一种生物质热解液制备高质燃油和/或化工原料的方法与流程
本发明涉及再生能源和生物质能源领域,尤其涉及一种生物质热解液制备高质燃油和/或化工原料的方法。
背景技术:
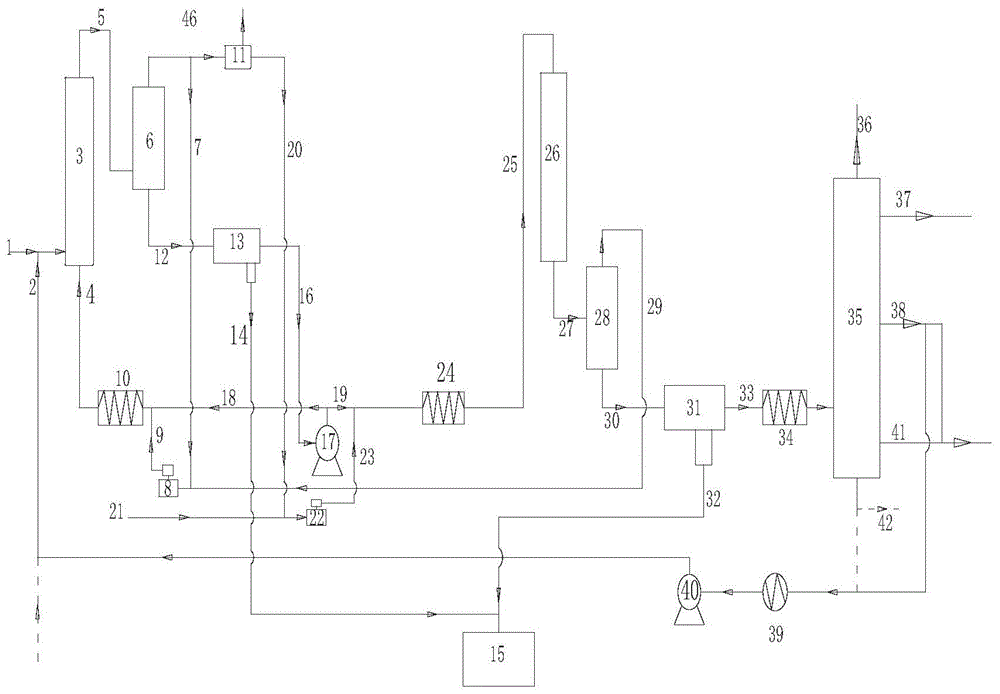
:能源是人类生存的基础物质,也是社会发展的标志之一。社会的发展对能源的需求不断增加,大量使用化石能源造成的化石能源危机和日益严峻的环境问题,迫使人类必须调整现有的能源结构。非常有必要寻找一种原料并将其转化为燃料和化工原料来替代现有化石能源,尤其是液态化石能源。生物质(广义上讲)是指利用大气、水、土地等通过光合作用而产生的各种有机体。生物质在生长过程中吸收水和二氧化碳将太阳能转化为生物质能并加以储存。生物质是一种可再生、可持续利用的一次能源,其潜力巨大又未被完全开发利用,而且生物质是唯一可以液态燃料的再生能源。合理利用大自然馈赠的生物质,在生产、消费、再加工过程保持碳的平衡,有助于改善大气环境、实现节能减排和调整能源结构。生物质转化为液态能源的方式主要包括:①热化学转化技术,如气化合成、干馏、热解液化(包括含催化剂和不含催化剂工艺)、加氢液化等;②生物转化技术,如通过发酵工艺制备各种醇类(甲醇、乙醇、丁醇等);③化学转化技术,如通过水解制备多元醇(可进一步制备烃类燃料)、油脂甲醇酯化反应制备生物柴油等。生物质热解液化技术具有反应速度快、收率高、操作简单、投资少等特点而得到迅速发展并被工业化。迄今为止,最大产能达到每年5万吨液体产品的装置已在加拿大建成。生物质热解液化技术从加热速度和停留时间的不同分为慢速热解、快速热解、闪速热解;从热解设备类型上分为旋转锥、固定床、移动床、流化床、重力下落式、真空热解等形式。另外,在生物质的炭化和气化过程中也会伴生一定的液体产物。以上方式得到的液体产物统称为生物质热解液。但生物质热解得到的液体产品(以下简称生物质热解液)不能够直接供应运输设备使用或直接作为化工原料使用,还需进一步转化处理制备成高质燃油(汽油、柴油和航空燃油混合物)和化工原料才能替代同类的石油产品。生物质热解液与石油及石油产品的性质有极大的区别,很多的文献资料都有详尽的描述(如avbridgewater,hhofbauer和svanloo,thermalbiomassconversion,cplpress,2009,37-78),归纳来看生物质热解液特点如下:含氧量较高(氧含量30-55%),高水含量和含有一定量的固体粉末,含醇类、醚类、酸类、醛类、酮类、脂类和酚类等有机物,极性强而不与石油(包括石油产品)互溶。较高的水分和大量容易发生缩合反应的带有酚基、羟基、羧基、羰基、醛基等官能团的物质,使得生物质热解液在加工处理过程中出现催化剂失活较快和极易结焦等现象,给生物质热解液深度加工制备高附加值燃油和化工产品带来了技术困难和较高的成本。venderbosch等stabilizationofbiomass-derivedpyrolysisoils,j.chem.technol.biotechnol.2010,85:674–686得出了“在生物油的加氢加工中,如果不存在h2和催化剂,则遵循热解油被进一步聚合的路径,最终成为焦化组分”。同时,生物质热解液中含有的木炭对深加工制备燃油工艺带来了困难,同时由于生物质热解液的粘度较高(常温下20-100cp),通过传统的过滤方式很难去除,ajavaid等的removalofcharparticlesfromfastpyrolysisbio-oilbymicrofiltration《journalofmembranescience》2010,363(1):120-127对此作了分析与说明。所以,很有必要发明一种具有经济性和工程适应性的方法来将生物质热解液制备成高品质的燃油和化工产品。中国专利申请cn201210153017.1首先对生物质热解油进行分级处理。得到的轻质组分进行初步氢化和深度氢化处理,以减少重整或催化缩合过程中的积碳。得到的重质组分以在贵金属铑铼催化剂作用下经过催化裂解处理制备富含酚类化合物的溶液。该方法需要进行预处理得到不同的组分并分项进行转化,增加了工艺的复杂性、投资成本和运行费用,而且使用了贵金属催化剂会增加催化剂的回收难度和成本。专利申请us20080053870a1、us005959167a、us7578927也是将生物质热解油分离水溶性物质和非水溶性物质,是将非水溶性物质作为反应原料,最终产率也很低。中国专利申请cn201210413417.1先将热解油直接加氢脱氧得到部分脱氧热解油,再与预热的来自于石油的烃类产物混合并雾化后进入提升管式催化裂化反应器制备包含一种或多种裂化产物。该方法直接将热解油加氢脱氧,仍无法避免加热过程中热解油结焦问题,同时热解油本身所含有的固体粉末(如中国专利申请cn201380024393.3专门提及)和加氢脱氧过程中产生的焦粉会对后续的雾化器造成严重的磨损,同时雾化耗能较高。另外,雾化后的物料经过催化裂化并不能将氧完全脱出,得到的产物不能直接作为运输用燃油使用,且在催化裂化过程中氧主要是通过发生缩合反应脱水而去除,由于热解液中氢相对于氧和碳比例偏低,生成的焦粉含量较高,大大降低产品的收率。专利申请cn20128002116.3和专利申请cn201410488593.0是将热解油直接加氢处理,由于没有其它辅助物质来稀释和减缓结焦,催化剂失活较快;专利申请us20090113787a1在此基础上进一步使用了转化活性较高的贵金属钯(pd)作为催化剂,仍发生聚合反应而使催化剂快速失去活性。中国专利申请cn201180023021.x是通过加氢处理将生物质热裂解油转化为更加稳定的产品,并把该产品与高硫含量的瓦斯油、重燃料油或渣油混合作为苛刻性相对低的环境的燃料。