一种CVOCs净化用催化剂及其制备方法与流程
一种cvocs净化用催化剂及其制备方法
技术领域
1.本发明涉及废气处理领域,具体涉及一种cvocs净化用催化剂及其制备方法。
背景技术:
2.目前,含氯挥发性有机物(chlorinated volatile organic compounds,cvocs)是挥发性有机物(volatile organic compounds,vocs)中的一类,在医药、皮革、涂料、印刷、染料、橡胶、化工等行业具有广泛地应用。cvocs具有化学稳定性高、生物可降解性低、脂溶性强等特性。cvocs具有严重的危害性,人体长时间暴露在cvocs条件下会引起免疫水平下降、中枢神经系统功能紊乱等病症;此外,cvocs也是引起大气中臭氧和pm2.5浓度超标的重要前驱物。
3.另一方面,氯苯或氯苯酚等cvocs是一级致癌物二噁英的重要前体物,普遍存在于生活垃圾和医疗废弃物焚烧、钢铁冶炼、发热供电等行业排放的烟气中。近年来,随着我国对于生态环境保护的逐年重视以及人民群众对于美好生活的向往,加快废气处理建设特别是cvocs治理工程建设是提高我国环境空气质量的重要手段之一。这不但能实现cvocs污染物的减量化,还能减少副产物二噁英的产生,达到无害化的目的,具有极高的环境、社会和经济价值。
4.催化氧化技术是治理cvocs最为有效和经济的方法之一,具体指在催化剂的作用下通过降低反应活化能,使目标污染物在催化剂表面发生氧化还原反应,实现对污染物的定向清除。该类技术具有能耗低、效率高、二次污染少等优点。
5.如cn110038407a公开了一种含氯化氢气体和有机氯化合物气体的废气净化处理方法,包括以下步骤:将废气与空气的混合气通入石墨预热器预热,然后通入换热器的壳程与固定床反应器出来的高温气体进行换热升温,然后通入固定床反应器进行催化氧化反应,反应后的高温气体通入换热器的管程进行换热冷却,冷却后的气体通入喷雾洗涤器吸收酸性气体,然后通入气液分离器后气体直接排出。该方法采用固定床反应器催化氧化的方法,高效、低成本地处理含氯化氢气体和有机氯化合物气体的废气,通过石墨预热器预热混合气,减少了hcl气体对管路、换热器的露点腐蚀,并将催化氧化后的高温尾气与待处理的废气通过换热器进行换热,充分利用了反应热,具有较好的经济效益。
6.cn102698751a一种用于含氯易挥发有机化合物低温催化燃烧消除的催化剂,该催化剂主要由过渡金属氧化物-氧化铈复合氧化物载体及其负载的氧化钌构成,其中过渡金属元素为ti、mn、co、fe、cu、ni。采用作为氧化剂的空气带入反应器,使含氯易挥发有机化合物转化为二氧化碳、氯化氢和氯气,完全燃烧尾气可以采用稀碱溶液吸收后放空。该催化剂的催化活性高,抗氯中毒能力强,催化剂寿命长,特别适用于低温催化燃烧消除含氯有机化合物。
7.但目前在治理cvocs时,该技术存在着以下挑战:一是由于催化剂性能较弱导致净化高浓度cvocs时cvocs转化率低、co2生成率不高,使处理后的尾气难以满足污染物排放标准;二是催化剂在高水汽含量下难以实现对cvocs的定向吸附、催化和清除,迫使净化效率
难以满足工业需求。
技术实现要素:
8.鉴于现有技术中存在的问题,本发明的目的在于提供一种cvocs净化用催化剂及其制备方法,解决了目前催化剂在高水汽含量下对有机含氯气体处理效果差,净化效率不达标的问题。
9.为达此目的,本发明采用以下技术方案:
10.第一方面,本发明提供了一种cvocs净化用催化剂的制备方法,所述制备方法包括如下步骤:将含硅助剂、钌聚乙烯咪唑配合物和催化助剂进行混合搅拌,之后经固液分离及第一焙烧得到所述催化剂;
11.所述含硅助剂为将甲基三乙氧基硅烷、正硅酸乙酯和醇在水相中进行第一搅拌反应,之后依次加入酸和钛酸正丁酯进行第二搅拌反应,之后经固液分离和第二焙烧得到。
