面积归一化法测定西红花药材中西红花苷类成分的方法与流程
本发明涉及中药活性成分分析技术领域,具体地说,是涉及一测多评法与面积归一化法相结合的测定西红花药材中西红花苷类成分的方法。
背景技术:
西红花(Crocus sativus L.)又称藏红花,仅以雌蕊上部三根红色柱头入药,人工采摘100000-150000朵花才能获得一公斤藏红花干品,因此价格昂贵,为名贵中药材,具有活血化瘀、凉血解毒、解郁安神之功效。主要有效成分为西红花苷(crocins),是一种红色的水溶性胡萝卜素类化合物,是西红花酸与葡萄糖或龙胆二糖结合而成的一系列酯苷类,目前已分离得到苷元均为西红花酸的16种不同顺反异构体的西红花苷。世界各地不同来源西红花中均以西红花苷-Ⅰ含量最高,其次为西红花苷-Ⅱ,西红花苷产地栽培、干燥与储存等对西红花苷含量影响很大,因此在西红花生产加工和质量控制方面均需要适当的藏红花苷含量测定方法,《中国药典》2015年版以西红花苷中的西红花苷-Ⅰ和苷-Ⅱ为指标对西红花质量控制,要求两者总含量不低于10%,但西红花苷-Ⅰ和苷-Ⅱ在某些样品中只占各西红花总苷的70%左右,且随着储存时间的增加,反式的西红花-Ⅰ和苷-Ⅱ会发生降解或异构化,部分转为其他顺式异构体,而西红花苷口服给药后,所有西红花苷均会水解为苷元西红花酸被吸收入血起效,因此西红花总苷含量的高低对药效的影响更大,《中国药典》根据西红花苷-Ⅰ和苷-Ⅱ的总含量为标准,不能全面反映西红花药材质量;西红花在国际上主要用作色素和香料,作为食品添加剂的国际标准ISO3632-2(2010版),用分光光度法于λmax 440nm测定西红花水提取液的色价来进行质量控制与分级,规定西红花苷色价大于190为一级品,该方法更不能精确定量西红花苷含量。因此,《中国药典》及国际标准ISO 3632-2对藏红花质量评价的标准均需进一步完善,需要建立适合生产加工和质量控制的西红花总苷含量测定方法。
一般认为分光光度法特征性不强,但西红花是一味很特殊的药材,药效成分均集中于柱头,含量高且相对简单,主要只有藏红花苷、藏花醛和苦藏花素三类成分,在可见光区440nm处只有西红花苷类有吸收,其他成分无干扰,用可见分光光度法(440nm)测定总西红花苷含量的方法,方法简便,易于推广,但该方法只适合生产加工者使用,若有人为掺假等行为,则不适合使用。
技术实现要素:
本发明所要解决的技术问题是针对《中国药典》仅测定西红花苷-Ⅰ和西红花苷-Ⅱ含量,不能全面体现总西红花苷的真实含量,《国际标准》测定的总西红花苷色价不能量化总西红花苷的含量的缺点。本发明将一测多评测定的两种西红花苷含量和面积归一化法测定的总西红花苷含量相结合,实现了一个对照品完成各西红花苷类成分的含量测定,降低了检测成本,并能全面评价西红花的质量,为西红花药材质量评价方法的完善提供了更好的技术参考。
本发明采用高效液相法,通过优选色谱条件,波长切换等技术测定药材中的西红花苷,可分离大部分西红花苷类成分,但西红花苷的苷元由七个共轭双键组成,易氧化不稳定,因此西红花苷对照品价格昂贵,精确定量所有西红花苷类成分含量,检测成本非常昂贵,本发明使用采用面积归一化法(ANM)对西红花药材中西红花总苷进行含量测定,解决了HPLC法含量测定时多个西红花苷对照品不易获得的问题。