该方法只是将生物质热裂解油部分脱氧,部分脱氧达到实现与传统燃料油参混的目的,不能生产车用高质燃油。中国专利申请cn201510007926.8为了解决生物质热解油的结焦难题,通过生物质热解油焦化,再将焦化得到液体油品加氢处理。专利申请cn201210413417.1先经过加氢处理得到部分脱氧油,再进行焦化。这两种方法都是期望通过焦化的方式来克服催化剂的失活问题,但由于生物质热解油在焦化过程中的聚合反应非常严重,形成大量的焦粉,最终油品收率很低。专利申请us4795841提出了一种用于生物质热解液两步加氢处理的方法,该生物质热解液在较低的温度下通过催化加氢预处理得到相对稳定的油品,然后再经过进一步加氢处理。但是该方法在较低温度下的脱氧量低,催化剂的快速失活。在预处理阶段所需相对较低的温度下反应速度缓慢,反映时空速率很低。此外,氢消耗高。专利申请us20090294324通过两段加氢的方法将生物质热解油转化成液体燃料,并且将最终烃产物中的重组分物质再循环回第一级反应,类似的专利申请cn201510224150.5将生物质热解油与减压蜡油混合加氢处理,但是这些方法都没有解决热解液与烃类的互溶性问题。即使专利申请cn200980120914.9(wo2009/126508)选择性的优化方案可先将热解油分相成水相和木质素相再混合后进料,也没有解决导致其反应速率低和催化剂寿命短的问题。专利申请us20110119994通过将生物质热解油加氢处理形成部分脱氧油,进一步与矿物类石油混合加氢处理。该方法在第一步加氢处理阶段没有添加采取任何减缓生物热解油聚合结焦的措施,其生成的部分脱氧油出现了上中下三层的分相想象,下层的脱氧油品还出现了焦粉等颗粒物,不仅给下游加工处理带来困难,同时也说明该方法没有解决生物热解油聚合结焦和催化剂失活问题。中国专利申请cn201280061563.0(wo2013/089839)通过将经过生物油加氢处理后形成的部分脱氧物质循环并加热后与生物油混合共同进入反应器的方式加氢处理。该方法虽然对生物油起到了一定的稀释作用,但部分脱氧油的溶氢能力不足以满足生物油的加氢所需,依然没有解决催化剂失活快的难题。另外该方法中要求加热后的部分脱氧油与生物油混合后需在较短时间与反应器中的催化剂接触并反应,这对增加管路和反应器内构件提出了很严格的要求,带来结焦的风险。中国专利申请cn201180054737.6(wo2012/035410)通过分散剂将生物油分散在烃液体中,分散后的混合物在催化剂存在的条件下加氢重整处理生成有机相物质,进一步加氢裂化处理得到烃混合物。该方法须借助于搅拌器和泵将分散剂、生物油和烃类物质先进行混合形成分散体后进行反应,该方法增加了操作的复杂性。该方法没有解决实际运行过程中反应物料预加热后的结焦问题(比如专利申请cn201280061563.0(wo2013/089839)方法中描述了形成混合相的物质加热后要在小于60秒(最好是小于10秒)的时间内与催化剂接触并发生反应),如果形成分散体后再加热,势必会出现生物油在加热炉和加热后的管道中发生聚合而结焦的现象,降低催化剂的寿命和运行周期。专利申请cn201280061563.0(wo2013/089839)方法中也是将裂解油与加热的加氢脱氧裂解油混合后,在较短时间内进入反应器,也依然没有解决裂解油预加热后的结焦问题,导致得到的低氧生物质衍生裂解油的氧含量在5-20%,会影响下游的第二加氢处理过程的长周期运行。类似的专利申请cn201380024393.3(wo2013/135986a1)将生物油与生物油经过加氢重整形成的有机相混合后经过加氢重整反应,再进一步加氢处理得到烃类物质。但是该方法循环的加氢重整有机相只是对生物油进行稀释和溶解一定量的氢气,加氢脱氧效果很低,得到的加氢重整有机相中的氧含量和水分含量较高,通过密度差异的方式分离加氢重整流出物中的有机相和水相困难,尤其是在有机相中还含有大量含氧官能团物质的情况下,高水分和高氧含量有机相物质加速下一步加氢裂化催化剂的失活。按照传统的加氢处理方法,生物质热解液聚合反应比加氢处理反应快得多,最终聚合形成焦炭导致堵塞管道和设备以及催化剂失活。虽然出现了很多的生物质热解液处理方案,但仍没有发现一种有效的加氢处理方法通过加氢脱氧速度远远大于聚合速度的方式来克服生物质热解液体快速热聚合而导致催化剂快速失活问题和结焦问题。技术实现要素:为了解决现有技术存在的由生物质热解液快速热聚合而导致的催化剂快速失活问题和结焦问题,本发明提高了一种生物质热解液制备高质燃油和/或化工原料的方法,该方法可以有效解决现有生物质热解液处理过程中存在的问题,通过加氢的方式将生物质热解液转化为高质燃油和化工原料。本发明提供的生物质热解液制备高质燃油和/或化工原料的方法,包括如下步骤:a)使生物质热解液在沸腾床反应器内的催化剂全混流循环体系中进行加氢脱氧反应得到脱氧油;b)来自步骤a)的脱氧油在固定床反应器内进行加氢裂化反应得到高质燃油和/或化工原料。所述催化剂全混流循环是指催化剂颗粒的宏观运动表现为由反应器底部流化至反应器料面位置,而后又返回至反应器底部的运动形式。所述催化剂为球形催化剂。所述催化剂全混流循环体系是在生物质热解液、供氢剂、循环油、氢气、催化剂、加氢脱氧产物和内构件的共同作用下形成的;具体地说,催化剂全混流循环体系是通过循环油和氢气提供流化动能促使催化剂处于流化状态、循环油与氢气混合物以及流化催化剂的高速扰动促使生物质热解液与供氢剂快速混合与稀释、以及沸腾床反应器内构件对气液固三相混合物的导向、分流及旋流这三方面共同作用形成的。