一种清肺消炎丸质量标准检测方法与流程
本发明涉及属于中药技术领域,具体是一种清肺消炎丸质量标准检测方法。
背景技术:
清肺消炎丸是一种传统的中药制剂,它是由麻黄、石膏、地龙、牛蒡子、葶苈子、人工牛黄、苦杏仁(炒)、羚羊角组成。主要用于清肺化痰,止咳平喘。用于痰热阻肺,咳嗽气喘,胸胁胀痛,吐痰黄稠,上呼吸道感染、急性支气管炎和慢性支气管炎的急性发作,以及肺部感染引起的咳嗽痰稠、喘息气急等病症。
清肺消炎丸的水丸为天津中新药业集团股份有限公司达仁堂制药厂生产的药品,其质量标准收载于国家食品药品监督管理局标准ybz00662010。原标准麻黄含量测定项下的测定方法为高效液相色谱法,该方法采用蒸馏方法提取,步骤繁琐、周期长,且过程不好控制;而且仅测定盐酸麻黄碱的含量,《中国药典》麻黄药材已经对盐酸麻黄碱和盐酸伪麻黄碱的总量进行了控制。
清肺消炎丸的水蜜丸为天津中新药业集团股份有限公司达仁堂制药厂生产的药品,其质量标准收载于《中国药典》2015年版。原标准麻黄含量测定项下的测定方法为薄层扫描法,该方法提取步骤繁琐、周期长、检测方法精度低,而且仅测定盐酸麻黄碱的含量,《中国药典》麻黄药材已经对盐酸麻黄碱和盐酸伪麻黄碱的总量进行了控制。
为了简化提取步骤,提高检测方法精度,并且将清肺消炎丸水丸与水蜜丸的含量检测控制方法进行统一,天津中新药业集团股份有限公司达仁堂制药厂开展研究制定了方法,此方法不仅能适用于清肺消炎丸的水丸也适用于水蜜丸,检测起来更加方便,快捷。
技术实现要素:
有鉴于此,本发明旨在提供一种清肺消炎丸质量标准检测方法。
为达到上述目的,本发明的技术方案是这样实现的:一种清肺消炎丸质量标准检测方法,包括如下步骤:
(1)对照样品溶液的制备:取盐酸麻黄碱、盐酸伪麻黄碱对照品适量,精密称定,加甲醇制成每1ml含盐酸麻黄碱、盐酸伪麻黄碱各40μg的混合溶液,即得;
(2)供试样品溶液的制备:取样品适量,研细,取约2.0g,精密称定,精密加入甲醇25ml,称定重量,超声处理,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得;
(3)阴性样品溶液的制备:按处方称取除麻黄的各味药材,按处方工艺制得样品,再按照所述步骤(2)供试样品溶液的制备方法,制得阴性样品溶液;
(4)分别精密吸取供试样品溶液、对照品溶液和阴性样品溶液注入高效液相色谱仪进行测定,即得。
进一步地,所述步骤(4)的色谱条件如下:
色谱柱:phenomenexlunac18,250mm×4.6mm,5μm;
流动相:乙腈-0.02mol/l磷酸二氢钾溶液,两者体积比为4:96;
柱温:40℃;
流速:1.0ml/min;
检测波长:210nm;
理论板数按盐酸麻黄碱峰计算应不低于8000。
进一步地,所述步骤(4)中的样品为清肺消炎丸的水丸或水蜜丸。
进一步地,所述步骤(4)中注入高效液相色谱的溶液为5~10ul。
进一步地,所述流动相0.02mol/l磷酸二氢钾溶液中含0.2%三乙胺,用磷酸调节至ph2.7。
进一步地,所述样品每1g含麻黄以盐酸麻黄碱和盐酸伪麻黄碱的总量计,水丸不得少于0.56mg,水蜜丸不得少于0.34mg。
相对于现有技术,本发明具有以下优势:
(1)本发明中化学成分复杂,实现了盐酸麻黄碱与盐酸伪麻黄碱的有效分离,并有效排除了其他杂质的干扰,稳定性好、精密度高、重现性好、便捷且易于掌握。