本发明以西红花苷-Ⅰ为内参物,建立了西红花苷-Ⅰ对西红花苷-Ⅱ的相对校正因子,利用相对校正因子计算西红花苷-Ⅱ的含量(一测多评);并与药典采用的外标法测定的32批西红花中西红花苷-Ⅰ和苷-Ⅱ含量进行比较,以验证一测多评法的可行性。结果西红花苷-Ⅰ相对西红花苷-Ⅱ的相对校正因子重复性好,西红花中西红花苷-Ⅱ的一测多评法的计算值与药典外标法测定结果基本一致,解决了药典测定方法中西红花苷-Ⅱ对照品性质不稳定、价格昂贵的问题。
为解决上述技术问题,本发明提供了一种面积归一化法测定西红花药材中西红花苷类成分的方法,包括步骤:
a.确定色谱条件;
b.制备对照品溶液:
混合对照品溶液:精密称取西红花苷-Ⅰ对照品和西红花苷-Ⅱ对照品,置于棕色量瓶中,用稀乙醇溶解并定容至刻度,得混合对照品溶液,摇匀,备用;
西红花苷-Ⅰ对照品溶液:精密称取西红花苷-Ⅰ对照品置棕色量瓶中,用稀乙醇溶解并定容至刻度,得西红花苷-Ⅰ对照品溶液;
c.制备供试品溶液:参照中国药典2015年版西红花项下,精密称取西红花样品粉末,置于棕色量瓶中,加稀乙醇40mL,冰浴超声处理20min,放置室温,加乙醇定容至刻度,摇匀,微孔滤膜过滤,取续滤液,得供试品溶液;
d.根据2015年版中国药典的外标法定量西红花苷-Ⅰ和苷-Ⅱ的含量;
e.一测多评法QMSA测定西红花苷-Ⅰ和苷-Ⅱ的含量;f.紫外分光光度法测定总西红花苷的含量;
g.面积归一化法测定总西红花苷的含量。
优选地,所述步骤a中,色谱条件为Ultimate XB C18色谱柱,色谱柱为250mm×4.6mm,5μm;柱温:30℃;流动相:水-甲醇,梯度洗脱,0-60min,甲醇:20%-100%;DAD检测器波长范围200-600nm,检测波长440nm,流速:1.0mL·min-1;进样量:10μL。
优选地,所述步骤c中,所述冰浴超声为250W、40KHz处理20min。
优选地,所述步骤c中,微孔滤膜的孔径为0.22μm。
优选地,所述步骤d,进一步为,取步骤b中西红花苷-Ⅰ和苷-Ⅱ的混合对照品溶液1、2、4、8、10、20μL,按步骤a中色谱条件进样分析,测定峰面积,分别以西红花苷-Ⅰ和苷-Ⅱ质量浓度(X,μg)对其相应峰面积(Y)进行线性回归,得回归方程,精密量取步骤c中的供试品溶液10μL,注入液相色谱仪测定,相应峰面积代入回归方程计算苷-Ⅰ和苷-Ⅱ含量。
优选地,所述步骤e,进一步为,取步骤b中混合对照品溶液,分别进样1、2、4、8、10、15、20μL,记录峰面积,应用多点校正法,以西红花苷-Ⅰ为内参物,计算西红花苷-Ⅰ对西红花苷-Ⅱ的相对校正因子fk/s,fk/s计算公式:fk/s=(Cs×Ak)/(Ck×As),待测成分(西红花苷-Ⅱ)质量浓度计算公式:Ck′=(Cs×Ak′)/(fk/s×As),
其中Cs为内参物(西红花苷-Ⅰ)质量浓度,As为内参物色谱峰峰面积,Ck为西红花苷-Ⅱ对照品质量浓度,Ak为西红花苷-Ⅱ对照品色谱峰峰面积,Ck′为西红花样品中西红花苷-Ⅱ质量浓度,Ak′为西红花样品中西红花苷-Ⅱ色谱峰峰面积。