所述生物质热解液在进入沸腾床反应器前先与供氢剂混合,在供氢剂保护下、于室温~80℃优选室温~50℃下进入反应器。生物质热解液包括由各种生物质经过慢速热解、快速热解、闪速热解、炭化或气化过程得到的液态物质。所述供氢剂为加氢裂化得到的烃类物质、石油类烃类物质、煤焦油加氢处理得到的烃类物质和有机质加氢脱氧得到的烃类物质中的至少一种,所述烃类物质沸点范围为160~260℃。所述催化剂为第viii族金属单独或第viii族金属添加第ivb、第vb、第vib、第viib、第ib和第iib族金属中的一种或两种作为活性组分担载于活性炭或多孔性的炭或表面进行了碳化的金属氧化物形成的催化剂。所述催化剂由沸腾床反应器底部排出后,经醇类物质、烃类物质或四氢呋喃溶剂洗涤进行再生,恢复活性。所述加氢脱氧反应的温度为200℃~400℃,压力为10~20mpa,反应体积空速为0.6~2.0h-1,氢油比为400:1~1000:1,循环比为1:4~4:1,供氢剂与生物质热解液的质量比为0.2:1~4:1;加氢裂化反应温度为150℃~420℃,压力为12~20mpa,反应体积空速为1.0~4.0h-1,氢油比为400:1~1200:1。需要说明的是,本发明中提到的氢油比均指体积比,循环比均指质量比。所述脱氧油可以直接进行加氢裂化处理,也可以与重质柴油、蜡油和煤焦油中的至少一种掺混后进入固定床反应器进行加氢裂化处理,经加氢裂化处理后均得到高质燃油和/或化工原料。所述沸腾床反应器可以采用单级形式,也可以采用两级或多级串联形式;固定床反应器可以采用单级形式,也可以采用两级或多级串联形式;沸腾床反应器可称为加氢脱氧反应器,固定床反应器可称为加氢裂化反应器或加氢精制反应器。本发明是将未经预处理的全组分生物质热解液在供氢剂的保护下直接加入到加氢脱氧沸腾床反应器的反应区域;生物质热解液在热循环油、供氢剂、催化剂、催化剂、氢气和内构件共同作用形成的催化剂全混流循环体系的沸腾床反应器中进行加氢脱氧反应,加氢脱氧反应流出物经分离得到脱氧油和水相物质;所述脱氧油(水分含量小于0.1%)分为两部分,一部分脱氧油作为循环油与氢气混合后被加热至高于250℃后由沸腾床反应器底部返回反应器,经过分布器后进入加氢脱氧反应床层;另一部分脱氧油与氢气混合进入加氢裂化反应器(固定床反应器)进行加氢裂化反应,加氢裂化反应流出物经过分离后得到油品物质和水相物质,油品物质进入分馏塔经分馏得到高质燃油和化工产品。本发明所用生物质热解液不用经过任何预处理,即使生物质热解液中含有微量的颗粒物质也会随着催化剂的外排系统移出。在接触催化剂之前,生物质热解液维持常温,防止生物质热解液的聚合。生物质热解液之所以容易发生聚合反应而结焦是因为其组分中富含大量的醛类、酮类、醚类、酚类等物质,这些物质在发生聚合反应的速度随着温度的升高而加剧,另外生物质热解过程中纤维素和半纤维素裂解生成的糖类物质受热也会发生脱水反应而得到大分子的聚合物甚至进一步脱水得到焦炭,这些聚合物和焦炭会引起催化剂的失活。本发明中是将生物质热解液直接加入到反应床层区域发生反应,同时通过供氢剂裹挟包裹生物质热解液的方式防止生物质热解液在管路中被加热,确保生物质热解液进入反应床层之前的温度小于80℃,优选小于50℃。供氢剂选用本发明中得到的高质燃油中的中间馏分油或石油类烃类物质产品、煤焦油加氢处理得到的烃类物质以及其它有机质加氢脱氧得到的烃类物质,烃类物质沸点范围优选160~260℃。由于生物质热解液是极性物质,相同条件下氢气在供氢剂中的溶解度比在生物质热解液中高几百倍。供氢剂的加入还大大增强了反应器内物质的溶氢能力,从而加快了生物质热解液脱氧反应速度,降低了聚合反应速度。同时供氢剂也作为生物质热解液不被过热的保护剂。脱氧油和氢气(包括循环氢和加氢裂化反应器分离出来的富氢气体)经过加热炉加热后经沸腾床反应器底部的分布器的分配后进入反应器,为加氢脱氧反应提供热源和催化剂流化的动能,并通过控制脱氧油和氢气混合物温度来控制加氢脱氧反应温度。脱氧油是生物质热解液中容易发生聚合的组分经过加氢转化后得到的热稳定的油相组分,在氢气存在的条件下被加热到450℃甚至更高的温度也不易发生聚合反应。此外,脱氧油和氢气的混合物给沸腾床催化剂提供了流化的动能,并通过控制脱氧油的流量来控制催化剂的流化状态。另外,脱氧油与氢气混合物以及流化催化剂的高速扰动对生物质热解液与供氢剂进行加热、混合与稀释,并在生物质热解液升温的过程中对其进行逐级加氢脱氧处理,进一步防止了聚合反应的发生。本发明采用的球形催化剂在反应床层区域是全混流循环状态,有利于脱氧反应的传质与传热,避免了强放热的加氢脱氧反应中局部温度过高而引起的聚合和结焦现象。催化剂全混流循环是指催化剂颗粒的宏观运动表现为由反应器底部流化至反应器料面位置,而后返回至反应器底部。该球形催化剂采用了致密外壳层和高孔隙的内部骨架结构,具有高强度高耐磨性的特性,确保了在流化状态下也不会发生催化剂破碎和骨架坍塌现象。催化剂在全混流循环状态,尤其是配合专有的能让物料高速旋转的三相分离器的情况下,催化剂颗粒的旋转与摩擦对催化剂外表面的聚合物形成自洁效果,进一步防止聚合物聚集而引起催化剂的失活。直接加入的生物质热解液与供氢剂,在全混流循环状态体系中,被快速的稀释并与氢气和脱氧油混合。沸腾床反应器流出的油水混合物呈现乳化并在油水界面明显出现大量消失缓慢的泡沫,说明脱氧油中含氧官能团物质具备表面活性剂的性能。在全混流循环体系中,这些含氧官能团物质可以将极性的生物质热解液、非极性的供氢剂和弱极性的脱氧油形成均相,促使了供氢剂携带的自由氢和氢气形成的自由氢与生物质热解液的快速反应,防止聚合反应发生,防止催化剂的快速失活。沸腾床反应器产物进入冷高分进行气液分离。