(2)本发明重新制定了本品的含量限度为“每1g含麻黄以盐酸麻黄碱和盐酸伪麻黄碱的总量计,水丸不得少于0.56mg,水蜜丸不得少于0.34mg”。
(3)本发明简化提取步骤,提高检测方法精度,并且将清肺消炎丸的水丸与水蜜丸的含量检测控制方法进行了统一,此方法不仅能适用于清肺消炎丸的水丸也适用于水蜜丸,检测起来更加方便,快捷。
附图说明
图1为清肺消炎丸(水丸)对照品溶液色谱图(菲罗门色谱柱)。
图2为清肺消炎丸(水丸)供试样品溶液色谱图(菲罗门色谱柱)。
图3为清肺消炎丸(水丸)阴性样品溶液色谱图(菲罗门色谱柱)。
图4为清肺消炎丸(水丸)对照品溶液色谱图(迪马色谱柱)。
图5为清肺消炎丸(水丸)供试品溶液色谱图(迪马色谱柱)。
图6为清肺消炎丸(水丸)对照品溶液色谱图(资生堂色谱柱)。
图7为清肺消炎丸(水丸)供试品溶液色谱图(资生堂色谱柱)。
图8为清肺消炎丸(水蜜丸)对照品溶液色谱图(菲罗门色谱柱)。
图9为清肺消炎丸(水蜜丸)供试样品溶液色谱图(菲罗门色谱柱)。
图10为清肺消炎丸(水蜜丸)阴性样品溶液色谱图(菲罗门色谱柱)。
图11为清肺消炎丸(水蜜丸)对照品溶液色谱图(迪马色谱柱)。
图12为清肺消炎丸(水蜜丸)供试品溶液色谱图(迪马色谱柱)。
图13为清肺消炎丸(水蜜丸)对照品溶液色谱图(资生堂色谱柱)。
图14为清肺消炎丸(水蜜丸)供试品溶液色谱图(资生堂色谱柱)。
具体实施方式
实施例一清肺消炎丸(水丸)质量标准检测方法
1.1仪器和试剂
仪器:agilent1200高效液相色谱仪。
试剂:乙腈(色谱纯,merck公司),甲醇(色谱纯,天津市康科德科技有限公司),磷酸(分析纯,天津市赢达稀贵化学试剂厂),磷酸二氢钾(分析纯,天津市赢达稀贵化学试剂厂),三乙胺(分析纯,天津市康科德科技有限公司),氨水(分析纯,天津市赢达稀贵化学试剂厂),水为去离子水。
对照品:盐酸麻黄碱(中国食品药品检定研究院购置,批号:171241-201007,供含量测定用,纯度为99.7%),盐酸伪麻黄碱(中国食品药品检定研究院购置,批号:171237-201208,供含量测定用,纯度为99.9%)。
样品(水丸):天津中新药业集团股份有限公司达仁堂制药厂(批号:9008789、9008790、9008791、9008877、9008878、9008879)。
1.2对照样品溶液的制备:取盐酸麻黄碱、盐酸伪麻黄碱对照品适量,精密称定,加甲醇制成每1ml含盐酸麻黄碱40.96μg、盐酸伪麻黄碱各41.44μg的混合溶液,进样,测定,即得,出峰顺序依次为:盐酸麻黄碱、盐酸伪麻黄碱,结果见图1。
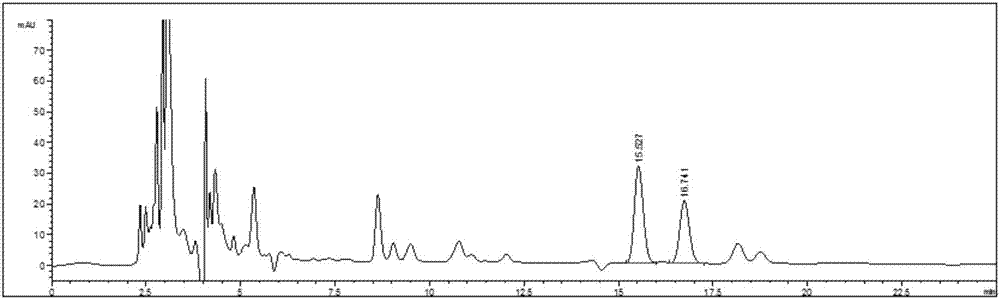
1.3供试样品溶液的制备:取同一批号(9008789)样品适量,研细,取约2.0g,精密称定,精密加入甲醇25ml,称定重量,超声处理45min(功率500w,频率40khz),放冷,再称定重量,用甲醇补足减失的重量,摇匀,用0.45μm微孔滤膜滤过,取续滤液,进样,测定,即得,出峰顺序依次为:盐酸麻黄碱、盐酸伪麻黄碱,结果见图2。
1.3.1提取方式的确定
(1)超声提取:取同一批号(9008789)样品适量,研细,取2.0g,精密称定,精密加入甲醇25ml,称定重量,超声处理45min(功率500w,频率40khz),放冷,再称定重量,用甲醇补足减失的重量,摇匀,用0.45μm微孔滤膜滤过,取续滤液,进样,测定,即得,结果见表1-1。