12.本发明提供的制备方法,通过采用特定的制备过程,实现了高抗水性催化剂的制备,能持续、高效地吸附、催化目标化合物,防止催化剂失活失效;同时,本发明所提供的的制备方法还能抑制贵金属离子在高温焙烧过程中迁移、聚集和长大。
13.本发明中,所述高水汽含量为有机含氯气体中水汽的含量≥5%,优选为5-10%。
14.本发明中,所述固液分离中具体可以使依次进行的陈化和烘干等操作。
15.本发明中,所述酸可以是盐酸、硝酸或硫酸等。
16.作为本发明优选的技术方案,所述混合搅拌中所述含硅助剂和所述钌聚乙烯咪唑配合物中的钌前驱体质量比为(50-200):1,例如可以是50:1、60:1、70:1、80:1、90:1、100:1、110:1、120:1、130:1、140:1、150:1、160:1、170:1、180:1、190:1或200:1等,但不限于所列举的数值,该范围内其他未列举的数值也符合要求。
17.优选地,所述混合搅拌中所述催化助剂和所述钌聚乙烯咪唑配合物中的钌前驱体质量比为(3-8):1,例如可以是3:1、3.2:1、3.4:1、3.6:1、3.8:1、4:1、4.2:1、4.4:1、4.6:1、4.8:1、5:1、5.2:1、5.4:1、5.6:1、5.8:1、6:1、6.2:1、6.4:1、6.6:1、6.8:1、7:1、7.2:1、7.4:1、7.6:1、7.8:1或8:1,但不限于所列举的数值,该范围内其他未列举的数值也符合要求。
18.优选地,所述催化助剂包括氧化铜、四氧化三钴、二氧化铈或氧化锰中的1种或至少2种的组合,如可以是氧化铜和四氧化三钴的组合,二氧化铈和氧化锰的组合或氧化铜和二氧化铈的组合等。
19.优选地,所述混合搅拌的时间为1-3h,例如可以是1h、1.1h、1.2h、1.3h、1.4h、1.5h、1.6h、1.8h、1.9h、2h、2.1h、2.2h、2.3h、2.4h、2.5h、2.6h、2.7h、2.8h、2.9h或3h等,但不限于所列举的数值,该范围内其他未列举的数值也符合要求。
20.作为本发明优选的技术方案,所述第一焙烧的温度300-500℃,例如可以是300℃、310℃、320℃、330℃、340℃、350℃、360℃、370℃、380℃、390℃、400℃、410℃、420℃、430℃、440℃、450℃、460℃、470℃、480℃、490℃或500℃等,但不限于所列举的数值,该范围内其他未列举的数值也符合要求。
21.优选地,所述第一焙烧的时间为2-5h,例如可以是2h、2.1h、2.2h、2.3h、2.4h、2.5h、2.6h、2.7h、2.8h、2.9h、3h、3.1h、3.2h、3.3h、3.4h、3.5h、3.6h、3.7h、3.8h、3.9h、4h、4.1h、4.2h、4.3h、4.4h、4.5h、4.6h、4.7h、4.8h、4.9h或5h等,但不限于所列举的数值,该范
围内其他未列举的数值也符合要求。
22.作为本发明优选的技术方案,所述含硅助剂的制备过程中甲基三乙氧基硅烷和正硅酸乙酯的质量比为(3-6):1,例如可以是3:1、3.1:1、3.2:1、3.3:1、3.4:1、3.5:1、3.6:1、3.7:1、3.8:1、3.9:1、4:1、4.1:1、4.2:1、4.3:1、4.4:1、4.5:1、4.6:1、4.7:1、4.8:1、4.9:1、5:1、5.1:1、5.2:1、5.3:1、5.4:1、5.5:1、5.6:1、5.7:1、5.8:1、5.9:1或6:1等,但不限于所列举的数值,该范围内其他未列举的数值也符合要求。
23.优选地,所述含硅助剂的制备过程中醇与甲基三甲氧基的质量比为(5-10):1,例如可以是5:1、5.5:1、6:1、6.5:1、7:1、7.5:1、8:1、8.5:1、9:1、9.5:1或10:1等,但不限于所列举的数值,该范围内其他未列举的数值也符合要求。