优选地,所述步骤f,进一步为,精密吸取西红花苷-Ⅰ对照品溶液0.1,0.2,0.4,0.6,0.8,1.0mL置于10mL量瓶中,用稀乙醇稀释至刻度,以稀乙醇作空白对照,采用紫外-可见分光光度法,于440nm波长处测定吸光度,得标准曲线;精密吸取步骤c中的供试品溶液5mL,置50mL棕色量瓶中,用稀乙醇稀释至刻度,用可见分光光度法于440nm波长处测定吸光度,以西红花苷-Ⅰ为对照标准品,按标准曲线计算总西红花苷含量。
优选地,所述步骤g,进一步为,称取西红花样品粉末约10mg,精密称定,置50mL棕色量瓶中,加稀乙醇40mL,冰浴超声(250W,40KHz)处理20min,放置室温,加乙醇定容至刻度,摇匀,0.22μm微孔滤膜滤过,精密吸取续滤液及步骤b中对照品溶液各10μL,注入液相色谱仪测定,测定时间为不少于60分钟,同时进行质谱分析,质谱条件为:离子源:大气压电喷雾离子源(ESI);检测模式:正负离子;全扫描质核比(m/z):50-1200;干燥气温度:350℃,干燥气流量:12.0L·min-1,雾化气压力:35.0psi;毛细管电压:4.5kV;碰撞电压1.0V;进入质谱的流动相流速分流至0.4mL·min-1。以西红花苷-Ⅰ为对照,按C总苷=(C苷-Ⅰ×A总苷)/A苷-Ⅰ计算总西红花苷的含量,
其中A苷-Ⅰ为样品中西红花苷-Ⅰ色谱峰峰面积;A总苷为西红花样品440nm下12个西红花苷类色谱峰峰面积之和;C苷-Ⅰ为外标法测定的西红花样品中西红花苷-Ⅰ的质量浓度;C总苷为西红花样品中总西红花苷质量浓度。
与现有技术相比,本发明所述的一测多评法与面积归一化法相结合的测定西红花药材中西红花苷类成分的方法,达到了如下效果:
本申请采用“一测多评”法,以西红花苷-Ⅰ为内参物,同时检测《中国药典》规定的两种西红花苷的含量,与中国药典方法进行比较,以减少价格昂贵的西红花苷-Ⅱ对照品的使用。并根据各西红花苷结构相似(均为西红花酸与不同数目葡萄糖结合的不同顺反异构体),最大紫外吸收相同,同时以西红花苷-Ⅰ为对照,采用面积归一化法计算总西红花苷的含量,并与课题组前期已建立的440nm下分光光度法检测总西红花苷的含量进行比较分析,建立能与西红花苷-Ⅰ和苷-Ⅱ同步检测总西红花苷含量的方法。相较于《中国药典》方法,将一测多评测定的两种西红花苷含量和面积归一化法测定的总西红花苷含量相结合,实现了一个对照品完成西红花苷的含量测定,降低了检测成本,并能全面评价西红花的质量,为西红花药材质量评价方法的完善提供了更好的技术参考。
藏红花中含有16种不同的藏红花苷,其中西红花苷-Ⅰ和西红花苷-Ⅱ含量占70%以上,口服给药后,各西红花苷均以去糖基的苷元西红花酸吸收入血发挥药效,但《中国药典》2015版仅测定西红花苷-Ⅰ和西红花苷-Ⅱ含量,不能体现总西红花苷的含量,而西红花在国际上无药用标准,只有作为食品添加剂的质量标准ISO 3632-2,用分光光度法测定西红花水溶液440nm的色价对其进行质量控制与分级,规定西红花苷色价大于190为Ⅰ级品,150-190为Ⅱ级品,100-150为Ⅲ级品,但测定色价不能给出总西红花苷的含量。本发明以西红花苷-Ⅰ为内参物,建立西红花苷-Ⅰ对西红花苷-Ⅱ的相对校正因子,利用相对校正因子计算西红花苷-Ⅱ的含量,实现一测多评(QMSA);并同时以西红花苷-Ⅰ为对照,峰面积归一化(ANM)法检测西红花中总西红花苷的含量。结果西红花苷-Ⅱ的QMSA法的计算值与药典的外标法测定结果基本一致。ANM计算的总西红花苷含量与UV法测定的总西红花苷含量无显著性差异。实现了一个对照品完成西红花苷的含量测定,降低了检测成本,并能全面评价西红花的质量。
