有效地补偿第一阳极在25ma/cm2不能生成自由基。在50ma/cm2,sbsn/coti/ir阳极产生更多的fc,伴随有足够的自由基,以实现完全的cod去除和大于50%toc去除。在这种情况下,氯和自由基介导的氧化对cod去除的贡献被计算为94%和6%(图11a-图11e)。
对于nh4+去除,sbsn/coti/ir阳极优于sbsn/coti/ir*阳极和商业阳极(图10b),因为在电化学处理期间的nh4+去除经由断点氯化(breakpointchlorination)实现,断点氯化又是cer活性的间接测量。大多数nh4+被转化成n2,其中较小的级分被氧化成no3-(图12a-图12b)。在l60和h30操作的sbsn/coti/ir阳极能够在电解四小时后去除约74%的总氮(图12a-图12b)。
在sbsn/coti/ir*阳极和sbsn/coti/ir阳极的情况下,在电化学废水处理期间(30mmcl-,25ma/cm2),总氯(tc)浓度和游离氯(fc)浓度低(<2mm)。氯在断点氯化期间或通过电双层中的废水有机物质降解被消耗,并且因此不能扩散到本体溶液相(bulksolutionphase)中。tc和fc两者的生成仅在sbsn/coti/ir阳极的情况下在完全nh4+去除和cod去除后被观察到(图13a-图13c)。在两种类型的阳极的情况下,在电解四小时(30mmcl-;25ma/cm2)后,产生多达5mmclo3-。增加电流或cl-浓度增加了clo3-产生(即10mm-11mm最大值)。这些趋势与nacl溶液中的实验一致,尽管浓度低约50%。
在低cl-和低电流条件下(即30mmcl-;25ma/cm2;图13a-图13c)在电解废水四小时后,通过sbsn/coti/ir*阳极产生显著的clo4-浓度(6mm)。然而,sbsn/coti/ir阳极在高电流条件下(即30mmcl-;50ma/cm2)形成相对低的clo4-浓度(0.85mm)。
在60mmcl-和25ma/cm2操作的sbsn/coti/ir阳极比sbsn/coti/ir*阳极消耗更少的能量用于cod去除和nh4+去除(370kwh/kgcod;383kwh/kgnh4+)。这些值仍然高于浸出液和反渗透浓缩物的eo中报告的值,这可能是由于人类废水的较低电导率(3.2ms/cm)。减小电极间距或增加废水电导率可以能够进一步降低能量消耗。
sbsn/coti/ir阳极在耐久性、反应性物质生成、污染物去除、副产物形成和能量消耗方面是显著改进的电解阳极。与非活性阳极(sbsn/coti/ir*)相比,通过活性阳极(sbsn/coti/ir*)提供的更有效的废水处理突出了非活性阳极由于通过cl-和fc的·oh猝灭对于废水处理的限制。此外,非活性阳极产生大量的clo4-。在适当的条件下,由电化学产生的fc介导的废水电解可以能够优于自由基辅助的电解。
在配备有sbsn/coti/ir阳极或sn/coti/ir阳极的适当设计的反应器中处理的废水可以适合于非饮用水再利用(例如,作为基于颜色和cod去除的循环厕所冲洗水),以及适合于消毒,消毒通过残余的fc提供。本文公开的半导体电解反应器可以被容易地自动化(例如,达到用于nh4+氯化的断点可以被用作结束批量处理(endbatchtreatment)的信号)。它们可以非常适合于用于分散化废水处理。
公开的阳极可以被用于太阳能供能厕所和废物处理系统,例如,美国公布的专利申请2014/0209479中公开的那些,该专利申请通过引用以其整体并入本文。例如,本文图1的源38可以是光伏源。并且,电解可以在人类废物上进行,例如美国公布的专利申请2014/0209479的图17c中描绘的尿的电解。
公开的阳极还可以用于氯-碱工业中。氯-碱工艺是用于nacl盐水的电解的工业工艺。其是用于产生氯和氢氧化钠(碱液(lye)/苛性钠)的技术,氯和氢氧化钠是工业所需的商品化学品。为了进行氯-碱工艺,公开的任何阳极可以被放置在用于电解nacl盐水的反应器中并且在用于电解nacl盐水的的反应器中使用,例如图1和图2示出的那些中的一个。反应器填充有合适的nacl盐水。当被放置在反应器中时,阳极接触nacl盐水。然后,足以在阳极处生成反应性氯的阳极电位通过源被施加至阳极,如图1中示出的。如图2中示出的,多个阳极和阴极可以被用于在该过程中。
前述描述是说明性的并且不是限制性的。尽管已经描述了某些示例性实施方案,但鉴于前述教导,本领域普通技术人员将容易地想到涉及本发明的其他实施方案、组合和修改。因此,本发明将仅由所附权利要求来限制,所附权利要求覆盖当结合以上说明书和附图阅读时的公开的实施方案,以及所有其他这样的实施方案和修改。
- 还没有人留言评论。精彩留言会获得点赞!