一种大黄的质量评价方法与流程
本发明属于中药领域,具体的涉及到一种大黄的质量评价方法,包括了含量测定、指纹图谱、质谱定性的多指标评价方法。
背景技术:
大黄是中国传统四大中药之一,为蓼科植物掌叶大黄rheumpalmatuml.、唐古特大黄rheumtanguticummaxim.exbalf.、药用大黄rheumofficinalebaill.的根和根茎,具有泻下攻积、清热泻火、凉血解毒、逐瘀通经及利湿退黄的功效。大黄化学成分复杂,主要包括蒽醌类、蒽酮类、二苯乙烯类、鞣质类、色酮类、苯丁酮苷类等各类型化合物,药理学及分子生物学研究说明番泻苷及蒽醌苷类为大黄泻下的主要成分,游离蒽醌类为大黄抑菌、抗肿瘤的有效成分;苯丁酮类成分中的莲花掌苷和异莲花掌苷具有良好的抗炎镇痛作用;鞣质类成分中没食子酰葡萄糖苷和没食子酰原花青素具有降血脂作用,rg-tannin有振奋精神作用,d-儿茶素和没食子酸有止血作用。目前对大黄及其制剂的质量控制,多以蒽醌类化合物的含量为标准,但大黄的药效成分不只是蒽醌类化合物,中药的多成分决定了应采用多指标的质量评价模式。指纹图谱能较全面体现中药成分的整体性,是目前符合中药特点的质量控制模式之一,但其各共有峰代表什么、含量多少,难以说清。近年来国内外对于大黄有较多研究,药材的指纹图谱和多成分含量测定也已见诸报道,但通过建立指纹图谱与多成分含量测定、化学成分定性分析相结合对大黄进行质量控制并以1-o-没食子酰-2-o-肉桂酰-β-d-葡萄糖苷为含量测定指标的研究尚未见报道,因此目前仍缺乏整体的综合评价大黄质量的方法。掌叶大黄是《中华人民共和国药典》中收载的品种且主产地资源丰富,采收较容易,资源易得到保证。
技术实现要素:
本发明主要对大黄进行质量控制研究。通过建立大黄uplc指纹图谱,并结合多成分含量测定和系统的化学成分鉴定,为大黄质量综合评价提供可行方法,并为进一步药效物质基础研究提供依据。
本发明提供一种大黄质量评价方法,包括多成分含量测定、指纹图谱、化学成分定性分析相结合进行质量评价。
本发明提供的大黄质量评价方法,其中多成分含量测定为儿茶素、1-o-没食子酰-2-o-肉桂酰-β-d-葡萄糖苷、大黄素、大黄酚和大黄素甲醚5种成分的含量测定,指纹图谱为大黄药材指纹图谱,化学成分定性分析为采用超高效液相色谱-飞行时间质谱法定性分析。
本发明提供的的大黄质量评价方法,其中多成分含量测定、大黄指纹图谱、化学成分定性分析采用相同的色谱条件,具体包括以下步骤:
(1)供试品溶液的制备:取大黄药材,粉碎,过四号筛,取粉末约0.1-1g,精密称定,置具塞锥形瓶中,精密加入50%-80%甲醇或者甲醇50ml,密塞,称定重量,超声处理10-30min,称重,补足减失的重量,摇匀,即得;
(2)对照品溶液的制备:分别取儿茶素、1-o-没食子酰-2-o-肉桂酰-β-d-葡萄糖苷、大黄素、大黄酚和大黄素甲醚对照品适量,精密称定,加甲醇溶解并稀释成质量浓度分别为0.8820、0.9500、0.0799、0.2390、0.0482mg/ml的对照品溶液;