24.优选地,所述含硅助剂的制备过程中醇和水的质量比为(2-5):1,例如可以是2:1、2.2:1、2.4:1、2.6:1、2.8:1、3:1、3.2:1、3.4:1、3.6:1、3.8:1、4:1、4.2:1、4.4:1、4.6:1、4.8:1或5:1等,但不限于所列举的数值,该范围内其他未列举的数值也符合要求。
25.优选地,所述含硅助剂的制备过程中酸的添加量为所述水添加量的0.5-2%,例如可以是0.5%、0.6%、0.7%、0.8%、0.9%、1%、1.1%、1.2%、1.3%、1.4%、1.5%、1.6%、1.7%、1.8%、1.9%或2%等,但不限于所列举的数值,该范围内其他未列举的数值也符合要求。
26.本发明中,所述酸可以是有机酸或无机酸,如可以是乙酸,硫酸,盐酸或硝酸等。
27.优选地,所述含硅助剂的制备过程中酸的摩尔浓度为9-12mol/l,例如可以是9mol/l、9.2mol/l、9.4mol/l、9.6mol/l、9.8mol/l、10mol/l、10.2mol/l、10.4mol/l、10.6mol/l、10.8mol/l、11mol/l、11.2mol/l、11.4mol/l、11.6mol/l、11.8mol/l或12mol/l等,但不限于所列举的数值,该范围内其他未列举的数值也符合要求。
28.优选地,所述含硅助剂的制备过程中钛酸正丁酯与甲基三乙氧基硅烷的质量比为(1-5):1,例如可以是1:1、1.2:1、1.4:1、1.6:1、1.8:1、2:1、2.2:1、2.4:1、2.6:1、2.8:1、3:1、3.2:1、3.4:1、3.6:1、3.8:1、4:1、4.2:1、4.4:1、4.6:1、4.8:1或5:1等,但不限于所列举的数值,该范围内其他未列举的数值也符合要求。
29.作为本发明优选的技术方案,所述含硅助剂的制备过程中第一搅拌反应的时间为0.5-1h,例如可以是0.5h、0.55h、06.h、0.65h、0.7h、0.75h、0.8h、0.85h、0.9h、0.95h或1h等,但不限于所列举的数值,该范围内其他未列举的数值也符合要求。
30.优选地,所述含硅助剂的制备过程中第二搅拌反应的时间为0.5-1h,例如可以是0.5h、0.55h、06.h、0.65h、0.7h、0.75h、0.8h、0.85h、0.9h、0.95h或1h等,但不限于所列举的数值,该范围内其他未列举的数值也符合要求。
31.作为本发明优选的技术方案,所述含硅助剂的制备过程中第二焙烧的温度为300-600℃,例如可以是300℃、320℃、340℃、360℃、380℃、400℃、420℃、440℃、460℃、480℃、500℃、520℃、540℃、560℃、580℃或600℃等,但不限于所列举的数值,该范围内其他未列举的数值也符合范围。
32.优选地,所述含硅助剂的制备过程中第二焙烧的时间为2-6h,例如可以是2h、2.2h、2.4h、2.6h、2.8h、3h、3.2h、3.4h、3.6h、3.8h、4h、4.2h、4.4h、4.6h、4.8h、5h、5.2h、5.4h、5.6h、5.8h或6h等,但不限于所列举的数值,该范围内其他未列举的数值也符合要求。
33.作为本发明优选的技术方案,所述钌聚乙烯咪唑配合物为将聚乙烯咪唑、钌前驱
体和液相进行第三搅拌反应得到。
34.本发明中,所述聚乙烯咪唑可参考文献:jose-maria,sansinena,jerzy,et al.effects of axial coordination of the metal center on the activity of iron tetraphenylporphyrin as a nonprecious catalyst for oxygen reduction[j].journal of physical chemistry c,2014,118(33),19139-19149的制备方法进行制备。