分离出来的气体中一部分气体作为循环氢供沸腾床加氢脱氧反应利用,一部分气体作为外排废氢并通过氢提纯设备后返回新氢管路,通过调节循环氢与外排废氢的比例来调节沸腾床加氢脱氧反应的氢分压。分离出的液体经过油水分离设备进行分离形成脱氧油和水相物质,水相物质进入下游的水处理设施,脱氧油一部分作为循环油返回沸腾床反应器,另一部分进入下一级加氢裂化反应器。由于脱氧油品中的含氧官能团是表面活性剂物质,在高压到低压过程中,随着大量气体释放,油水混合物发生乳化现象并伴随着大量泡沫,该乳化的油水混合物在静止条件下很难通过密度差的方式实现分离,同时满足工艺所需分离设备体积也巨大。但是,随着脱氧油的含氧量(本发明中氧含量控制在5%~15%)的大幅降低,脱氧油的极性大幅降低为接近非极性(<10μs/cm),尤其是供氢剂的加入,同时得到的水相由于溶解羧酸和醇类物质的缘故而具有较强极性,本发明通过两相的极性差异实现分离。williamspt,nugranadn.在comparisonofproductsfromthepyrolysisandcatalyticpyrolysisofricehusks[j]energy,2000,25(6):493-513中也提到高温下主要以co和co2的型式脱氧、低温下主要以h2o的型式脱氧。加氢脱氧反应温度越低,反应速率降低,同时水的产率增加,氢气的消耗增加;加氢脱氧反应温度越高,生物质热解液聚合反应的速率增加导致结焦的风险增大。为了防止生物质热解液聚合反应结焦而引起催化剂的失活,所以,在采用单一高性能催化剂的情况下,沸腾床加氢脱氧反应器可以选用单级反应器,因为高性能催化剂(如钌、钯等)能减低加氢反应的活化能,促使加氢脱氧反应在相对较低的温度下达到所需要的脱氧目标;在采用不同性能催化剂的情况下和/或采用相同催化剂但需要不同操作温度的情况下沸腾床加氢脱氧反应器可以选用两级或多级串联反应器,其主要目的是分别对容易发生聚合反应的物质进行分质处理。脱氧油接近非极性的燃料油,两者可以以任何比例进行互溶而不会发生分层现象,且脱氧油硫氮含量很低,同时有一定的氧含量(通常<10%,优选<8%)有助于完全燃烧,所以该脱氧油可以作为低硫低氮燃料油使用,尤其是在高海拔地区,燃烧排放的颗粒物、硫化物和氮氧化物均低于传统化石类燃料油。但由于脱氧油作为燃料油的经济价值远低于深加工后高质燃油(汽油、柴油和航空燃油)和化工原料,所以将脱氧油与氢气加热后进行固定床加氢裂化反应。加氢裂化反应流出物经过气液分离器分离出的气体与循环氢混合后,再与循环油混合并经过加热进入沸腾床加氢脱氧反应器。加氢裂化反应流出物经过气液分离器分离后的液体经过油水分离设备进行分离形成油品物质和水相物质,水相物质进入水处理设施,油品物质进入分馏塔。该油水分离原理也主要是利用油品物质和水相物质的极性差异实现分离。由于脱氧油是相对稳定的物质,从减少投资和运行费用的角度考虑,加氢裂化反应器选用固定床为优先选择。但脱氧油中仍然含有少量易发生聚合反应的烯烃,为了保证装置的长周期运行,在采用单一复合型催化剂的情况下,加氢裂化反应器可以选用单级;在采用不同性能催化剂和/或需要不同反应温度的情况下,可以选用两级或多级串联对脱氧油进行加氢裂化和/或加氢精制反应。加氢裂化反应后油品物质加热后进入分馏塔分离为顶部的轻烃、上部的石脑油、中部的供氢剂、下部的柴油和塔底的重柴油馏分。供氢剂选用沸点范围在120~300℃(优选160~260℃)的馏分。不建议选用更低沸点馏分的原因在于更低沸点的石脑油馏分在循环回加氢脱氧反应器后更易汽化从而降低反应器中的氢分压。另外,本发明得到的加氢裂化油品物质的终馏点小于420℃,高沸点物质可以作为重柴油直接使用,为了增加高质燃油和化工原料的产量也可以将更高沸点馏分的油品物质与供氢剂混合后循环回加氢脱氧反应器。本发明的方法具有如下有益效果:1)将生物质热解液转化为车辆可以直接使用的高质燃油和化工产品。2)运行成本低,包括氢耗低、能使用非贵重金属催化剂,操作条件温和。3)阻止了生物质热解液受热缩合和结焦。4)解决了催化剂失活快的问题,防止焦炭的形成,以满足工业化长周期运行的要求。5)生物质热解液的适用性广,能直接使用,不需过滤和分相等预处理。6)为了形成规模效应,可以将生物质热解液与重柴、蜡油或煤焦油共同加氢处理(简称共炼)制备高质燃油和/或化工原料。附图说明图1为本发明制备高质燃油和化工原料的方法的工艺流程示意图;图2为生物质热解液gcms分析图谱;图3为脱氧油gcms分析图谱;图4为高质燃油与干基生物质热解液、汽油和柴油的分子量分布图谱;图5为生物质热解液与重质柴油、蜡油和煤焦油共炼制备高质燃油和化工原料的方法的工艺流程示意图。具体实施方式下面结合附图和实施例对本发明作进一步说明。实施例一如附图1所示,生物质热解液1在供氢剂2的裹挟和保护下在常温下直接进入单级的沸腾床加氢脱氧反应器3的反应床层区域,生物质热解液与供氢剂的质量比例为1:0.5。供氢剂为沸点在200~240℃的石油类烃类物质。循环氢7和加氢裂化反应的气体物质29通过循环氢压缩机8增压后形成的混合气体9与循环油18混合并经加热炉10加热后形成气液混合物4从沸腾床加氢脱氧反应器3底部进入并给加氢脱氧反应提供流化的动能,并通过调节气液混合物4的温度来控制加氢脱氧反应的温度在280℃。生物质热解液在循环油与氢气混合物以及全混流循环催化剂的高速扰动下迅速地被加热、混合与稀释并进行加氢脱氧反应。反应流出物5经过冷高分气液分离器6后,气体物质一部分作为循环氢7通过循环回加氢脱氧反应,另一部分气体物质经氢提纯设施11提纯后的再生氢20并入新氢21进入加氢裂化反应。冷高分流出的液体物质12经油水分离器13分离后,水相物质14进入水处理设施15。