(3)色谱参数:c18色谱柱,柱温25-40°c,流速0.2-0.4ml·min-1,检测波长270-290nm,进样量0.5-2μl;流动相a为乙腈,b为0.1%甲酸水溶液,梯度洗脱程序为0~30min,4%~25%a;30~45min,25%~70%a;45~50min,70%a;质谱条件为:采用电喷雾离子源,负离子mse模式,雾化气体为高纯度氮气,碰撞气体为高纯度氩气,质量扫描范围m/z100~1500;毛细管电压为2.5kv;锥孔电压为40v;离子源温度为100°c;雾化温度为400°c;载气体积流量为800l/h,碰撞能量20~40v;采用亮氨酸-脑啡肽进行精确质量校正[m-h]-为554.2615。
本发明提供的的大黄质量评价方法,其中步骤(1)供试品溶液的制备:取大黄药材,粉碎,过四号筛,取粉末约0.5g,精密称定,置具塞锥形瓶中,精密加入甲醇50ml,密塞,称定重量,超声处理20min,超声功率250w,频率40khz,放冷,再称定重量,用甲醇补足减失的重量,摇匀,用0.2μm滤膜滤过,取续滤液,即得。
本发明提供的的大黄质量评价方法,其中多成分含量测定、大黄指纹图谱、化学成分定性分析采用相同的色谱条件,具体包括以下步骤:
(1)供试品溶液的制备:取大黄药材,粉碎,过四号筛,取粉末约0.5g,精密称定,置具塞锥形瓶中,精密加入甲醇50ml,密塞,称定重量,超声处理20min,超声功率250w,频率40khz,放冷,再称定重量,用甲醇补足减失的重量,摇匀,用0.2μm滤膜滤过,取续滤液,即得;
(2)对照品溶液的制备:分别取儿茶素、1-o-没食子酰-2-o-肉桂酰-β-d-葡萄糖苷、大黄素、大黄酚和大黄素甲醚对照品适量,精密称定,加甲醇溶解并稀释成质量浓度分别为0.8820、0.9500、0.0799、0.2390、0.0482mg/ml的对照品溶液;
(3)色谱参数:c18色谱柱,柱温25-40°c,流速0.2-0.4ml·min-1,检测波长280nm,进样量0.5-2μl;流动相a为乙腈,b为0.1%甲酸水溶液,梯度洗脱程序为0~30min,4%~25%a;30~45min,25%~70%a;45~50min,70%a;质谱条件为:采用电喷雾离子源,负离子mse模式,雾化气体为高纯度氮气,碰撞气体为高纯度氩气,质量扫描范围m/z100~1500;毛细管电压为2.5kv;锥孔电压为40v;离子源温度为100°c;雾化温度为400°c;载气体积流量为800l/h,碰撞能量20~40v;采用亮氨酸-脑啡肽进行精确质量校正[m-h]-为554.2615。
本发明的大黄质量评价方法,其中多成分含量测定、大黄指纹图谱、化学成分定性分析采用相同的色谱条件,具体包括以下步骤:
(1)供试品溶液的制备:取大黄药材,粉碎,过四号筛,取粉末约0.5g,精密称定,置具塞锥形瓶中,精密加入甲醇50ml,密塞,称定重量,超声处理20min,超声功率250w,频率40khz,放冷,再称定重量,用甲醇补足减失的重量,摇匀,用0.2μm滤膜滤过,取续滤液,即得;
(2)对照品溶液的制备:分别取儿茶素、1-o-没食子酰-2-o-肉桂酰-β-d-葡萄糖苷、大黄素、大黄酚和大黄素甲醚对照品适量,精密称定,加甲醇溶解并稀释成质量浓度分别为0.8820、0.9500、0.0799、0.2390、0.0482mg/ml的对照品溶液;
