一种基于金属有机框架材料复合光子晶体薄层的高效薄层色谱分离方法与流程
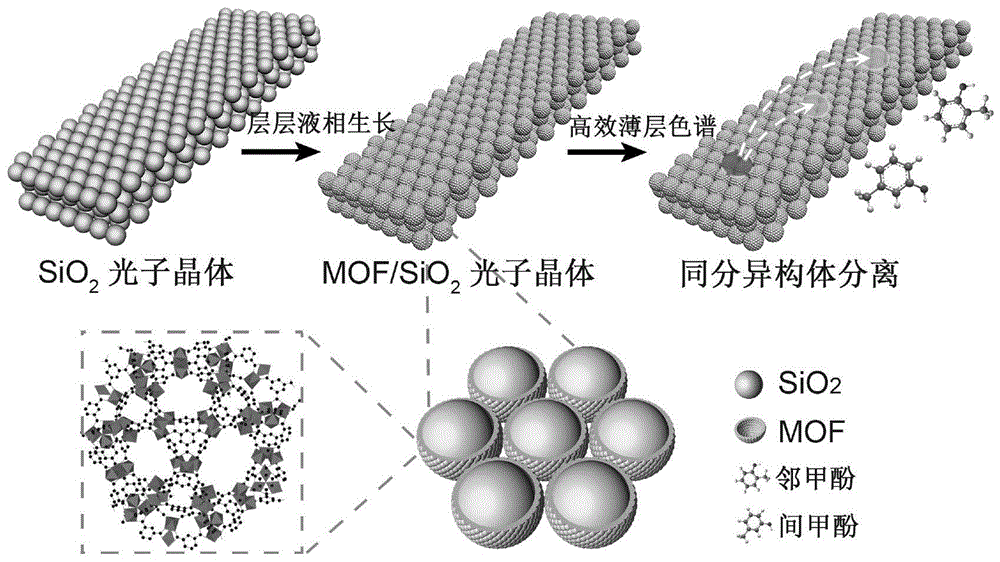
本发明属于光子晶体材料应用领域以及薄层色谱分离技术领域,具体涉及一种以金属有机框架材料复合光子晶体薄层板为固定相的高效薄层色谱技术。
背景技术:
薄层色谱是以涂布在玻璃板上的颗粒薄层为固定相,以特定溶剂为流动相,利用化学物质与固定相之间的差异性吸附脱附作用,对混合样品进行分离、鉴定的一种层析分离技术。自20世纪50年代提出以来,该技术因其便捷的操作、简单的设备要求等优势被广泛应用于食品安全检测、药物鉴定及代谢产物分析、有机合成、化工产品鉴定、毒品分析、农残检测等相关专业领域。在实际应用中,薄层色谱分离包括制板、点样、展开、显色等操作;根据分离后样品点与参照物比移值的比对即可定性样品点的化学组成,结合紫外可见及荧光扫描,还能定量确定混合物中各组分的含量,因而具有重要的实用价值。
虽然薄层色谱具有快速、简便、高效、经济等多种优势,但该技术因其固有特征而存在分离效率低、样品检出条件多样复杂这两大劣势,限制了薄层色谱在多种场合中的应用。众所周知,薄层色谱分离中待分析物在固定相中的迁移距离通常为2至4厘米,远远低于气相、液相色谱柱中10至20米的迁移距离;这就造成薄层色谱分离的理论塔板数必然数量级的低于后者的塔板数,由此导致物质与薄层之间的吸附脱附次数非常有限,使得薄层色谱的分离效率较低,无法分离结构相近的化学物质。为了提高薄层色谱的分离效率,科研工作者先后发展了棒状薄层色谱、加压薄层色谱、离心薄层色谱、胶束薄层色谱、包合薄层色谱、二维薄层色谱等多种“高效”薄层色谱技术。虽然这些技术有各自擅长的应用场合,但都揭示了采用粒度更小、更均匀的改性硅胶颗粒,提高固定相的选择性是提高薄层色谱分离效率的有效途径。
另一方面,薄层色谱分离的样品检出条件多样而又复杂。对于有色物质而言,分离后可直接在白色硅胶板上将其辨识出来;对于无色但有荧光特性的物质,分离后可在紫外灯辐照下将其辨识出来。目前使用最为广泛的是荧光硅胶板,主要用于无色但有紫外可见吸收物质的分离,在紫外灯辐照下利用荧光淬灭辨识样品点。对于许多无色、无紫外可见吸收、无荧光特性物质的识别检出,则必须借助硫酸、高锰酸钾染色,不仅增加了薄层色谱分离的操作步骤,也增大了操作者使用危险化学品的风险。目前,在薄层色谱样品检出中,除了联用各种光谱仪,上述检出问题并无简单统一的解决方案。
胶体光子晶体可能是同时满足高效分离和可视化检出的理想薄层色谱固定相材料。这是因为胶体晶中的纳米胶粒约0.2微米,远小于商品化的硅胶颗粒,具有更高的分离效率;同时胶体晶具有结构色,在不借助紫外辐照、染色的情况下即可肉眼识别出样品点。本发明申请人已于2017年公开发表了有关“基于介孔sio2光子晶体的薄层色谱分离”的学术论文,但上述工作中薄层色谱固定相的分离效率相比于商品化的硅胶板提高有限,无法分离本发明中提出的结构相近化学物质,如甲酚异构体。除上述工作,在申请人大量调研基础上,并未发现光子晶体薄层色谱分离技术的相关报道。因此,发展一种具有更强分离能力并兼具结构色检出的胶体光子晶体薄层色谱新技术,具有重要的意义和价值。
技术实现要素:
本发明针对现有薄层色谱技术的不足,提出了一种基于金属有机框架材料(mof)复合光子晶体(mof-pc)薄层(薄层板)的高效薄层色谱技术。作为色谱固定相,mof-pc薄层具有高比表面积及独特的孔道结构,因而在分离中展现出更强的吸附脱附效应与选择性,有效弥补了薄层色谱塔板数低导致分离效果不佳的固有缺点,确保混合物质的高效分离,大幅提高了分离效率;所述mof-pc薄层还具有光子晶体特有的结构色和反射信号,因而可以利用样品点与薄层板之间的结构色差异及反射信号变化直接确定样品的具体位置,从而获取比移值、选择因子、分离度等色谱参数。同时利用光子晶体结构色实现物质的可视化检出,简化了检出过程并提高操作的安全性。与现有技术相比,本发明所述技术具有显著提高的分离效果,能够分离传统薄层色谱无法分离的结构相近物质。此外,该技术依靠结构色直接识别样品点,无需紫外辐照、层板染色等额外操作,为样品检出提供了绿色、便捷、安全的新途径。