(2)回流提取:取同一批号(9008789)样品适量,研细,取2.0g,精密称定,精密加入甲醇25ml,称定重量,置水浴上加热回流45min,放冷,再称定重量,加甲醇补足减失的重量,摇匀,用0.45μm微孔滤膜滤过,取续滤液,进样,测定,即得,结果见表1-1。
表1-1提取方式的比较
结论:采用超声提取与加热回流提取方式,测得样品中盐酸麻黄碱与盐酸伪麻黄碱的总量差别不大,考虑到超声提取方法简单,因此采用超声提取。
1.3.2提取时间的考察
取同一批号(9008789)样品适量,研细,取2.0g,精密称定,分别精密加入甲醇25ml,称定重量,分别超声提取15、30、45、60、90min(功率500w,频率40khz),放冷,再称定重量,用甲醇补足减失的重量,摇匀,用0.45μm微孔滤膜滤过,取续滤液,进样,测定,即得,结果见表1-2。
表1-2提取时间的比较
结论:超声提取45min以后,样品中盐酸麻黄碱和盐酸伪麻黄碱的总量基本不变,因此选择提取时间为45min。
1.3.3提取溶剂的选择
取同一批号(9008789)样品适量,研细,取2.0g,精密称定,分别精密加入甲醇、70%甲醇、50%甲醇、水、乙醇各25ml,称定重量,分别超声提取45min(功率500w,频率40khz),放冷,再称定重量,分别用各自的溶剂补足减失的重量,摇匀,用0.45μm微孔滤膜滤过,取续滤液,进样,测定,即得,结果见表1-3。
表1-3提取溶剂的比较
结论:计算样品中盐酸麻黄碱和盐酸伪麻黄碱的总量,结果以甲醇、70%甲醇和50%作为溶剂提取的总量基本一致;同时,甲醇提取液供试品色谱中杂质较少,因此选择甲醇为提取溶剂。
1.3.4提取溶剂量的考察
取同一批号(9008789)样品适量,研细,取2.0g,精密称定,分别精密加入甲醇15ml、25ml、50ml和100ml,称定重量,超声提取45min(功率500w,频率40khz),放冷,再称定重量,用甲醇补足减失的重量,摇匀,用0.45μm微孔滤膜滤过,取续滤液,进样,测定,即得,结果见表1-4。
表1-4提取溶剂量的比较
结论:提取溶剂量为15ml时测得的总量偏低,以后随着提取溶剂量的增加,测得盐酸麻黄碱和盐酸伪麻黄碱的总量没有明显差别,因此提取溶剂量确定为25ml。
1.4阴性样品溶液的制备:按处方称取除麻黄的各味药材,按处方工艺制得样品,再按照1.3供试样品溶液的制备方法,制得阴性样品溶液,进样,测定,即得,出峰顺序依次为:盐酸麻黄碱、盐酸伪麻黄碱,结果见图3。
1.5色谱条件
色谱柱:phenomenexlunac18,250mm×4.6mm,5μm;
流动相:乙腈-0.02mol/l磷酸二氢钾溶液,两者体积比为4:96;
柱温:40℃;
流速:1.0ml/min;
检测波长:210nm;
理论板数按盐酸麻黄碱峰计算应不低于8000。
1.5.1流动相的选择
采用1.5项下的色谱条件进行测定,样品中盐酸麻黄碱和盐酸伪麻黄碱色谱峰与杂质峰分离较好,出峰顺序依次为:盐酸麻黄碱、盐酸伪麻黄碱,见图1-2。
1.5.2不同色谱柱的比较
三个厂家生产的色谱柱(菲罗门,资生堂,迪马),采用1.5项下的流动相,样品分离效果均较好,出峰顺序依次为:盐酸麻黄碱、盐酸伪麻黄碱,见图1、2、4、5、6、7。
1.6标准曲线的制备