脱氧油16经过循环泵17增压,一部分增压后脱氧油作为循环油18回到加氢脱氧反应,另一部分脱氧油19进入加氢裂化反应。通过控制循环油18的量来控制加氢脱氧反应催化剂的流化状态,并使其满足沸腾床反应器的操作域。本实施例中的循环油与生物质热解液1重量比例为4:1。通过控制循环氢的量来控制加氢脱氧反应的氢油比,本实例中氢油体积比为500:1,氢油比过低会引起反应的不完全和聚合结焦的现象,氢油比过高引起能耗增加的同时给沸腾床反应器的三相分离带来困难(超出沸腾床反应器操作域的范围)。本发明的高强度高耐磨性微球形催化剂在全混流循环体系下,相比于条形催化,其优势在于:避免了强放热的加氢脱氧反应中局部温度过高而引起的聚合和结焦现象。催化剂颗粒的旋转与轻微摩擦对催化剂外表面的聚合物形成自洁效果,进一步防止聚合物聚集而引起催化剂的失活。本实例中的加氢脱氧催化剂采用pd/c(碳基材料担载钯)。本实例的液时体积空速为1.2h-1,反应压力为13.0mpa。本实例采用的生物质热解液为松树木屑经过快速热裂解得到的液体产品。本实例加氢脱氧反应运行1000小时后,其物料平衡数据见表1,生物质热解液与脱氧油的物料性质见表2和组成分析见表3。生物质热解液与脱氧油的gcms分析结果分别见附图2和附图3,表明热解液经过加氢脱氧大部分的氧被转化为水、co、co2而被脱出,生成的脱氧油可以作为加氢裂化原料。加氢脱氧反应中生成的气体组成结果见表4。表1加氢脱氧反应的物料平衡数据表注1:氢气为反应过程中氢气的消耗量,是通过检测分析进出加氢脱氧反应器的气体流量和氢气含量,两者中氢气量的差值作为加氢脱氧反应的氢耗。注2:气相为加氢脱氧反应中生成的气相物质,不包括循环氢中氢气和其他气体组份的量。注3:产品产率为各部分产品占中总消耗的质量百分比(包括氢气消耗)。表2生物质热解液与加氢脱氧反应生成的脱氧油性质比较表3生物质热解液和脱水加氢脱氧油品gcms注4分析结果对比生物质热解液(干基)加氢脱氧油醇类13.062.48脂类7.944.73酮类12.931.15醛类15.330.00醚类0.662.05羧酸类16.460.01酚类18.7515.15烷烃类4.3247.65芳香烃类10.1625.28不饱和烃类0.391.50注4:gcms分析是在升温最高至到350℃的条件下得出的,不包括更高沸点物质。表4加氢脱氧反应生成气体的组成体积百分比(不包括氢气)在加氢脱氧反应中,生物质热解液中容易发生聚合反应的醛类物质和糖类物质以及部分羧酸类物质被转化为稳定的醇类和烃类物质,部分醇类和羧酸类等极性物质在反应中转化为烃类物质、氢气、水、co和co2,未反应的部分醇类和羧酸类物质在油水分离设备中进入水相,油相中主要是性能比较稳定的酚类物质和烃类等物质。脱氧油的馏分范围大幅减低至最高沸点495℃,这是因为生物质热解液中大分子组分主要是依靠氧原子连结,加氢脱氧的过程中氧以水、co和co2的形式脱出,将生物质热解液中大分子剪裁为小分子的烃类、醇类、羧酸类等物质。实验中还发现,相对于生物质热解液的质量而言,总氧含量从49.4%减低到7.4%,扣除生物质热解液所含水分中的24.89%氧含量,加氢脱氧反应中脱除的氧的量占比为17.11%。加氢脱氧反应中以co和co2的方式脱除的氧的量为5.10%,主要是因为发生了脱羰基反应和脱羧基反应,也有部分醇类物质在水蒸汽存在情况下发生裂解制氢的可能,但氢气生成过程中伴随着co和co2的生成,不会改变总体的氢氧比例。加氢脱氧反应中12.01%的氧是以水的形式脱除,理论上脱除这些氧需要消耗氢的质量约为生物质热解液的1.50%,实际消耗氢气的量为生物质热解液质量的1.54%,说明该加氢脱氧反应主要以脱氧为主,只有少部分碳碳键的断裂。另外,从反应过程中生成的气体的组成来看,生成的气体中co和co2的质量占70%左右,再次印证了加氢脱氧反应主要以脱氧为主,只有较少比例的轻组分烃类物质生成,而且其中还有一部分是因为甲酯和乙酯类物质脱氧而来。本实例加氢脱氧反应运行2000小时后,沸腾床加氢脱氧反应器的催化剂被取出并用呋喃溶液充分洗涤,烘干后的催化剂重量与反应前加入的催化剂的重量比较,未显示出重量增加,这意味着很少或没有焦炭形成。新氢21与再生氢气20混合并经过氢气压缩机22增压后的氢气23与脱氧油19混合再经过加热炉24加热后的气液混合物25作为原料进入单级固定床加氢裂化反应器26中,并通过调节气液混合物25的温度来控制加氢裂化反应温度为360℃。反应流出物27经过冷高分气液分离器28后,气体物质29作为补充氢提供给加氢脱氧反应使用,液体物质30经油水分离器31分离后,水相物质32进入水处理设施15,生产的高质燃油33经加热炉34加热后进入分馏塔35。轻烃馏分物质36从分馏塔塔顶排出,石脑油馏分物质37从分馏塔上部抽出,沸点范围在200-240℃的馏分38从分馏塔中部的侧线抽出,一部分经冷却器39冷却并经过供氢剂泵40增压后作为供氢剂2循环回加氢脱氧反应器,另一部分外排并入从塔下部的轻质柴油41管线。塔底的重柴油馏分42可以外排作为产品销售,为了得到更大产量比例的石脑油馏分和汽柴油时,也可以将塔底的重柴油馏分41与供氢剂2混合后循环回加氢脱氧反应器,再次经过加氢脱氧和加氢裂化两级反应器得到更低沸点的馏分物质。通过氢气压缩机22控制氢气23的流量来控制加氢裂化反应氢油比,本实例中加氢裂化反应氢油比为700:1。本实例中的加氢裂化催化剂采用cow/al2o3,本实施例的液时体积空速为2.0h-1,反应压力为15.0mpa。加氢裂化反应相比于加氢脱氧反应压力更高,因为更高压力有利于提高加氢裂化反应中的氢分压,从而更好的促进反应的进行;同时,加氢裂化反应气液分离得到的气体物质29需要循环回加氢脱氧反应,较高压力有利于系统压力平衡;另外,本实验中还发现较低浓度的氢含量也能满足于本发明中加氢脱氧反应的要求。