附图说明
此处所说明的附图用来提供对本发明的进一步理解,构成本发明的一部分,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。在附图中:
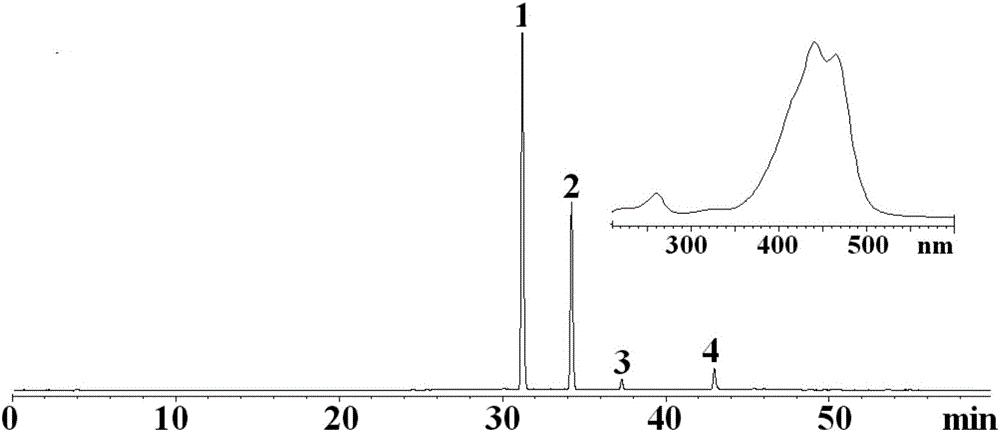
图1为混合对照品(A)440nm波长下HPLC图;其中:1-西红花苷-Ⅰ;2-西红花苷-Ⅱ;
图2为样品440nm波长下HPLC图;其中:1-西红花苷-Ⅰ;2-西红花苷-Ⅱ;3-西红花苷-Ⅲ;4-西红花苷-Ⅳ;
图3为样品波长切换HPLC图;
图4为样品质谱总离子流图。
具体实施方式
如在说明书及权利要求当中使用了某些词汇来指称特定组件。本领域技术人员应可理解,硬件制造商可能会用不同名词来称呼同一个组件。本说明书及权利要求并不以名称的差异来作为区分组件的方式,而是以组件在功能上的差异来作为区分的准则。如在通篇说明书及权利要求当中所提及的“包含”为一开放式用语,故应解释成“包含但不限定于”。“大致”是指在可接收的误差范围内,本领域技术人员能够在一定误差范围内解决所述技术问题,基本达到所述技术效果。说明书后续描述为实施本发明的较佳实施方式,然所述描述乃以说明本发明的一般原则为目的,并非用以限定本发明的范围。本发明的保护范围当视所附权利要求所界定者为准。
以下结合附图对本发明作进一步详细说明,但不作为对本发明的限定。
实施例1:
a.确定色谱条件
Ultimate XB C18色谱柱(250mm×4.6mm,5μm);柱温:30℃;流动相:水(A)-甲醇(B),梯度洗脱,0-60min,B:20%-100%;DAD波长范围200-600nm,检测波长440nm,流速:1.0mL·min-1;进样量:10μL。在该色谱条件下,各色谱峰有较好的基线分离;
b.制备对照品溶液
精密称取西红花苷-Ⅰ对照品5.96mg,西红花苷-Ⅱ对照品2.96mg,置100mL棕色量瓶中,用稀乙醇溶解并定容至刻度。得浓度为59.6μg·mL-1和29.6μg·mL-1的混合对照品溶液,摇匀,备用。
西红花苷-Ⅰ对照品溶液:精密称取西红花苷-Ⅰ对照品置棕色量瓶中,用稀乙醇溶解并定容至刻度,得西红花苷-Ⅰ对照品溶液;
c.制备供试品溶液
参照中国药典2015年版西红花项下,称取西红花样品粉末约10mg,精密称定,置50mL棕色量瓶中,加稀乙醇40mL,冰浴超声(250W,40KHz)处理20min,放置室温,加乙醇定容至刻度,摇匀,0.22μm微孔滤膜滤过,取续滤液,即得。
d.2015年版《中国药典》外标法(external standard method,ESM)定量西红花苷-Ⅰ和苷-Ⅱ的含量