1.6.1盐酸麻黄碱标准曲线
取盐酸麻黄碱对照品适量,精密称定,加甲醇制成浓度为每1ml中分别含盐酸麻黄碱10.1345、20.2690、30.4035、40.5380、81.0760、121.6141μg的对照品溶液,分别精密吸取5μl,注入液相色谱仪,按照1.5色谱条件分析,分别测定各自峰面积,以对照品进样的量(μg)为横坐标,峰面积值为纵坐标,求得回归方程,结果见表1-5
表1-5盐酸麻黄碱标准曲线
结论:求得盐酸麻黄碱回归方程为y=2160.6x+1.5568,r=0.9999,结果表明盐酸麻黄碱在0.050675~0.608100μg范围内线性良好。
1.6.2盐酸伪麻黄碱标准曲线:
取盐酸伪麻黄碱对照品适量,精密称定,加甲醇制成浓度为每1ml中分别含盐酸伪麻黄碱10.3047、20.6094、30.9141、41.2187、82.4375、123.6562μg的对照品溶液,分别精密吸取5μl,注入液相色谱仪,按照1.5色谱条件分析,分别测定各自峰面积,以对照品进样的量(μg)为横坐标,峰面积值为纵坐标,求得回归方程,结果见表1-6。
表1-6盐酸伪麻黄碱标准曲线
结论:求得盐酸伪麻黄碱回归方程为y=2182.7x+1.1705,r=0.9999,结果表明盐酸伪麻黄碱在0.051525~0.618300μg范围内线性良好。
1.6.3进样重复性试验
取同一批号(9008789)适量,研细,取2.0g,精密称定,精密加入甲醇25ml,按照1.3供试样品溶液制备操作,按照1.5色谱条件分析,连续进样6次,测定盐酸麻黄碱和盐酸伪麻黄碱峰面积,结果见表1-7。
表1-7进样重复性试验
结论:样品中盐酸麻黄碱和盐酸伪麻黄碱峰面积平均值分别为515、357,rsd分别为0.44%、0.29%,均符合要求。
1.6.4重现性试验
取同一批号(9008789)样品适量,研细,取2.0g,精密称定,共6份,分别精密加入甲醇25ml,按照1.3供试样品溶液制备操作,按照1.5色谱条件分析,测定,结果见表1-8。
表1-8重现性试验
结论:样品中盐酸麻黄碱、盐酸伪麻黄碱、以及两者总量的平均值分别0.6047、0.4143、1.0190mg/g,rsd分别为0.44%、0.39%、0.36%,均符合要求。
1.6.5稳定性试验
取同一批号(9008789)样品适量,研细,取2.0g,精密称定,精密加入甲醇25ml,按照1.3供试样品溶液制备操作,按照1.5色谱条件分别在0、4、8、12、18、24h进样,测定,结果见表1-9。
表1-9稳定性试验
结论:样品中盐酸麻黄碱和盐酸伪麻黄碱峰面积平均值分别为518、359,rsd分别为0.29%、0.46%,均符合要求。
1.6.6回收率试验
取同一批号(9008789)样品适量,研细,取1.0g,精密称定,共6份,分别精密加入含有盐酸麻黄碱、盐酸伪麻黄碱浓度分别为24.8851、16.7193μg/ml的甲醇混合对照品溶液25ml,按照1.3供试样品溶液制备操作,制得供回收率用供试样品溶液,按照1.5色谱条件分析,结果见表1-10、1-11。
表1-10盐酸麻黄碱回收率试验
表1-11盐酸伪麻黄碱回收率试验
结论:计算回收率,结果盐酸麻黄碱、盐酸伪麻黄碱平均回收率分别为100.04%、100.15%,rsd分别为0.50%、0.52%,结果均符合规定。
1.7样品测定