[0035]
优选地,所述钌聚乙烯咪唑配合物的制备过程中聚乙烯咪唑和钌前驱体的质量比为(5-20):1,例如可以是5:1、5.5:1、6:1、6.5:1、7:1、7.5:1、8:1、8.5:1、9:1、9.5:1、10:1、10.5:1、11:1、11.5:1、12:1、12.5:1、13:1、13.5:1、14:1、14.5:1、15:1、15.5:1、16:1、16.5:1、17:1、17.5:1、18:1、18.5:1、19:1、19.5:1或20:1等,但不限于所列举的数值,该范围内其他未列举的数值也符合要求。
[0036]
优选地,所述钌聚乙烯咪唑配合物的制备过程中液相包括质量比为(1-3):1的水和醇,例如可以是1:1、1.1:1、1.2:1、1.3:1、1.4:1、1.5:1、1.6:1、1.7:1、1.8:1、1.9:1、2:1、2.1:1、2.2:1、2.3:1、2.4:1、2.5:1、2.6:1、2.7:1、2.8:1、2.9:1或3:1等,但不限于所列举的数值,该范围内其他未列举的数值也符合要求。
[0037]
本发明中,所述醇可以使甲醇、乙醇、丙三醇或乙二醇等。
[0038]
优选地,所述钌聚乙烯咪唑配合物的制备过程中醇与聚乙烯咪唑的质量比为(5-10):1,例如可以是5:1、5.2:1、5.4:1、5.6:1、5.8:1、6:1、6.2:1、6.4:1、6.6:1、6.8:1、7:1、7.2:1、7.4:1、7.6:1、7.8:1、8:1、8.2:1、8.4:1、8.6:1、8.8:1、9:1、9.2:1、9.4:1、9.6:1、9.8:1或10:1等,但不限于所列举的数值,该范围内其他未列举的数值也符合要求。
[0039]
优选地,所述钌聚乙烯咪唑配合物的制备过程中钌前驱体包括氯化钌、亚硝酰基硝酸合钌或三氯化六铵合钌中的1种或至少2种的组合。
[0040]
优选地,所述钌聚乙烯咪唑配合物的制备过程中第三搅拌反应的时间为0.5-2h,例如可以是0.5h、0.6h、0.7h、0.8h、0.9h、1h、1.1h、1.2h、1.3h、1.4h、1.5h、1.6h、1.7h、1.8h、1.9h或2h等,但不限于所列举的数值,该范围内其他未列举的数值也符合要求。
[0041]
作为本发明优选的技术方案,所述制备方法还包括将所述催化剂、溶胶、涂覆助剂及水进行混合并球磨,得到涂覆料,之后将所述涂覆料涂覆至载体表面,然后经干燥和第三焙烧得到整体式催化剂。
[0042]
优选地,所述溶胶包括纳米硅溶胶。
[0043]
优选地,所述涂覆助剂包括十六烷基三甲基溴化铵、聚乙二醇、十二烷基硫酸钠中的1种或至少2种的组合。
[0044]
本发明中,所述聚乙二醇的分子量≤400。
[0045]
优选地,所述催化剂与溶胶的质量比为(9.5-10.5):1,例如可以是9.5:1、9.55:1、9.6:1、9.65:1、9.7:1、9.75:1、9.8:1、9.85:1、9.9:1、9.95:1、10:1、10.05:1、10.1:1、10.15:1、10.2:1、10.25:1、10.3:1、10.35:1、10.4:1、10.45:1或10.5:1等,但不限于所列举的数值,该范围内其他未列举的数值也符合要求。
[0046]