取步骤b中西红花苷-Ⅰ和苷-Ⅱ混合对照品溶液1、2、4、8、10、20μL,按A项色谱条件进样分析,测定峰面积。分别以西红花苷-Ⅰ和苷-Ⅱ质量浓度(X,μg)对其相应峰面积(Y)进行线性回归,分别得苷-Ⅰ和苷-Ⅱ的回归方程Y=941.16X-4.0829(r=0.9999)和Y=1108.9X-0.8237(r=0.9999)。结果表明西红花苷-Ⅰ和苷-Ⅱ分别在0.0596-1.192μg和0.0296-0.592μg与其各自峰面积积呈良好线性关系。精密吸取步骤c中供试品溶液10μL,注入液相色谱仪测定,相应峰面积代入回归方程计算苷-Ⅰ和苷-Ⅱ含量。
e.一测多评法(QMSA)测定西红花苷-Ⅰ和苷-Ⅱ的含量
应用多点校正法,以西红花苷-Ⅰ为内参物,计算西红花苷-Ⅰ对西红花苷-Ⅱ的相对校正因子fk/s,以一个标准品西红花苷-Ⅰ,测定两个西红花苷-Ⅰ和西红花苷-Ⅱ的含量。取步骤b中下混合对照品溶液,分别进样1、2、4、8、10、15、20μL,记录峰面积,应用多点校正法,以西红花苷-Ⅰ为内参物,计算西红花苷-Ⅰ对西红花苷-Ⅱ的相对校正因子fk/s,fk/s计算公式:fk/s=(Cs×Ak)/(Ck×As)。待测成分(西红花苷-Ⅱ)质量浓度计算公式:Ck′=(Cs×Ak′)/(fk/s×As)。两式中Cs为内参物(西红花苷-Ⅰ)质量浓度,As为内参物色谱峰峰面积,Ck为西红花苷-Ⅱ对照品质量浓度,Ak为西红花苷-Ⅱ对照品色谱峰峰面积,Ck′为西红花样品中西红花苷-Ⅱ质量浓度,Ak′为西红花样品中西红花苷-Ⅱ色谱峰峰面积。
f.UV法测定总西红花苷的含量
精密吸取步骤c中下的供试品溶液5mL,置50mL棕色量瓶中,用稀乙醇稀释至刻度,用可见分光光度法于440nm波长处测定西红花药材吸光度,以西红花苷-Ⅰ为对照标准品,按标准曲线分别计算总西红花苷含量。
g.面积归一化法测定总西红花苷的含量
称取西红花样品粉末约10mg,精密称定,置50mL棕色量瓶中,加稀乙醇40mL,冰浴超声(250W,40KHz)处理20min,放置室温,加乙醇定容至刻度,摇匀,0.22μm微孔滤膜滤过,精密吸取续滤液及步骤b中对照品溶液各10μL,注入液相色谱仪测定,由于西红花中的西红花苷、苦藏花素和藏花醛三类主要化合物在440nm、250nm和330nm下有特征性吸收,但吸收强度相差很大,所以采用了波长切换技术。按上述实验条件,取对照品和西红花样品溶液进样分析,得对照品和西红花样品高效液相色谱图和质谱总离子流色谱图。在440nm下共分离鉴定出12个不同的西红花苷类成分,另有4中西红花苷类成分低于HPLC法的检测限。以西红花苷-Ⅰ为对照,按C总苷=(C苷-Ⅰ×A总苷)/A苷-Ⅰ计算总西红花苷的含量。式中A苷-Ⅰ为样品中西红花苷-Ⅰ色谱峰峰面积;A总苷为西红花样品440nm下所有色谱峰峰面积之和;C苷-Ⅰ为外标法测定的西红花样品中西红花苷-Ⅰ的质量浓度;C总苷为西红花样品中总西红花苷质量浓度。
实施例2:国内外32批西红花药材中西红花苷类成分的测定
1.仪器
Agilent 1200高效液相色谱仪(美国Agilent公司);Agilent 6330离子阱质谱仪,配有电喷雾离子源(ESI)(美国Agilent公司);Lambda 45紫外-可见分光光度仪(美国PerkinElmer公司)。
试剂与供试品
标准品:西红花苷-Ⅰ(批号:111588-201202),西红花苷-Ⅱ(批号:111589-201304)均购于中国食品药品检定研究院;甲醇和甲酸均为色谱纯(美国Tedia公司),其余化学试剂均为分析纯。