(3)色谱参数为:c18色谱柱,色谱柱规格为100mm×2.1mm,1.7μm,柱温30°c,流速0.3ml·min-1,检测波长280nm,进样量1μl;流动相a为乙腈,b为0.1%甲酸水溶液,梯度洗脱程序为0~30min,4%~25%a;30~45min,25%~70%a;45~50min,70%a,质谱条件为:采用电喷雾离子源,负离子mse模式,雾化气体为高纯度氮气,碰撞气体为高纯度氩气,质量扫描范围m/z100~1500;毛细管电压为2.5kv;锥孔电压为40v;离子源温度为100°c;雾化温度为400°c;载气体积流量为800l/h,碰撞能量20~40v;采用亮氨酸-脑啡肽进行精确质量校正[m-h]-为554.2615。
为了证实本发明方法的稳定性、准确性、可行性进行了以下实验研究:
1仪器与试药
1.1仪器
watersuplc超高效液相色谱仪(配备样品管理器ftn、四元溶剂管理器、pda检测器),acquityuplc-synaptg2ms色谱-质谱联用仪(waters公司)milli-q超纯水净化系统(美国millipore公司);kq250db型超声波清洗器(昆山市超声仪器有限公司);mettleral204型和ab135-s型电子分析天平(瑞士mettlertoledo公司)。
1.2药品与试剂
儿茶素(lot#bcbr0996v)购自sigma公司,大黄素(110756-201512,含量以98.7%计)、大黄酚(110796-201319,含量以99.6%计)、大黄素甲醚(110758-201415,含量以99.1%计),大黄酸(110757-200206,供含量测定用),芦荟大黄素(110795-201308,含量以97.8%计),均购自中国食品药品检定研究院。番泻苷a(20120514,hplc≥98%),番泻苷b(20120605,hplc≥98%)购自上海源叶生物科技有限公司。没食子酸(bcbk2434v,91215-100mg)购自sigma公司。1-o-没食子酰-2-o-肉桂酰-β-d-葡萄糖苷为自制,并经uplc-pda检测,质量分数大于98%。芦荟大黄素-8-o-β-d-葡萄糖苷,1,6-二-o-没食子酰l-2-o-肉桂酰-β-d-葡萄糖苷,大黄酚-1-o-β-d-葡萄糖苷,大黄素-8-o-β-d-葡萄糖苷,大黄素甲醚-8-o-β-d-葡萄糖苷,1-o-没食子酰-2-o-肉桂酰-β-d-葡萄糖苷由本实验室制备、鉴定所得,并经uplc-pda检测,质量分数大于95%。色谱级甲醇、乙腈和质谱级乙腈购自美国thermofisher公司,质谱级甲酸(lot.sfasi-fq)购自梯希爱(上海)化成工业发展有限公司。水为超纯水(milli-q制备)。
1.3药材样本采集
10批大黄药材经石家庄以岭药业股份有限公司药材资源部田清存主任鉴定为为蓼科植物掌叶大黄(rheumpalmatuml.)。样本存放于北京以岭药业有限公司,药材来源见表1。
2方法
2.1色谱条件和质谱条件
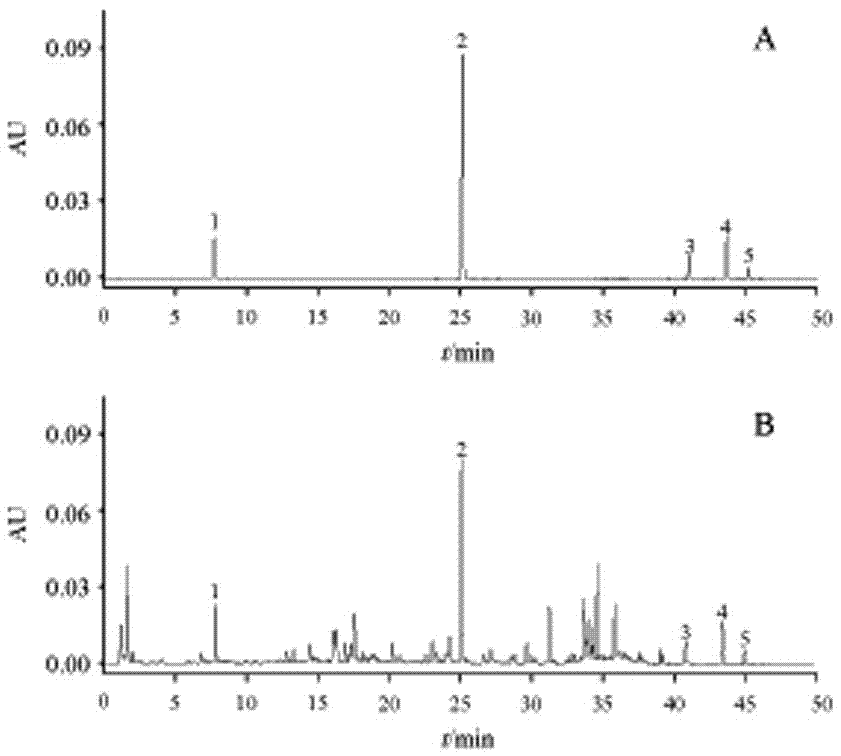
2.1.1色谱条件watersbehc18色谱柱(100mm×2.1mm,1.7μm),柱温30°c,流速0.3ml·min-1,检测波长280nm,进样量1μl。流动相为乙腈(a)-0.1%甲酸水溶液(b),梯度洗脱程序为0~30min,4%~25%a;30~45min,25%~70%a;45~50min,70%a,色谱采集时间为50min。此色谱条件检测时间适中,信息量丰富,亦用于指纹图谱的分析,对照品及供试品图谱见图1。