优选地,所述催化剂与涂覆助剂的质量比为(5-10):1,例如可以是5:1、5.2:1、5.4:1、5.6:1、5.8:1、6:1、6.2:1、6.4:1、6.6:1、6.8:1、7:1、7.2:1、7.4:1、7.6:1、7.8:1、8:1、8.2:1、8.4:1、8.6:1、8.8:1、9:1、9.2:1、9.4:1、9.6:1、9.8:1或10:1等,但不限于所列举的数值,该范围内其他未列举的数值也符合要求。
2%;所述酸的质量浓度为9-12mol/l;所述钛酸正丁酯与甲基三乙氧基硅烷的质量比为(1-5):1;所述第一搅拌反应的时间为0.5-1h;所述第二搅拌反应的时间为0.5-1h;所述第二焙烧的温度为300-600℃,时间为2-6h;
[0056]
所述钌聚乙烯咪唑配合物为将聚乙烯咪唑、钌前驱体和液相进行第三搅拌反应得到;所述聚乙烯咪唑和钌前驱体的质量比为(5-20):1;所述液相包括质量比为(1-3):1的水和醇;所述醇与聚乙烯咪唑的质量比为(5-10):1;所述钌前驱体包括氯化钌、亚硝酰基硝酸合钌或三氯化六铵合钌中的1种或至少2种的组合;所述第三搅拌反应的时间为0.5-2h。
[0057]
第二方面,本发明提供了一种cvocs净化用催化剂,所述催化剂采用如第一方面所述制备方法得到,用于处理水汽含量≥5%的有机含氯气体。
[0058]
与现有技术方案相比,本发明具有以下有益效果:
[0059]
(1)本发明提供的制备方法中,引入聚乙烯咪唑能有效抑制贵金属组分在高温焙烧过程中发生的迁移、聚集、长大,确保焙烧后的钌组分以微颗粒形式均匀分布在催化剂表面,保证在单位体积内拥有足够数量的催化活性位点,从而确保催化剂具有较高的催化效率和稳定性。
[0060]
(2)本发明提供的制备方法,通过引入硅钛物相并和聚乙烯咪唑相配使得催化剂具有高抗水性,防止出现水中毒,促使催化剂表面能持续、高效地吸附、催化目标化合物。
[0061]
(3)本发明提供的整体式催化剂能有效针对高浓度cvocs废气难处理等问题,实现对目标污染物的定向清除,并保证较高的cvocs转化率和co2生成率,cvocs转化率≥98%,co2生成率≥98%。
具体实施方式
[0062]
为更好地说明本发明,便于理解本发明的技术方案,本发明的典型但非限制性的实施例如下:
[0063]
实施例1
[0064]
本实施例提供了一种cvocs净化用催化剂的制备方法,所述制备方法包括如下步骤:将含硅助剂、钌聚乙烯咪唑配合物和催化助剂进行混合搅拌,之后经固液分离及第一焙烧得到所述催化剂,将所述催化剂、溶胶、涂覆助剂及水进行混合并球磨,得到涂覆料,之后将所述涂覆料涂覆至载体表面,然后经干燥和第三焙烧得到整体式催化剂;
[0065]
具体采用:(1)将30g甲基三乙氧基硅烷和10g正硅酸乙酯溶解在乙醇(180g),然后加入50g水并充分混合,缓慢滴加0.5g盐酸(12mol/l),搅拌0.5h,加入60g钛酸正丁酯,缓慢搅拌0.5h,之后陈化12h,收集产物,置于烘箱在120℃下干燥5h,最后置于马弗炉中在450℃下焙烧3h,制得含硅助剂。
[0066]
(2)将10g聚乙烯咪唑和1g三氯化钌充分溶解于100g乙醇中,搅拌30分钟,然后再加入200g去离子水,继续搅拌1小时,制得钌-聚乙烯咪唑配合物溶液。
[0067]
(3)将200g含硅助剂加入至所得步骤(2)钌-聚乙烯咪唑配合物溶液,并加入4g二氧化铈,搅拌2小时,之后在150℃下干燥5小时,在450℃下焙烧3小时,制得粉末催化剂。
[0068]
(4)将20g粉末催化剂加入至100g去离子水中,并加入2g纳米硅溶胶、1g十二烷基硫酸钠、1g聚乙二醇(分子量为300),球磨2小时,制成浆液;随后将蜂窝状堇青石载体浸没至浆液中,取出用洗耳球吹尽孔道中残留的浆液,在120℃下干燥4小时,随后在450℃下焙
烧3小时,得到能净化cvocs的整体式催化剂。
[0069]
实施例2
[0070]