32批西红花药材由浙江省建德市三都西红花专业合作社和不同种植基地提供,经浙江中医药大学陈锡林教授鉴定为番红花(Crocus Sativus L.)的干燥柱头。
3.对照品溶液的制备
精密称取西红花苷-Ⅰ对照品6.20mg,置25mL棕色量瓶中,用稀乙醇溶解并定容至刻度。从中精密移取4.0mL置10mL量瓶中,稀乙醇稀释至刻度,摇匀。得浓度为99.2μg·mL-1的对照品溶液。
4.供试品溶液制备
参照2010年版《中国药典》西红花项下,称取西红花样品粉末约10mg,精密称定,置50mL棕色量瓶中,加稀乙醇40mL,冰浴超声处理20min,放置室温,加乙醇定容至刻度,摇匀,0.22μm微孔滤膜滤过,取续滤液,精密吸取5mL,置50mL棕色量瓶中,用稀乙醇稀释至刻度。
5.HPLC色谱条件
Ultimate XB C18色谱柱(250mm×4.6mm,5μm);柱温:30℃;流动相:水(A)-甲醇(B),梯度洗脱,0-60min,B:20%-100%;DAD波长范围200~600nm,检测波长440nm,流速:1.0mL·min-1;进样量:10μL。在该色谱条件下,各色谱峰有较好的基线分离。
6.测定波长的选择
对照品溶液及供试品溶液分别于200-700nm进行波长扫描,发现两者均在440nm处有最大吸收,故确定检测波长为440nm。与西红花苷类成分的最大吸收波长相一致。
7.UV法标准曲线及线性关系考察
分别精密吸取对照品溶液0.1,0.2,0.4,0.6,0.8,1.0mL置于10mL量瓶中,用稀乙醇稀释至刻度,以稀乙醇作空白对照,照紫外-可见分光光度法,于440nm波长处测定吸光度,得回归方程为y=0.1130x+0.0012(r=0.9999)。结果表明,西红花苷-Ⅰ在0.992-9.92μg·mL-1浓度范围内与吸光度呈良好线性关系。
8.UV法测定样品中总西红花苷含量
取32批西红花药材,分别按步骤4下制备供试品溶液,以稀乙醇作空白对照,照紫外-可见分光光度法,于440nm波长处测定吸光度,按标准曲线分别计算总西红花苷含量,测定结果见表1。
9.《中国药典》方法测定西红花苷-Ⅰ和西红花苷-Ⅱ含量
取32批西红花药材,分别按步骤4下制备供试品溶液,按《中国药典》色谱条件进样。32批西红花样品中西红花苷-Ⅰ和西红花苷-Ⅱ总含量的测定结果见表1。
10.一测多评法(QMSA)测定西红花苷-Ⅰ和苷-Ⅱ的含量
取步骤3下对照品溶液,分别进样1、2、4、8、10、15、20μL,记录峰面积,应用多点校正法,以西红花苷-Ⅰ为内参物,计算西红花苷-Ⅰ对西红花苷-Ⅱ的相对校正因子fk/s,fk/s计算公式:fk/s=(Cs×Ak)/(Ck×As)。待测成分(西红花苷-Ⅱ)质量浓度计算公式:Ck′=(Cs×Ak′)/(fk/s×As)。两式中Cs为内参物(西红花苷-Ⅰ)质量浓度,As为内参物色谱峰峰面积,Ck为西红花苷-Ⅱ对照品质量浓度,Ak为西红花苷-Ⅱ对照品色谱峰峰面积,Ck′为西红花样品中西红花苷-Ⅱ质量浓度,Ak′为西红花样品中西红花苷-Ⅱ色谱峰峰面积。
