一种同时测定玛咖中11种玛咖酰胺含量的方法与流程
本发明属于分析化学技术领域,具体涉及一种同时测定玛咖中11种玛咖酰胺含量的方法。
背景技术:
玛咖(Lepidium meyenii)为十字花科独行菜属的一种一年生草本植物,最早生长在海拔超过4000米的秘鲁中央的安第斯山区,具有2000年的食用历史。玛咖的根茎部分比地上部分具有更高的营养价值,根形似小圆萝卜,外形颜色有紫、黑、黄等。研究表明玛咖具有丰富的营养价值和药用价值,玛咖还具有许多传统的疗效,如作为泻药、治疗风湿病、呼吸道疾病、痛经及女性更年期综合征等。另外,玛咖还具有改善性功能、提高人畜生育能力、缓解疲劳及抗氧化等功能。基于以上特点,玛咖被称为“秘鲁人参”。玛咖中的玛咖酰胺被视为主要的活性功能成分,是评价玛咖质量的重要标准。
目前对玛咖酰胺的检测方法很多,高效液相色谱(high performance liquid chromatography,HPLC)技术应用于玛咖酰胺的含量检测也有报道,但已有报道的方法中同时测定玛咖酰胺的种类较少。
技术实现要素:
本发明提供的采用HPLC法能同时测定玛咖中11种玛咖酰胺的含量,并利用建立的方法测定了20个不同样品的含量,检测结果准确可靠,简便快捷,是一种分析玛咖酰胺类成分的有效途径,可为玛咖中玛咖酰胺的开发利用提供理论依据,为玛咖品种选育及优良品种推广奠定理论基础。
本发明的技术方案如下:一种同时测定玛咖中11种玛咖酰胺含量的方法,包括以下几个步骤:
(1)待测溶液的制备
粉碎,过筛,精密称取玛咖粉末,以1g:10mL料液比加入石油醚,超声提取30min,过滤,滤渣再重复提取两次,合并滤液,浓缩,用甲醇定容制得待测溶液;
(2)对照品混合液的制备
分别精密称取N-(间-甲氧基苄基)-亚麻酰胺、N-苄基亚麻酰胺、N-(间甲氧基苄基)-亚油酰胺、N-苄基亚油酰胺、N-苄基十五碳酰胺、N-(间-甲氧基苄基)-十六碳酰胺、N-苄基十六烷酰胺、N-苄基-9Z-十八碳烯酰胺、N-苄基十七烷酰胺、N-(间-甲氧基苄基)-十八碳酰胺、N-苄基十八烷酰胺对照品,用甲醇分别溶解后混合,配制成多组对照品混合液,所述对照品混合液中各对照品的质量浓度介于0.5~1000μg/mL范围内;
(3)标准曲线的建立
分别吸取步骤(2)中多组对照品混合溶液,进行液相色谱测定;
色谱条件为:色谱柱Xtimate XB-C18色谱柱4.6×250mm,5μm;流动相:流动相A为水、流动相B为乙腈;梯度:0~24min,80%流动相B~100%流动相B;检测器G4212-60008二极管阵列检测器,检测波长210nm、280nm;流速0.8mL/min;柱温40℃;进样量10μL;
以步骤(2)中各对照品的质量浓度为横坐标,峰面积为纵坐标做标准曲线;
(4)待测溶液中玛咖酰胺含量的测定
按步骤(3)的色谱条件对待测溶液进行测定,并利用标准曲线计算待测样品中每种玛咖酰胺的含量。
进一步的,步骤(3)中所述的多组对照品混合溶液中各玛咖酰胺的浓度范围分别为:N-(间-甲氧基苄基)-亚麻酰胺0.5~35μg/mL、N-苄基亚麻酰胺5~300μg/mL、N-(间甲氧基苄基)-亚油酰胺0.5~100μg/mL、N-苄基亚油酰胺4~1000μg/mL、N-苄基十五碳酰胺1~30μg/mL、N-(间-甲氧基苄基)-十六碳酰胺5~50μg/mL、N-苄基十六烷酰胺1~1000μg/mL、N-苄基-9Z-十八碳烯酰胺5~150μg/mL、N-苄基十七烷酰胺1~40μg/mL、N-(间-甲氧基苄基)-十八碳酰胺1~10μg/mL、N-苄基十八烷酰胺5~60μg/mL。
进一步的,该方法对11种玛咖酰胺的检出限如下:
N-(间甲氧基苄基)-亚麻酰胺:2.57;
N-苄基亚麻酰胺:1.46;
N-(间甲氧基苄基)-亚油酰胺:2.06;
N-苄基亚油酰胺:1.64;
N-苄基十五碳酰胺:10.35;
N-(间甲氧基苄基)-十六碳酰胺:2.91;
N-苄基十六烷酰胺:2.82;
N-苄基-9Z-十八碳烯酰胺:1.29;
N-苄基十七烷酰胺:7.85;
N-(间甲氧基苄基)-十八碳酰胺:4.93;
N-苄基十八烷酰胺:3.41。
本发明的有益效果如下:
(1)本发明的提取方法,解决了现有的回流或索氏提取等方法的操作麻烦、提取率低、成本高、提取时间较长的问题;
(2)本发明的检测方法为梯度洗脱方法,分离度好,可以同时检测玛咖中11种玛咖酰胺;本发明只用乙腈和水作为流动相,解决了三氟乙酸对色谱柱损耗大、清洗麻烦等问题,流动相配置方便,简便了操作;