2.1.2质谱条件采用电喷雾离子源(esi),负离子mse模式,雾化气体为高纯度氮气(n2),碰撞气体为高纯度氩气(ar)。质量扫描范围m/z100~1500;毛细管电压为2.5kv;锥孔电压为40v;离子源温度为100°c;雾化温度为400°c;载气体积流量为800l/h,碰撞能量(ce)20~40v;采用亮氨酸-脑啡肽进行精确质量校正([m-h]-为554.2615)。
2.2溶液的制备
2.2.1对照品溶液的制备分别取儿茶素、1-o-没食子酰-2-o-肉桂酰-β-d-葡萄糖苷、大黄素、大黄酚和大黄素甲醚对照品适量,精密称定,加甲醇溶解并稀释成质量浓度分别为0.8820、0.9500、0.0799、0.2390、0.0482mg/ml的对照品储备液。
2.2.2供试品溶液的制备取大黄药材,粉碎,过四号筛。取粉末约0.5g,精密称定,置具塞锥形瓶中,精密加入甲醇50ml,密塞,称定重量,超声处理(功率250w,频率40khz)20min,放冷,再称定重量,用甲醇补足减失的重量,摇匀,用0.2μm滤膜滤过,取续滤液,即得。
3结果与分析
3.1大黄5种成分测定
3.1.1线性关系考察精密吸取混合对照品储备液适量,以甲醇逐级稀释,稀释倍数分别为0、10、20、40、100、200倍,摇匀。分别按“2.1.1”项下色谱条件进样分析,进样量均为1μl,测定峰面积。以质量浓度为横坐标(x),峰面积为纵坐标(y),绘制各标准曲线,得到线性回归方程和线性范围,结果见表2,5种成分在各浓度范围内的线性关系良好。
3.1.2精密度试验分别精密吸取混合对照品溶液1μl,在“2.1.1”项下色谱条件重复进样6次,计算儿茶素、1-o-没食子酰-2-o-肉桂酰-β-d-葡萄糖苷、大黄素、大黄酚、大黄素甲醚峰面积rsd值,各成分峰面积rsd(%)均小于2%,表明仪器精密度良好。
3.1.3稳定性试验取s1样品0.5g,按“2.2.2”项下方法制备供试品溶液,按“2.1.1”项下色谱条件分别在0、2、4、6、8、12和24h进样分析,计算5个成分峰面积的rsd值,各成分峰面积rsd(%)均小于2%,表明供试品溶液在24h内稳定性良好。
3.1.4重复性试验取s1样品6份,每份约0.5g,按“2.2.2”项方法制备供试品溶液,按“2.1.1”项下色谱条件进样分析,计算化合物儿茶素、2-o-cinnamoyl-1-o-galloyl-β-d-glucose、大黄素、大黄酚、大黄素甲醚质量分数rsd值,各成分质量分数rsd(%)均小于3%,表明该方法重复性良好。
3.1.5加样回收率试验取s1样品6份,每份0.25g,精密称定,置具塞锥形瓶中,分别精密加入相当于样品成分含量100%的各对照品溶液,按“2.2.2”项方法制备供试品溶液,“2.1.1”项下色谱条件进样分析,计算5种成分的加样回收率,平均回收率分别为97.23%、100.21%、99.10%、98.62%、99.97%,各成分回收率的rsd(%)小于3%,表明该方法准确度良好。
3.1.6样品测定按“2.2.2”项方法制备10批供试品溶液,“2.1.1”项下色谱条件分别测定10批大黄药材中的5种成分含量,结果见表3。不同批次的大黄药材中5个成分均有不同程度的差异。