相比于将气体物质29排入废氢管线经过氢气提纯设施再生,需额外增加氢气压缩机增压(由于压力平衡的原因)后才能并入循环氢9的管线的方法,本发明中将加氢裂化反应的气体物质29并入循环氢9的管线,共用循环氢压缩机8的方法大幅节省了投资和能耗。本实施例加氢裂化反应的物料平衡数据见表5,加氢裂化反应原料与生成高质燃油的物料性质见表6,加氢裂化反应的气体组分分析见表7。表5加氢裂化反应的物料平衡数据表注5:氢气为加氢裂化反应过程中氢气的消耗量,是通过检测分析进出加氢裂化反应器的气体流量和氢气含量,两者中氢气量的差值作为反应的氢耗。注6:气相为加氢裂化反应中生成的气相物质,不包括氢气。注7:产品产率为各部分产品占中总消耗的百分比(包括氢气消耗)。表6脱氧油与加氢裂化反应生成的燃油性质比较表7加氢裂化气相分析结果在加氢裂化反应中,氧被进一步脱出的同时大分子物质被裂解为小分子物质,氧基本被完全脱出,硫含量小于10ppm,终馏点从495℃降低到420℃。重柴油含量约为2%,可以作为产品销售。干基物质热解液、高质燃油、柴油和汽油的分子量分布(gpc)分析结果见图4。说明热解液经过加氢脱氧和加氢裂化后得到的混合物包括石脑油、汽油、柴油和煤油馏分,该混合物与汽油柴油相近。本实施例中还发现:为了不生产重柴油,将塔底重柴油与供氢剂混合后循环回沸腾床加氢脱氧反应器并经过加氢脱氧和加氢裂化两级反应,经过长周期的运行发现,该部分沸点范围产品的量并没有增加,说明该部分物质能够被本发明的工艺和催化剂加氢处理为低沸点的物质。实施例二本实施例中是采用两级加氢脱氧反应器串联和两级加氢裂化反应器串联的方式。供氢剂为本发明得到的沸点范围为180~240℃的高质燃油产品。在第一级加氢脱氧反应中采用与实施例一相同的催化剂pd/c,第二级加氢脱氧反应中采用催化剂为nimo/al2o3,第一级加氢脱氧反应的温度控制在280℃,第二级加氢脱氧反应的温度控制在330℃。在固定床反应中采用不同的催化剂,第一级精制裂化反应使用的催化剂为como/al2o3,反应温度控制在180℃、液时体积空速为4.0h-1;第二级加氢裂化反应的催化剂为niw/al2o3,第二级加氢裂化反应的反应器其尺寸规格、催化剂装填量以及液时体积空速与实施例一相同,温度控制在350℃。其它操作条件和工艺流程与实施例一相同。表8加氢脱氧反应运行3000h,实施例二与实施例一生成的脱氧油性质比较表9加氢裂化反应运行3000h,实施例二与实施例一生成的高质燃油性质比较通过与实施例一的单级加氢脱氧反应和单级加氢裂化反应的比较发现,两级沸腾床加氢脱氧和固定床加氢精制与加氢裂化串联反应得到高质燃油收率与实施例一基本相同,因为所有反应主要是以加氢脱氧为目的,在脱氧的同时将易聚合的组分转化为稳定性的组分。同时由于生物质热解液中的大分子物质主要是由氧原子连结而成,在脱氧的过程中,这些大分子的物质变成小分子物质,而且生物质热解液中含三环及三环以上稠环芳烃的量极少,所以生产的燃油的终馏点低于420℃,且重柴油比例小于2%。在本实施例的加氢脱氧反应中,由于经过两级反应,脱氧油中的氧含量由7.4%降到了6.2%,脱氧效果明显提高,具体数据见表8。本实施例中还发现,通过两级固定床加氢精制与加氢裂化串联,虽然生成的高质燃油的收率及产品性质相差不明显(具体数据见表9),但催化剂的寿命可延长,具体参见表10。表10单级和两级加氢裂化反应的对催化剂寿命影响比较实施例一实施例二连续运行时间,h60006000反应器压降增加,mpa0.420.22燃油收率,%85.6585.68本实施例中发现,第一级固定床加氢反应主要是对加氢脱氧油中未被完全处理而留存的易发生聚合的不饱和烃在较低温度下进一步加氢处理得到更稳定的物质,具体数据见表11,在第二级固定床加氢裂化反应时发生聚合反应的速率大幅降低,所以在第二级加氢裂化反应条件与实施例一中加氢裂化反应完全相同的情况下,第二级加氢裂化反应器压降增加速度更低,催化剂的使用寿命大幅延长。表11实施例二中脱氧油品与第一级加氢裂化反应后油品gcms分析结果对比实施例三本实施例中采用单级加氢脱氧反应器,生物质热解液为小麦秆经过快速热裂解得到的液体产品,供氢剂为轻质柴油,加氢脱氧反应中采用的催化剂为ru/c(碳基材料担载钌),其他操作条件与实施例一相同。加氢裂化反应采用三级固定床串联方式,第一级和第二级加氢裂化反应使用的催化剂与实施例二相同,其他操作条件与实施例二相同,第三级加氢裂化反应操作温度为380℃,第三级加氢裂化反应使用催化剂和其它操作条件与第二级加氢裂化反应相同。连续运行2000小时后,本实施例中得到的高质燃油性质与实施例一和实施例二比较见表12。表12实施例三与实施例一、实施例二生成的高质燃油性质比较通过比较发现,单级加氢脱氧反应器和三级固定床加氢裂化反应器串联方式得到的高质燃油收率和燃料油性质与实施例一和实施例二基本相同。说明在加氢脱氧达到一定的程度后,加氢裂化反应对产品的性能具有较大影响。另外,还发现通过两级加氢裂化反应器串联的方式会延长加氢裂化催化剂寿命,同实施例二类似。实施例四本实施例中采用三级沸腾床加氢脱氧反应器串联,生物质热解液为杨树树枝和树皮的混合物经过慢速热裂解得到的液体产品,供氢剂为煤焦油加氢处理得到的沸点范围为180~240℃的物质。第一级加氢脱氧反应采用催化剂为nimn/c(碳基材料担载镍锰),第二级加氢脱氧反应中采用催化剂为como/al2o3与实施例二中第二级加氢脱氧反应相同,其他操作条件与实施例二相同,第三级加氢脱氧反应操作温度为370℃,第三级加氢脱氧反应使用催化剂和其它操作条件与第二级加氢裂化反应相同。