本实施例提供了一种cvocs净化用催化剂的制备方法,所述制备方法包括如下步骤:将含硅助剂、钌聚乙烯咪唑配合物和催化助剂进行混合搅拌,之后经固液分离及第一焙烧得到所述催化剂,将所述催化剂、溶胶、涂覆助剂及水进行混合并球磨,得到涂覆料,之后将所述涂覆料涂覆至载体表面,然后经干燥和第三焙烧得到整体式催化剂;
[0071]
具体采用:(1)将40g甲基三乙氧基硅烷和10g正硅酸乙酯溶解在乙醇(300g),然后加入80g水并充分混合,缓慢滴加0.8g盐酸(12mol/l),搅拌0.5h,加入100g钛酸正丁酯,缓慢搅拌0.5h,之后陈化12h,收集产物,置于烘箱在120℃下干燥5h,最后置于马弗炉中在450℃下焙烧3h,制得含硅助剂。
[0072]
(2)将10g聚乙烯咪唑和1g三氯化六铵合钌充分溶解于100g乙醇中,搅拌30分钟,然后再加入100g去离子水,继续搅拌1小时,制得钌-聚乙烯咪唑配合物溶液。
[0073]
(3)将100g含硅助剂加入至步骤(2)所得钌-聚乙烯咪唑配合物溶液中,并加入4g氧化铜,搅拌2小时,之后在150℃下干燥5小时,在450℃下焙烧3小时,制得粉末催化剂。
[0074]
(4)将20g粉末催化剂加入至100g水中,并加入2g纳米硅溶胶、2g十二烷基硫酸钠,球磨2小时,制成浆液;随后将蜂窝状堇青石载体浸没至浆液中,取出用洗耳球吹尽孔道中残留的浆液,在120℃下干燥4小时,随后在450℃下焙烧3小时,得到能净化cvocs的整体式催化剂。
[0075]
实施例3
[0076]
本实施例提供了一种cvocs净化用催化剂的制备方法,所述制备方法包括如下步骤:将含硅助剂、钌聚乙烯咪唑配合物和催化助剂进行混合搅拌,之后经固液分离及第一焙烧得到所述催化剂,将所述催化剂、溶胶、涂覆助剂及水进行混合并球磨,得到涂覆料,之后将所述涂覆料涂覆至载体表面,然后经干燥和第三焙烧得到整体式催化剂;
[0077]
具体采用:(1)将50g甲基三乙氧基硅烷和15g正硅酸乙酯溶解在乙醇(300g),然后加入80g水并充分混合,缓慢滴加1g盐酸(12mol/l),搅拌0.5h,加入120g钛酸正丁酯,缓慢搅拌0.5h,之后陈化12h,收集产物,置于烘箱在120℃下干燥5h,最后置于马弗炉中在450℃下焙烧3h,制得含硅助剂。
[0078]
(2)将10g聚乙烯咪唑和2g亚硝酰基硝酸合钌充分溶解于100g乙醇中,搅拌30分钟,然后再加入100g去离子水,继续搅拌1小时,制得钌-聚乙烯咪唑配合物溶液。
[0079]
(3)将100g含硅助剂加入至步骤(2)所得钌-聚乙烯咪唑配合物溶液中,并加入8g氧化锰,搅拌2小时,之后在150℃下干燥5小时,在480℃下焙烧2小时,制得粉末催化剂。
[0080]
(4)将20g粉末催化剂加入至100g去离子水中,并加入2g纳米硅溶胶、2g聚乙二醇(分子量为400),球磨2小时,制成浆液;随后将蜂窝状莫来石载体浸没至浆液中,取出用洗耳球吹尽孔道中残留的浆液,在100℃下干燥5小时,随后在500℃下焙烧1小时,得到能净化cvocs的整体式催化剂。
[0081]
实施例4
[0082]
于实施例1的区别仅在于所述钌聚乙烯咪唑配合物的制备过程在一氧化碳气氛下进行。
[0083]
实施例5
[0084]
与实施例1的区别仅在于混合搅拌中不添加所述钌聚乙烯咪唑配合物。
[0085]
实施例6
[0086]
与实施例1的区别仅在于将钌前驱体替换为羰基钌。
[0087]
实施例7
[0088]
与实施例1的区别仅在于不添加钛酸正丁酯。
[0089]
实施例8
[0090]
与实施例1的区别仅在于混合搅拌中不添加含硅助剂。
[0091]
实施例9
[0092]
与实施例1的区别仅在于钌聚乙烯咪唑配合物制备过程中添加与钌前驱体等量的硝酸钴。
[0093]
应用例1
[0094]
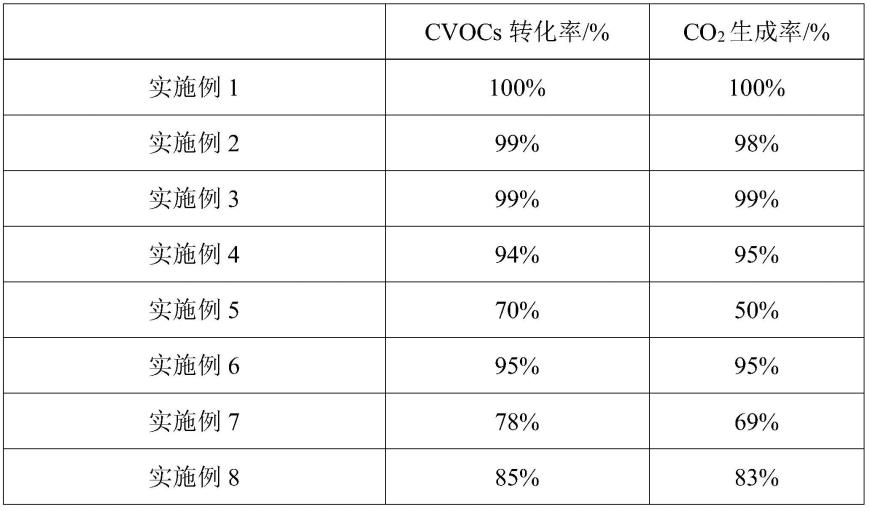