3.2大黄的指纹图谱研究
3.2.1共有峰的确定及方法学验证采用国家药典委员会开发的中药色谱指纹图谱相似度评价系统2012版软件对10批大黄药材uplc指纹图谱进行数据处理,确定共有峰21个,由于1-o-没食子酰-2-o-肉桂酰-β-d-葡萄糖苷在各批次大黄中均有,保留时间适中,分离度良好且峰面积较大,故选定其作为参照峰(s),以相似度进行指纹图谱方法学考察,测定方法与“3.1”项相同。结果显示,同一样品溶液连续进样6次,相似度高于0.990,精密度良好。重复性考察相似度高于0.990,稳定性考察相似度高于0.900,表明方法学验证符合指纹图谱技术要求。
3.2.2相似度计算及对照指纹图谱的生成采用国家药典委员会开发的中药色谱指纹图谱相似度评价系统2012版软件对10批大黄样品进行分析,采用中位数法对各指纹图谱色谱峰进行了多点校正和自动匹配,以s1为参照指纹图谱,计算相似度并生成对照指纹图谱,10批大黄样品uplc指纹图谱见图2,生成对照指纹图谱见图3。计算得到的10批样品与对照指纹图谱的相似度结果见表3。结果表明各批次药材之间有较好的一致性,所建立的uplc指纹图谱可用于大黄的质量评价。
成分鉴定
采用uplcsynaptg2-sms对甲醇超声提取的样品(s1)进行化学成分定性分析,负离子模式下基峰图见图4,共检出80化合物。通过对照品、保留时间和质谱信息,并结合相关文献数据比对,进行化学成分确认,初步鉴定了其中71化合物,另有9个未知化合物,其中15个化合物通过对照品指认,结果见表4。
注:带*的是已经用对照品指认的化学成分,a~u对应指纹图谱1~21个共有峰
4小结
4.1含量测定及指纹图谱实验条件优化
本实验比较了用不同提取溶剂,如甲醇、70%甲醇、50%甲醇、乙醇、70%乙醇、50%乙醇在超声条件下提取大黄样品。通过对比得到的色谱峰峰容量和峰面积,最终确定用甲醇作为提取溶剂,又分别考察了超声20、30、40min和回流0.5、1、2h的不同提取方式和时间,结果表明提取方式和提前时间对色谱峰容量及峰面积影响不大,故选取超声20min作为样品处理方式。
4.2检测波长的确定
大黄中极性较大的成分在280nm下吸收较强,极性较小的游离蒽醌类如大黄素、大黄酚等在254nm下吸收较强,但在280nm下也有较强吸收,且280nm下指纹图谱峰容量较多,色谱信息丰富,出于含量测定和指纹图谱同时考虑,故选择280nm作为检测波长。
4.3本研究建立了大黄药材多成分含量测定和指纹图谱、定性分析相结合的质量评价方法,指纹图谱结合定性分析初步鉴定了大黄中71个化学成分,结构类型分别为蒽醌类22个、蒽酮类13个、二苯乙烯类3个、鞣质类28个、苯丁酮类3个和色原酮类2个。含量测定方法包含大黄中3个蒽醌和2个鞣质类成分,10批样品中儿茶素、1-o-没食子酰-2-o-肉桂酰-β-d-葡萄糖苷、大黄素、大黄酚和大黄素甲醚的含量范围分别为3.165~23.099、4.323~13.874、0.07~0.971、0.120~2.058、0.042~0.492mg·g-1,由结果可见,儿茶素和1-o-没食子酰-2-o-肉桂酰-β-d-葡萄糖苷在样品中含量较高,文献报道没食子酰葡萄糖苷具有降血脂作用,因此有必要对大黄中没食子酰葡萄糖苷进行质量控制。大黄药材的化学成分复杂,仅以几种成分来控制药材质量较为片面,因此本实验采用指纹图谱进一步对大黄药材质量进行表征和研究,同时结合物质基础,完善质量评价体系,为用药提供科学的依据。