加氢裂化反应采用单级固定床,采用与实施例一相同的催化剂niw/al2o3,其他操作条件与实施例一加氢裂化反应相同。连续运行2000小时后,本实施例中得到的脱氧油油品的性质与实施例一比较见表13。表13加氢脱氧反应运行2000h,实施例四与实施例一得到的脱氧油性质比较通过试验结果分析发现,采用非贵金属催化剂通过分级处理的情况下也可以达到采用贵金属催化剂的脱氧效果,同时得到的脱氧油性质相似。将加氢脱氧反应得到的脱氧油品经过加氢裂化反应,得到的高质燃油与实施例一和实施例二得到的高质燃油比较见表14。表14实施例四与实施例一、实施例二生成的高质燃油性质比较通过比较发现,本实施例得到的高质燃油收率和燃料油性质与实施例一和实施例二基本相似。说明在加氢脱氧反应的脱氧效果达到一定的程度后,加氢裂化产品的性能也基本相同。本实施例说明,在加氢脱氧采用非贵重金属催化剂、多级沸腾床反应器串联的情况下,得到的脱氧油品通过加氢裂化反应后,也能够得到理想的高品质的混合燃油。实施例五本实施例中采用单级沸腾床加氢脱氧反应器,生物质热解液为玉米秆炭化过程中得到的液体产品,采用催化剂为como/c,反应的温度为350℃,生物质热解液与供氢剂的质量比例分别为1:2、2:1和4:1,其它操作条件与实施例一相同。连续运行2000小时后得到的脱氧油性质见表15。表15不同生物质热解液与供氢剂比例下加氢脱氧反应生成的脱氧油性质比较从结果可以看出,随着加氢脱氧反应器中生物质热解液浓度的增加,加氢脱氧效果降低,得到的脱氧油中氧含量增加、热值降低、总酸值和重组分物质的量增加。所以本发明加入供氢剂的目的除了增加氢的溶解度之外,还进一步降低了生物质热解液在沸腾床加氢脱氧反应器中的浓度,促进了脱氧的发生,减缓了聚合反应的速度,增加了催化剂的寿命。实施例六本实施例中采用单级沸腾床加氢脱氧反应器,采用采用催化剂为nicr/c,加氢脱氧反应的温度分别为280℃、310℃、340℃和370℃,其它操作条件与实施例一相同。连续运行1000小时后生成的脱氧油性质见表16。表16不同加氢脱氧反应温度下生成的脱氧油性质比较从结果比较可以看出,加氢脱氧效果随着反应温度的增加,得到的脱氧油中的氧含量降低、热值增加。但是,当反应温度超过340℃以后,脱氧油氧含量降低不明显、热值降低、总酸值和粘度反而升高。这是因为当温度过高,加氢脱氧反应受限于传质速度而不能进一步增大,但高温促使聚合反应加快,致使一部分未及时反应的物质发生了聚合反应。实施例七本实施例中采用单级沸腾床加氢脱氧反应器,采用催化剂为conb/al2o3,加氢脱氧反应的温度为330℃,反应压力分别为11.0mpa、13.0pa、15.0mpa和18.0mpa,其它操作条件与实施例一相同。连续运行1000小时后生成的脱氧油性质见表17。表17不同加氢脱氧反应压力下生成的脱氧油性质比较从结果比较可以看出,加氢脱氧效果随着反应压力的增加,得到的脱氧油中的氧含量降低、热值增加、总酸值和粘度降低。这是因为压力增加,反应系统的氢分压增大,有利于脱氧反应的发生,但压力增加会增加设备的投资和运行费用。实施例八本实施例中采用单级沸腾床加氢脱氧反应器,采用催化剂为nicezr/al2o3,反应温度为330℃,加氢脱氧反应中循环油与生物质热解液的质量比例(简称循环比)分别为2:1、3:1、4:1、5:1和6:1,其它操作条件与实施例一相同。连续运行1000小时后生成的脱氧油性质见表18。表18在不同循环比条件下加氢脱氧反应生成的脱氧油性质比较从实验结果可以看出,加氢脱氧反应循环比的变化对脱氧效果有明显的影响。因为使用循环油的主要目的有:1.对生物质热解液进行稀释,减缓聚合反应的速度;2.促使反应器内的催化剂处于的流化状态,与生物质热解液、循环油、供氢剂、氢气、催化剂和内构件共同作用形成催化剂全混流循环体系。当循环比过低时,催化剂处于的流化效果不理想,甚至可能超出沸腾床反应器的操作域范围;另外对原料的稀释效果也大幅降低,最终导致脱氧不是很明显。当循环比增加,处于沸腾床反应器的操作域范围时,能够达到理想的脱氧效果,进一步增加循环比对生成的脱氧油性质影响较小,而且当循环比过高超出沸腾床反应器的操作域范围时,对下游的工艺设备带来损伤。所以根据反应器内构件和催化剂特性选用合适的循环比很重要。本发明的循环比范围在2:1-7:1之间为优先选择。实施例九本实施例中采用单级沸腾床加氢脱氧反应器,催化剂为niru/c,反应温度为330℃,加氢脱氧反应中的氢气与生物质热解液的标准状态下体积比分别为300:1、500:1、700:1、900:1和1100:1,其它操作条件与实施例一相同。连续运行1000小时后生成的脱氧油性质见表19。表19不同氢油比下加氢脱氧反应生成的脱氧油性质比较从脱氧油性质比较可以看出,加氢脱氧效果随着氢油比的增加,得到的脱氧油中的氧含量降低、热值增加、总酸值和粘度降低。但是当氢油比达到一定数值以后,脱氧油氧含量降低不明显、热值升高以及总酸值降低和粘度降低变化不明显。这是因为当氢油比较小时,反应器内氢分压较小,加氢脱氧反应不完全;当氢分压达到一定程度后,脱氧反应受限于传质速度和温度其他因素,继续增大氢油比对脱氧反应的效果变化不明显,同时高氢油比会增加运行成本。实施例十本实施例中采用单级沸腾床加氢脱氧反应器,催化剂为ni/c,反应温度为330℃,加氢脱氧反应的液时体积空速分别为0.4h-1、0.6h-1、0.8h-1、1.0h-1和1.4h-1,其它操作条件与实施例一相同。连续运行1000小时后生成的脱氧油性质见表20。表20不同液时体积空速下加氢脱氧反应生成的脱氧油性质比较从脱氧油性质比较可以看出,加氢脱氧效果随着液时体积空速的增加,得到的脱氧油中的氧含量升高、热值降低、总酸值和粘度增加。