取6个不同批号的样品,按照1.3供试样品溶液制备操作,按照1.5色谱条件分析,测定,计算样品中盐酸麻黄碱、盐酸伪麻黄碱的含量,结果见表1-12。
表1-12样品含量测定结果
1.8不同色谱柱对含量测定的影响
取批号为9008789的供试样品溶液,分别采用菲罗门phenomenexlunac18(2)色谱柱(见图1-2),迪马diamonsil-c18色谱柱(见图1-5),资生堂capcellpak-c18色谱柱(见图1-7),用agilent1200高效液相色谱仪,按照1.5色谱条件分析,测定,出峰顺序依次为:盐酸麻黄碱、盐酸伪麻黄碱,计算样品中盐酸麻黄碱和盐酸伪麻黄碱的含量,结果见表1-13。
表1-13不同色谱柱对含量测定的影响
结论:结果表明不同色谱柱含量测定结果基本一致。
1.9含量限度的制定
取多批次清肺消炎丸样品,按照1.3供试样品溶液制备操作,按照1.5色谱条件分析,测定并计算样品中盐酸麻黄碱、盐酸伪麻黄碱的含量。根据对应使用的麻黄药材含量,计算在清肺消炎丸(水丸)实际生产过程指标成分的转移率,结果见表1-14。
表1-14六批清肺水丸样品含量测定结果
结论:批转移率计算结果表明,在清肺消炎丸水丸生产过程,盐酸麻黄碱和盐酸伪麻黄碱总量的转移率在63.2%~75.2%之间,平均转移率达69.4%。清肺消炎丸水丸的法定生产工艺:八味药材,除人工牛黄外,羚羊角粉碎成极细粉,其余麻黄等六味粉碎成细粉,与羚羊角及人工牛黄粉末配研,混匀,过筛。用水泛丸,制成水丸,干燥,即得。生产工艺流程主要包括药材粉碎、混合、灭菌、泛丸、整形、干燥等工序。根据对实际生产过程的观察,分析指标成分转移率水平主要与麻黄药材检验时的取样代表性、粉碎过程中损失及干燥过程时间较长(干燥温度55℃,时间为72小时以上)等因素有关。
根据本品处方量和制法,经折算,本品每1g含麻黄药材100mg,本品每1g样品理论上含盐酸麻黄碱和盐酸伪麻黄碱的总量应为0.8mg。根据中国药典2015年版一部麻黄饮片项下含盐酸麻黄碱和盐酸伪麻黄碱的总量不得低于0.80%的要求,结合实际生产过程中70%的转移率情况计算,暂定本品的含量限度为“每1g含麻黄以盐酸麻黄碱(c10h15no·hcl)和盐酸伪麻黄碱(c10h15no·hcl)的总量计,不得少于0.56mg”。
实施例二清肺消炎丸(水蜜丸)质量标准检测方法
2.1仪器和试剂
仪器:agilent1200高效液相色谱仪。
试剂:乙腈(色谱纯,merck公司),甲醇(色谱纯,天津市康科德科技有限公司),磷酸(分析纯,天津市赢达稀贵化学试剂厂),磷酸二氢钾(分析纯,天津市赢达稀贵化学试剂厂),三乙胺(分析纯,天津市康科德科技有限公司),氨水(分析纯,天津市赢达稀贵化学试剂厂),水为去离子水。
对照品:盐酸麻黄碱(中国食品药品检定研究院购置,批号:171241-201007,供含量测定用,纯度为99.7%),盐酸伪麻黄碱(中国食品药品检定研究院购置,批号:171237-201208,供含量测定用,纯度为99.9%)。
样品(水蜜丸):天津中新药业集团股份有限公司达仁堂制药厂(批号:9210036、7870171、7870172、8370001、8370002、8510003、8510004、8510005、9210060、9210061、9210062、9210072、9210073、9210074)。