附图说明
图1:对照品(a)和大黄药材样品(b)的uplc色谱图,其中1、儿茶素2、1-o-没食子酰-2-o-肉桂酰-β-d-葡萄糖苷3、大黄素4、大黄酚5、大黄素甲醚;
图2:10批大黄药材的uplc指纹图谱;
图3:大黄药材的对照指纹图谱;
图4:大黄样品负离子模式下基峰图
实施例1:
大黄药材质量评价方法:
(1)供试品溶液的制备:取大黄药材,粉碎,过四号筛,取粉末约0.5g,精密称定,置具塞锥形瓶中,精密加入甲醇50ml,密塞,称定重量,超声处理20min,超声功率250w,频率40khz,放冷,再称定重量,用甲醇补足减失的重量,摇匀,用0.2μm滤膜滤过,取续滤液,即得;
(2)对照品溶液的制备:分别取儿茶素、1-o-没食子酰-2-o-肉桂酰-β-d-葡萄糖苷、大黄素、大黄酚和大黄素甲醚对照品适量,精密称定,加甲醇溶解并稀释成质量浓度分别为0.8820、0.9500、0.0799、0.2390、0.0482mg/ml的对照品溶液;
(3)色谱参数为:c18色谱柱,色谱柱规格为100mm×2.1mm,1.7μm,柱温30°c,流速0.3ml·min-1,检测波长280nm,进样量1μl;流动相a为乙腈,b为0.1%甲酸水溶液,梯度洗脱程序为0~30min,4%~25%a;30~45min,25%~70%a;45~50min,70%a,质谱条件为:采用电喷雾离子源,负离子mse模式,雾化气体为高纯度氮气,碰撞气体为高纯度氩气,质量扫描范围m/z100~1500;毛细管电压为2.5kv;锥孔电压为40v;离子源温度为100°c;雾化温度为400°c;载气体积流量为800l/h,碰撞能量20~40v;采用亮氨酸-脑啡肽进行精确质量校正[m-h]-为554.2615。
(4)结果:五种成分含量测定、大黄指纹图谱、化学成分定性分析均符合相关要求。
实施例2
大黄药材质量评价方法:
(1)供试品溶液的制备:取大黄药材,粉碎,过四号筛,取粉末约0.1g,精密称定,置具塞锥形瓶中,精密加入70%甲醇50ml,密塞,称定重量,超声处理30min,超声功率250w,频率40khz,放冷,再称定重量,用甲醇补足减失的重量,摇匀,用0.2μm滤膜滤过,取续滤液,即得;
(2)对照品溶液的制备:分别取儿茶素、1-o-没食子酰-2-o-肉桂酰-β-d-葡萄糖苷、大黄素、大黄酚和大黄素甲醚对照品适量,精密称定,加甲醇溶解并稀释成质量浓度分别为0.8820、0.9500、0.0799、0.2390、0.0482mg/ml的对照品溶液;
(3)色谱参数为:c18色谱柱,色谱柱规格为100mm×2.1mm,1.7μm,柱温25°c,流速0.5ml·min-1,检测波长280nm,进样量1μl;流动相a为乙腈,b为0.1%甲酸水溶液,梯度洗脱程序为0~30min,4%~25%a;30~45min,25%~70%a;45~50min,70%a,质谱条件为:采用电喷雾离子源,负离子mse模式,雾化气体为高纯度氮气,碰撞气体为高纯度氩气,质量扫描范围m/z100~1500;毛细管电压为2.5kv;锥孔电压为40v;离子源温度为100°c;雾化温度为400°c;载气体积流量为800l/h,碰撞能量20~40v;采用亮氨酸-脑啡肽进行精确质量校正[m-h]-为554.2615。