这是因为液时体积空速增加时,部分没来得及反应的物质在高温下发生聚合反应。实施例十一本实施例中采用单级沸腾床加氢脱氧反应器,催化剂为nicr/c,反应温度为330℃,其它操作条件与实施例一相同。连续运行3000小时后,发现催化剂的活性有所降低。用丁醇对催化剂进行旋流洗涤并进行还原后,将再生后的催化剂与原来新鲜催化剂按一定的比例投入沸腾床加氢脱氧反应器使用,运行1000小时后生成的脱氧油与未添加再生催化剂前生成的脱氧油的性质比较见表21。表21添加再生催化剂前后加氢脱氧反应生成的脱氧油性质比较将活性降低的催化剂再生后的实验结果与未添加再生催化剂的实验结果进行比较:未添加再生催化剂的实验运行1000小时时,加氢脱氧反应将生物质热解液中氧的质量含量从49.4%降低到7.2%;而添加80%再生催化剂的实验运行1000小时时,加氢脱氧反应将生物质热解液中氧的质量含量从49.4%降低到7.3%。比较发现脱氧率几乎没有发生改变,说明本发明的催化剂再生方法可以将催化剂活性恢复99%以上。实施例十二本实施例中采用单级固定床加氢裂化反应器,催化剂为como/al2o3,加氢裂化反应的温度分别为300℃、330℃、360℃和390℃,其它操作条件与实施例一加氢裂化反应相同。采用实施例一得到的脱氧油为原料,连续运行1000小时后生成的燃油性质见表22。表22不同加氢裂化反应温度下生成的燃油性质比较从实验结果可以看出,随着温度的增加得到的燃油中的密度降低、热值增加、终馏点降低。这是因为随着温度的增加,加氢裂化反应速度加快。但是,当温度过高,得到的轻烃组分比例增加,燃油的收率降低。实施例十三本实施例中采用单级固定床加氢裂化反应器,催化剂为niw/al2o3,加氢裂化反应的压力分别为12.0mpa、13.5mpa、15.0mpa和18.0mpa,其它操作条件与实施例一加氢裂化反应相同。采用实施例一得到的脱氧油为原料,连续运行1000小时后生成的燃油性质见表23。表23不同加氢裂化反应温度下生成的燃油性质比较从实验结果可以看出,随着反应压力的增加得到的燃油中的密度降低、热值增加、终馏点降低。这是因为随着压力的增加,加氢裂化反应速度加快。但是,当压力过高,得到的轻烃组分比例增加,燃油的收率略有降低。实施例十四本实施例中采用两级固定床加氢裂化反应器,采用与实施例二加氢裂化反应相同的催化剂,加氢裂化反应中的氢气与脱氧油的标准状态下体积比分别为300:1、500:1、700:1、900:1和1100:1,其它操作条件与实施例二加氢裂化反应相同。采用实施例二得到的脱氧油为原料,连续运行1000小时后生成的燃油性质见表24。表24不同氢油体积比下,加氢裂化反应生成的燃油性质比较从本实施例结果可以看出,随着氢油比的增加,反应得到的燃油的密度降低、热值增加、终馏点降低。这是因为随着氢油比的增加,氢分压提高,加氢裂化反应速度加快,得到的轻烃组分比例增加,燃油的收率略有降低。实施例十五本实施例中采用单级固定床加氢裂化反应器,采用与实施例一加氢裂化反应相同的催化剂,加氢裂化反应中的液时体积空速分别为1.0h-1、1.5h-1、2.0h-1、3.0h-1和4.0h-1,其它操作条件与实施例一相同。采用实施例一得到的脱氧油为原料,连续运行1000小时后生成的燃油性质见表25。表25不同液时体积空速下加氢裂化反应生成的燃油性质比较从本实施例结果可以看出,随着反应液时体积空速的增加,反应得到的燃油中的密度增加、热值降低、终馏点升高。实施例十六本实施例中采用单级固定床加氢裂化反应器,采用与实施例一加氢裂化相同的催化剂,加氢裂化反应原料中游离水质量含量分别为10ppm、100ppm、0.1%、0.5%和1%,其它操作条件与实施例一相同。连续运行1000小时后生成的高质燃油性质见表26。表26不同游离水含量下加氢裂化反应生成的高质燃油性质比较从脱氧油性质比较可以看出,加氢脱氧效果随着游离水质量含量的增加,得到的产品的总酸值升高、热值降低。这是因为加氢裂化催化剂在富含游离水的情况下会发生载体骨架的破坏。另外,使用硫化催化剂的情况下,游离水也会加速催化剂的失活。所以,加氢裂化反应原料中游离水含量的增加会加速催化剂的失活而影响反应的长周期运行。但是,游离水质量含量降低对脱氧油脱水的效率要求越高,会增加投资成本和运行费用。结合本发明的催化剂特性,本发明的加氢裂化反应原料中游离水含量在10~1000ppm为优先选择。实施例十七本实施例工艺流程示意参见图5。本实施例中采用两级固定床加氢裂化反应器串联,采用与实施例二加氢裂化反应相同的催化剂。脱氧油19与重质柴油、蜡油或煤焦油43经加热炉24加热后,进入第一级固定床加氢精制反应器44反应后,反应产物45经加热炉46加热再进入固定床加氢裂化反应器26进行共炼,其它操作条件与实施例二的加氢裂化反应相同。连续运行3000小时后得到的高质燃油性质见表27。表27脱氧油与重质柴油、蜡油或煤焦油共炼得到的高质燃油性质比较注:本实施例低温煤焦油采用长烟煤低温裂解(<500℃)得到的液态物质。从结果可以看出,使用脱氧油与重质柴油、蜡油或煤焦油部分共炼,得到的高质燃油同样能达到车用燃油的相关标准要求。因为本发明生成的脱氧油能够完全与重质柴油、蜡油或煤焦油以任意比例互溶,能满足本发明加氢裂化反应原料要求,且采用的固定床催化剂与传统重质柴油、蜡油或煤焦油加氢处理所使用催化剂类似。实施结果说明,本发明可以做到脱氧油与重质柴油、蜡油或煤焦油实现共炼,可以利用传统炼油装置实现共炼,节省设备投资,大幅提高装置的最终产品的产能,从而进一步增加本发明的经济性。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1