2.2对照样品溶液的制备:取盐酸麻黄碱、盐酸伪麻黄碱对照品适量,精密称定,加甲醇制成每1ml含盐酸麻黄碱40.54μg、盐酸伪麻黄碱各41.22μg的混合溶液,进样,测定,即得,出峰顺序依次为:盐酸麻黄碱、盐酸伪麻黄碱,结果见图8。
2.3供试样品溶液的制备:取同一批号(9210036)样品适量,研细,取约2.0g,精密称定,精密加入甲醇25ml,称定重量,超声处理30min(功率500w,频率40khz),放冷,再称定重量,用甲醇补足减失的重量,摇匀,用0.45μm微孔滤膜滤过,取续滤液,进样,测定,即得,出峰顺序依次为:盐酸麻黄碱、盐酸伪麻黄碱,结果见图9。
2.3.1提取方式的确定
(1)超声提取:取同一批号(9210036)样品适量,研细,取2.0g,精密称定,精密加入甲醇25ml,称定重量,超声处理30min(功率500w,频率40khz),放冷,再称定重量,用甲醇补足减失的重量,摇匀,用0.45μm微孔滤膜滤过,取续滤液,进样,测定,即得,结果见表2-1。
(2)回流提取:取同一批号(9210036)样品适量,研细,取2.0g,精密称定,精密加入甲醇25ml,称定重量,置水浴上加热回流30min,放冷,再称定重量,加甲醇补足减失的重量,摇匀,用0.45μm微孔滤膜滤过,取续滤液,进样,测定,即得,结果见表2-1。
表2-1提取方式的比较
结论:采用超声提取与加热回流提取方式,测得样品中盐酸麻黄碱与盐酸伪麻黄碱的总量差别不大,考虑到超声提取方法简单,因此采用超声提取。
2.3.2提取时间的考察
取同一批号(9210036)样品适量,研细,取2.0g,精密称定,分别精密加入甲醇25ml,称定重量,分别超声提取15、30、45、60、90min(功率500w,频率40khz),放冷,再称定重量,用甲醇补足减失的重量,摇匀,用0.45μm微孔滤膜滤过,取续滤液,进样,测定,即得,结果见表2-2。
表2-2提取时间的比较
结论:超声提取45min,样品中盐酸麻黄碱和盐酸伪麻黄碱的总量最高,因此选择提取时间为45min。
2.3.3提取溶剂的选择
取同一批号(9210036)样品适量,研细,取2.0g,精密称定,分别精密加入甲醇、氨水-甲醇-乙酸乙酯(4:10:90)、氨水-甲醇-乙酸乙酯(4:50:50)、氨水-甲醇-乙酸乙酯(4:80:20)各25ml,以及先用3ml氨水润湿再加精密加入乙酸乙酯25ml,称定重量,分别超声提取30min(功率500w,频率40khz),放冷,再称定重量,分别用各自的溶剂补足减失的重量,摇匀,用0.45μm微孔滤膜滤过,取续滤液,进样,测定,即得,结果见表2-3。
表2-3提取溶剂的比较
结论:计算样品中盐酸麻黄碱和盐酸伪麻黄碱的总量,结果以甲醇作为溶剂较好因此选择甲醇为提取溶剂。
2.3.4提取溶剂量的考察
取同一批号(9210036)样品适量,研细,取2.0g,精密称定,分别精密加入甲醇25ml、50ml和100ml,称定重量,超声提取30min(功率500w,频率40khz),放冷,再称定重量,用甲醇补足减失的重量,摇匀,用0.45μm微孔滤膜滤过,取续滤液,进样,测定,即得,结果见表2-4。
表2-4提取溶剂量的比较
结论:随着提取溶剂量的增加,测得盐酸麻黄碱和盐酸伪麻黄碱的总量没有明显差别,因此提取溶剂量确定为25ml。