将上述实施例1-9所得催化剂及依据cn111135816a中实施例a2和实施例b2所得催化剂对汽水含量为7%的二氯甲烷和含汽水含量为7%的氯苯进行燃烧处理,处理结果详见表1和表2,其中,表1为二氯甲烷的处理结果,表2为氯苯的处理结果。
[0095]
上述实施例中所用聚乙烯咪唑采用参考文献:jose-maria,sansinena,jerzy,et al.effects of axial coordination of the metal center on the activity of iron tetraphenylporphyrin as a nonprecious catalyst for oxygen reduction[j].journal of physical chemistry c,2014,118(33),19139-19149的制备方法进行制备。
[0096]
表1
[0097] cvocs转化率/%co2生成率/%实施例199%99%实施例2100%99%实施例3100%100%实施例493%92%实施例564%42%实施例694%93%实施例771%61%实施例880%78%实施例984%65%cn111135816a实施例a265%63%cn111135816a实施例b269%71%
[0098]
表2
[0099][0100][0101]
上表中,cvocs转化率的计算方式为(给入气体中含氯有机气体的含量-燃烧后气体中含氯有机气体的含量)/给入气体中含氯有机气体的含量,co2生成率的计算方式为燃烧后气体中二氧化碳含量/给入气体中含氯有机气体的含量。具体如本发明中应用例具体为针对含水汽的二氯甲烷进行燃烧处理,则cvocs转化率的计算方式为(给入气体中二氯甲烷的含量-燃烧后气体中二氯甲烷的含量)/给入气体中二氯甲烷的含量,co2生成率的计算方式为燃烧后气体中二氧化碳含量/给入气体中二氯甲烷的含量。当针对含水汽的氯苯进行燃烧处理,则cvocs转化率的计算方式为(给入气体中氯苯的含量-燃烧后气体中氯苯的含量)/给入气体中氯苯的含量,co2生成率的计算方式为燃烧后气体中二氧化碳含量*6/给入气体中氯苯的含量。
[0102]
通过上述结果可知,本发明提供的制备方法,通过采用特定的制备过程,实现了高抗水性催化剂的制备,能持续、高效地吸附、催化目标化合物,防止催化剂失活失效;同时,本发明所提供的的制备方法还能抑制贵金属离子在高温焙烧过程中迁移、聚集和长大。
[0103]
声明,本发明通过上述实施例来说明本发明的详细结构特征,但本发明并不局限于上述详细结构特征,即不意味着本发明必须依赖上述详细结构特征才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明所选用部件的等效替换以及辅助部件的增加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
[0104]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,这些简单变型均属于本发明的保护范围。
[0105]
另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛
盾的情况下,可以通过任何合适的方式进行组合,为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。
[0106]
此外,本发明的各种不同的实施方式之间也可以进行任意组合,只要其不违背本发明的思想,其同样应当视为本发明所公开的内容。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1