(3)本发明的方法线性关系良好、精密度高、重复性好、稳定性好、回收率高、简便、快捷并且准确可行,可作为玛咖中主要玛咖酰胺标准的定量检测方法。
附图说明
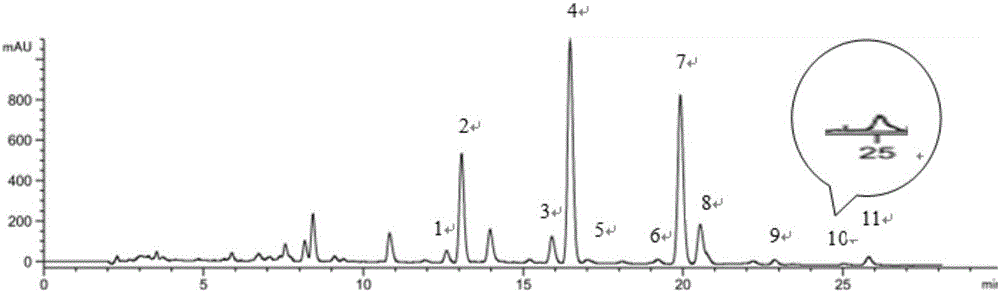
图1是实施例1中玛咖酰胺对照品混合液的液相色谱图;
图2是实施例1中S18拉萨紫玛咖样品的液相色谱图。
其中:1为编号C1,2为编号C2,3为编号C3,4为编号C4,5为编号C5,6为编号C6,7为编号C7,8为编号C8,9为编号C9,10为编号C10,11为编号C11。
具体实施方式
下面结合具体实施例对本发明的技术方案作进一步的说明,但本发明不以任何形式受限于实施例内容。实施例中所述试验方法如无特殊说明,均为常规方法;如无特殊说明,所述试剂和生物材料,均可从商业途径获得。
实施例1
本实施例选择编号为S1-S20的20种玛咖作为待测样品进行测定,具体步骤如下:
(1)待测溶液的制备
粉碎待测样品,过筛,精密称取5.0g玛咖粉末,加入50mL石油醚,超声提取30min,过滤,滤渣以上述方法再提取两次,合并滤液,浓缩,用甲醇定容至10mL,待HPLC定量分析。
(2)对照品混合液的制备
分别精密称取N-(间-甲氧基苄基)-亚麻酰胺、N-苄基亚麻酰胺、N-(间甲氧基苄基)-亚油酰胺、N-苄基亚油酰胺、N-苄基十五碳酰胺、N-(间-甲氧基苄基)-十六碳酰胺、N-苄基十六烷酰胺、N-苄基-9Z-十八碳烯酰胺、N-苄基十七烷酰胺、N-(间-甲氧基苄基)-十八碳酰胺、N-苄基十八烷酰胺对照品,用甲醇分别溶解后混合,配制成六组对照品混合液,所述对照品混合液中各对照品的质量浓度介于0.5~1000μg/mL范围内;各对照品的浓度具体如下表1:
表1
(3)标准曲线的建立
分别吸取步骤(2)中多组对照品混合溶液,进行液相色谱测定;
色谱条件为:色谱柱Xtimate XB-C18色谱柱4.6×250mm,5μm;流动相:流动相A为水和流动相B为乙腈;梯度:0~24min,80%流动相B~100%流动相B;检测器G4212-60008二极管阵列检测器,检测波长210nm、280nm;流速0.8mL/min;柱温40℃;进样量10μL;
以步骤(2)中各对照品的质量浓度为横坐标,峰面积为纵坐标做标准曲线;按信噪比3:1确定检测限,标准曲线方程及检出限见表2;
表2 11种玛咖酰胺对照品标准曲线方程
(4)待测溶液中玛咖酰胺含量的测定
色谱条件为:色谱柱Xtimate XB-C18色谱柱(4.6×250mm,5μm);流动相为A(水)和B(乙腈),梯度:0~24min,80%B~100%B;检测器G4212-60008二极管阵列检测器,检测波长210nm、280nm;流速0.8mL/min;柱温40℃;进样量10μL,并利用标准曲线计算待测样品中每种玛咖酰胺的含量。结果如下表3:
表3玛咖中玛咖酰胺的含量(单位:μg/g)
实施例2
(1)精密度实验
取玛咖酰胺对照品溶液,上述色谱条件连续进样3次,计算各玛咖酰胺峰面积的相对标准偏差(Relative Standard Deviation,RSD),结果所有玛咖酰胺的RSD值在0.94%~4.73%之间,表明所用仪器精密度良好。结果如表4所示。
表4精密度实验结果(n=3)
(2)重复性实验
取S18样品(产于拉萨的紫玛咖)提取溶液,上述色谱条件连续进样3次,计算各玛咖酰胺含量的相对标准偏差,结果所有玛咖酰胺的RSD值在0.42%~4.42%之间,表明该方法重复性良好。结果如表5所示。
表5重复性实验结果(n=3)
(3)稳定性实验
取同一玛咖样品提取液,按上述色谱条件,分别在放置0、2、4、8和12h后进样分析,计算含量,计算样品各玛咖酰胺的RSD,结果其RSD值在0.47%~4.40%之间,表明供试品溶液在室温条件下放置12h稳定性良好。结果如表6所示。
表6稳定性实验结果(n=5)
(4)加样回收率实验
分别取9份相同重量的已知各玛咖酰胺含量的玛咖样品按上述方法制备玛咖酰胺提取液,分别加入不同含量的玛咖酰胺对照品溶液,完全混匀后经过0.45μm微孔滤膜过滤,进样10μL。各玛咖酰胺的平均回收率在98.23%~100.82%之间,表明方法准确率高,结果见表7。
表7加样回收率实验结果(n=9)
综上所述,本发明的方法线性关系良好、精密度高、重复性好、稳定性好、回收率高、简便、快捷并且准确可行,可作为玛咖中主要玛咖酰胺标准的定量检测方法。
- 还没有人留言评论。精彩留言会获得点赞!