2.4阴性样品溶液的制备:按处方称取除麻黄的各味药材,按处方工艺制得样品,再按照2.3供试样品溶液的制备方法,制得阴性样品溶液,进样,测定,即得,出峰顺序依次为:盐酸麻黄碱、盐酸伪麻黄碱,结果见图10。
2.5色谱条件
色谱柱:phenomenexlunac18,250mm×4.6mm,5μm;
流动相:乙腈-0.02mol/l磷酸二氢钾溶液,两者体积比为4:96;
柱温:40℃;
流速:1.0ml/min;
检测波长:210nm;
理论板数按盐酸麻黄碱峰计算应不低于8000。
2.5.1流动相的选择
采用2.5项下的色谱条件进行测定,样品中盐酸麻黄碱和盐酸伪麻黄碱色谱峰与杂质峰分离较好,出峰顺序依次为:盐酸麻黄碱、盐酸伪麻黄碱,见图9。
2.5.2不同色谱柱的比较
三个厂家生产的色谱柱(菲罗门,资生堂,迪马),采用2.5项下的流动相,样品分离效果均较好,出峰顺序依次为:盐酸麻黄碱、盐酸伪麻黄碱,见图8、9、11、12、13、14。
2.6标准曲线的制备
2.6.1盐酸麻黄碱标准曲线
取盐酸麻黄碱对照品适量,精密称定,加甲醇制成浓度为每1ml中分别含盐酸麻黄碱10.1345、20.2690、30.4035、40.5380、81.0760、121.6141μg的对照品溶液,分别精密吸取5μl,注入液相色谱仪,按照2.5色谱条件分析,分别测定各自峰面积,以对照品进样的量(μg)为横坐标,峰面积值为纵坐标,求得回归方程,结果见表2-5
表2-5盐酸麻黄碱标准曲线
结论:求得盐酸麻黄碱回归方程为y=2160.6x+1.5568,r=0.9999,结果表明盐酸麻黄碱在0.050675~0.608100μg范围内线性良好。
2.6.2盐酸伪麻黄碱标准曲线:
取盐酸伪麻黄碱对照品适量,精密称定,加甲醇制成浓度为每1ml中分别含盐酸伪麻黄碱10.3047、20.6094、30.9141、41.2187、82.4375、123.6562μg的对照品溶液,分别精密吸取5μl,注入液相色谱仪,按照1.5色谱条件分析,分别测定各自峰面积,以对照品进样的量(μg)为横坐标,峰面积值为纵坐标,求得回归方程,结果见表2-6。
表2-6盐酸伪麻黄碱标准曲线
结论:求得盐酸伪麻黄碱回归方程为y=2182.7x+1.1705,r=0.9999,结果表明盐酸伪麻黄碱在0.051525~0.618300μg范围内线性良好。
2.6.3进样重复性试验
取同一批号(9210036)适量,研细,取2.0g,精密称定,精密加入甲醇25ml,按照2.3供试样品溶液制备操作,按照2.5色谱条件分析,连续进样6次,测定盐酸麻黄碱和盐酸伪麻黄碱峰面积,结果见表2-7。
表2-7进样重复性试验
结论:样品中盐酸麻黄碱和盐酸伪麻黄碱峰面积平均值分别为359、331,rsd分别为0.38%、0.82%,均符合要求。
2.6.4重现性试验
取同一批号(9210036)样品适量,研细,取2.0g,精密称定,共6份,分别精密加入甲醇25ml,按照2.3供试样品溶液制备操作,按照2.5色谱条件分析,测定,结果见表2-8。
表2-8重现性试验
结论:样品中盐酸麻黄碱、盐酸伪麻黄碱、以及两者总量的平均值分别0.4209、0.3869、0.8078mg/g,rsd分别为0.06%、0.80%、0.39%,均符合要求。
2.6.5稳定性试验
取同一批号(9210036)样品适量,研细,取2.0g,精密称定,精密加入甲醇25ml,按照2.3供试样品溶液制备操作,按照2.5色谱条件分别在0、2、4、8、12、24h进样,测定,结果见表2-9。