取32批西红花药材,分别按步骤4下制备供试品溶液,按步骤5下色谱条件进样。32批西红花样品中西红花苷-Ⅰ和西红花苷-Ⅱ总含量的测定结果见表1。
11.面积归一化法测定总西红花苷的含量
取32批西红花药材,分别按步骤4下制备供试品溶液,按步骤5下色谱条件进样。以西红花苷-Ⅰ为对照,按C总苷=(C苷-Ⅰ×A总苷)/A苷-Ⅰ计算总西红花苷的含量。式中A苷-Ⅰ为样品中西红花苷-Ⅰ色谱峰峰面积;A总苷为西红花样品440nm下所有色谱峰峰面积之和;C苷-Ⅰ为外标法测定的西红花样品中西红花苷-Ⅰ的质量浓度;C总苷为西红花样品中总西红花苷质量浓度。32批西红花样品中西红花总苷含量的测定结果见表1。
表1 ESM与QAMS测定的西红花中两种西红花苷类成分含量及ANM与UV测定总西红花苷含量结果(%,n=3)
西红花苷入血后以苷元西红花酸起效,因此以西红花药材总苷含量评价其质量更准确。中国药典(2015版)采用HPLC法测定西红花苷-Ⅰ和苷-Ⅱ含量,未测其它苷类成分面积归一化法含量,是基于对照品不易获得和测定时间不能太长的考虑;本发明采用面积归一化法测定的总西红花苷含量仅需使用西红花苷-Ⅰ一种对照品,测定时间在60min内有11个西红花苷类成分得到分离,32批国内外西红花样品测定结果,面积归一化法与已发表的UV法测定的总西红花苷含量无显著性差异,说明面积归一化法可用于总西红花苷含量的测定。本申请中国内样品为收购产地新鲜西红花花丝后同一厂家干燥加工生产,国外样品也为著名的大公司出品,经HPLC分析均不含掺假品,因此使用UV法测定更简便,但对于可能掺假产品则必须使用HPLC采用面积归一化法测定西红花总苷。
中国药典(2015版)为现行法定标准,要求西红花苷-Ⅰ和苷-Ⅱ的总量大于10%为合格品,但西红花苷-Ⅱ标准品较贵,而两者的苷元均是西红花酸,为脱辅基类胡萝卜类化合物,产生紫外吸收的官能团相同,均在440nm有最大吸收,为一测多评法的建立提供了最有利内源基础。本申请中不同仪器、不同色谱柱测得的西红花苷-Ⅱ的相对校正因子、相对保留时间的RSD均小于5%,符合一测多评法要求。采用中国药典外标法测定的西红花苷-Ⅱ含量与通过一测多评法计算得到的含量基本一致,以西红花苷-Ⅰ为内参物,苷-Ⅱ的相对校正因子为1.18,有较好的重现性和稳定性,因此使用一测多评法测定西红花苷-Ⅰ和苷-Ⅱ的总量可用于判别西红花药材是否合格,降低检测成本。
面积归一化法计算总西红花苷与一测多评计算西红花苷-Ⅰ和苷-Ⅱ含量两种方法结合,实现了一个对照品完成各西红花苷的含量测定,方法简便,结果可靠,样品中各西红花苷组分间比例量的差异,可以在一定程度上反映西红花生长发育,采收加工,储存等过程中西红花苷类成分的转化特点,为西红花生产加工及流通过程中提高西红花质量提供了更好的技术参考。
上述说明示出并描述了本发明的若干优选实施例,但如前所述,应当理解本发明并非局限于本申请中所披露的形式,不应看作是对其他实施例的排除,而可用于各种其他组合、修改和环境,并能够在本申请中所述发明构想范围内,通过上述教导或相关领域的技术或知识进行改动。而本领域人员所进行的改动和变化不脱离本发明的精神和范围,则都应在本发明所附权利要求的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!