(4)结果:五种成分含量测定、大黄指纹图谱、化学成分定性分析均符合相关要求。
实施例3
(1)供试品溶液的制备:取大黄药材,粉碎,过四号筛,取粉末约1g,精密称定,置具塞锥形瓶中,精密加入80%甲醇50ml,密塞,称定重量,超声处理10min,超声功率250w,频率40khz,放冷,再称定重量,用甲醇补足减失的重量,摇匀,用0.2μm滤膜滤过,取续滤液,即得;
(2)对照品溶液的制备:分别取儿茶素、1-o-没食子酰-2-o-肉桂酰-β-d-葡萄糖苷、大黄素、大黄酚和大黄素甲醚对照品适量,精密称定,加甲醇溶解并稀释成质量浓度分别为0.8820、0.9500、0.0799、0.2390、0.0482mg/ml的对照品溶液;
(3)色谱参数为:c18色谱柱,色谱柱规格为100mm×2.1mm,1.7μm,柱温35°c,流速0.4ml·min-1,检测波长280nm,进样量2μl;流动相a为乙腈,对照品溶液b为0.1%甲酸水溶液,梯度洗脱程序为0~30min,4%~25%a;30~45min,25%~70%a;45~50min,70%a,质谱条件为:采用电喷雾离子源,负离子mse模式,雾化气体为高纯度氮气,碰撞气体为高纯度氩气,质量扫描范围m/z100~1500;毛细管电压为2.5kv;锥孔电压为40v;离子源温度为100°c;雾化温度为400°c;载气体积流量为800l/h,碰撞能量20~40v;采用亮氨酸-脑啡肽进行精确质量校正[m-h]-为554.2615。
(4)结果:五种成分含量测定、大黄指纹图谱、化学成分定性分析均符合相关要求。
实施例4
(1)供试品溶液的制备:取大黄药材,粉碎,过四号筛,取粉末约0.8g,精密称定,置具塞锥形瓶中,精密加入75%甲醇50ml,密塞,称定重量,超声处理18min,超声功率250w,频率40khz,放冷,再称定重量,用甲醇补足减失的重量,摇匀,用0.2μm滤膜滤过,取续滤液,即得;
(2)对照品溶液的制备:分别取儿茶素、1-o-没食子酰-2-o-肉桂酰-β-d-葡萄糖苷、大黄素、大黄酚和大黄素甲醚对照品适量,精密称定,加甲醇溶解并稀释成质量浓度分别为0.8820、0.9500、0.0799、0.2390、0.0482mg/ml的对照品溶液对照品溶液;
(3)色谱参数为:c18色谱柱,色谱柱规格为100mm×2.1mm,1.7μm,柱温40°c,流速0.3ml·min-1,检测波长270nm,进样量0.5μl;流动相a为乙腈,b为0.1%甲酸水溶液,梯度洗脱程序为0~30min,4%~25%a;30~45min,25%~70%a;45~50min,70%a,质谱条件为:采用电喷雾离子源,负离子mse模式,雾化气体为高纯度氮气,碰撞气体为高纯度氩气,质量扫描范围m/z100~1500;毛细管电压为2.5kv;锥孔电压为40v;离子源温度为100°c;雾化温度为400°c;载气体积流量为800l/h,碰撞能量20~40v;采用亮氨酸-脑啡肽进行精确质量校正[m-h]-为554.2615。
(4)结果:五种成分含量测定、大黄指纹图谱、化学成分定性分析均符合相关要求。
- 还没有人留言评论。精彩留言会获得点赞!