表2-9稳定性试验
结论:样品中盐酸麻黄碱和盐酸伪麻黄碱峰面积平均值分别为360、333,rsd分别为0.58%、0.84%,均符合要求。
2.6.6回收率试验
取同一批号(9210036)样品适量,研细,取1.0g,精密称定,共6份,分别精密加入含有盐酸麻黄碱、盐酸伪麻黄碱浓度分别为16.2152、16.4875μg/ml的甲醇混合对照品溶液25ml,按照2.3供试样品溶液制备操作,制得供回收率用供试样品溶液,按照2.5色谱条件分析,结果见表2-10、2-11。
表2-10盐酸麻黄碱回收率试验
表2-11盐酸伪麻黄碱回收率试验
结论:计算回收率,结果盐酸麻黄碱、盐酸伪麻黄碱平均回收率分别为97.82%、98.29%,rsd分别为0.57%、0.64%,结果均符合规定。
2.7样品测定
取11个不同批号的样品,按照2.3供试样品溶液制备操作,按2.5色谱条件分析,测定,计算样品中盐酸麻黄碱、盐酸伪麻黄碱的含量,结果见表2-12。
表2-12样品含量测定结果
2.8不同色谱柱对含量测定的影响
取批号为9210036的供试样品溶液,分别采用菲罗门phenomenexlunac18(2)色谱柱(见图2-3),迪马diamonsil-c18色谱柱(见图2-5),资生堂capcellpak-c18色谱柱(见图2-7),用agilent1200高效液相色谱仪,按照2.5色谱条件分析,测定,出峰顺序依次为:盐酸麻黄碱、盐酸伪麻黄碱,计算样品中盐酸麻黄碱和盐酸伪麻黄碱的含量,结果见表2-13。
表2-13不同色谱柱对含量测定的影响
结论:结果表明不同色谱柱含量测定结果基本一致。
2.9含量限度的制定
取多批次清肺消炎丸样品,按照2.3供试样品溶液制备操作,按照2.5色谱条件分析,测定并计算样品中盐酸麻黄碱、盐酸伪麻黄碱的含量。根据对应使用的麻黄药材含量,计算在清肺消炎丸(水蜜丸)实际生产过程指标成分的转移率,结果见表2-14。
表2-14六批清肺水丸样品含量测定结果
结论:批转移率计算结果表明,在清肺消炎丸水蜜丸生产过程,盐酸麻黄碱和盐酸伪麻黄碱总量的转移率在62%~89%之间,平均转移率达到72.4%。清肺消炎丸(水蜜丸)的法定生产工艺为:八味药材,除人工牛黄外,羚羊角粉碎成极细粉,其余麻黄等六味粉碎成细粉,与羚羊角及人工牛黄粉末配研,混匀,过筛。每100g粉末用炼蜜60~80g加适量水制丸,制成水蜜丸,干燥,即得。生产工艺流程主要包括药材粉碎、混合、灭菌、机制水蜜丸、整形、干燥等工序。根据对实际生产过程的观察,分析指标成分转移率水平主要与麻黄药材检验时的取样代表性、粉碎过程中损失及干燥过程时间较长(干燥温度70-80℃,时间为36小时以上)等因素有关。
根据本品处方量和制法,经折算,本品每1g含麻黄药材60mg,本品每1g样品理论上含盐酸麻黄碱和盐酸伪麻黄碱的总量应为0.48mg。根据中国药典2015年版一部麻黄饮片项下含盐酸麻黄碱和盐酸伪麻黄碱的总量不得低于0.80%的要求,结合实际生产过程中70%的转移率情况计算,暂定本品的含量限度为“每1g含麻黄以盐酸麻黄碱(c10h15no·hcl)和盐酸伪麻黄碱(c10h15no·hcl)的总量计,不得少于0.34mg”。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!