本发明提出了一种mof-pc薄层的制备方法,具体包括如下步骤:
(1)将含有二氧化硅(sio2)胶粒的过饱和溶液涂布于基片上,加热,得到固态sio2光子晶体薄层。
(2)恢复上述固态sio2光子晶体薄层中sio2胶粒的表面羟基,然后对胶粒进行氨基修饰和羧基修饰,获得羧基修饰的sio2光子晶体薄层。
(3)将羧基修饰的sio2光子晶体薄层交替浸润在含有金属离子的溶液及含有配体的溶液中,通过层层液相生长法在sio2胶粒表面原位形成mof层,制得所述金属有机框架材料复合光子晶体(mof-pc)薄层。
步骤(1)中,所述含有sio2胶粒的过饱和溶液为体积分数为20%-40%的sio2胶体颗粒与体积分数为60%-80%的溶剂组成的胶粒过饱和溶液;优选地,为体积分数为35%的sio2胶体颗粒与体积分数为65%的乙二醇组成的胶体过饱和溶液。
其中,所述sio2胶体为单分散sio2胶体。
其中,所述溶剂为有机溶剂,选自乙二醇、二甲亚砜、二甲基甲酰胺、碳酸丙烯酯等中的一种或多种;优选地,为乙二醇。
步骤(1)中,所述基片为玻璃基片、塑料基片、硅晶片等中的一种或多种;优选地,为玻璃基片。
步骤(1)中,所述将sio2胶粒的过饱和溶液涂布于基片上形成的液膜厚度为100微米-300微米;优选地,液膜厚度为100微米。
步骤(1)中,所述sio2胶粒的尺寸为150纳米-1000纳米;优选地,为200纳米。
步骤(1)中,所述固态sio2光子晶体薄层为含有sio2胶粒的过饱和溶液涂布于基片上,在加热挥发溶剂后,形成的sio2胶粒有序堆积的颗粒膜。
步骤(1)中进行加热的目的为:挥发溶剂使薄膜干燥固化。
步骤(1)中,所述加热的温度为60℃-100℃;优选地,为90℃。
步骤(1)中,所述加热时间为10-120分钟;优选地,为30分钟。
步骤(1)中,在加热步骤后还包括高温煅烧的步骤,所述进行高温煅烧的目的为:增强薄膜机械强度。
其中,所述高温煅烧的温度为450℃-550℃;优选地,为500℃。
其中,所述高温煅烧的时间为1-4小时;优选地,为2小时。
所述高温煅烧的操作步骤优选为将sio2光子晶体薄层置于马弗炉中高温煅烧。
步骤(2)中,所述恢复sio2胶粒表面羟基的操作为将固态sio2光子晶体薄层浸泡在体积比为7:3的浓硫酸和质量分数为30%的过氧化氢的混合液中,使胶粒表面形成大量的羟基。
所述浸泡的时间为不小于2小时;优选地,为12小时。
所述固态sio2光子晶体薄层与混合液的体积比为1:(300-2000);优选地,为1:1000。
步骤(2)中进行表面羧基修饰的目的为:增强胶粒对金属离子的吸附能力。
步骤(2)中,所述氨基修饰为:将恢复表面羟基的sio2光子晶体薄层浸泡在含有氨丙基三乙氧基硅烷(aptes)的乙醇溶液或含有氨丙基三甲氧基硅烷(aptms)的乙醇溶液中进行反应。取出后用乙醇洗涤并干燥,得到表面富含氨基的sio2光子晶体薄层。
所述乙醇溶液中氨丙基三乙氧基硅烷aptes或氨丙基三甲氧基硅烷aptms的体积分数为0.03%-0.18%;优选地,体积分数为0.06%。
所述反应的温度为20-80℃;优选地,为室温。
所述反应的时间为不小于4小时;优选地,为12小时。
所述恢复表面羟基的sio2光子晶体薄层与溶液的体积比为1:(300-2000);优选地,为1:1000。所述溶液指含有氨丙基三乙氧基硅烷(aptes)的乙醇溶液或含有氨丙基三甲氧基硅烷(aptms)的乙醇溶液。
步骤(2)中,所述的羧基修饰为将氨基修饰的sio2光子晶体薄层浸泡在浓度为0.0025-0.01g/ml的丁二酸酐的二甲基甲酰胺溶液中进行反应;优选地,溶液浓度为0.005g/ml。
取出后用乙醇洗涤并干燥,可得表面富含羧基的sio2光子晶体薄层。
所述反应的温度为20-80℃;优选地,为室温。
所述反应的时间为4-24小时;优选地,为12小时。
所述氨基修饰的sio2光子晶体薄层与溶液的体积比为1:(300-2000);优选地,为1:1000。所述溶液指含有丁二酸酐的二甲基甲酰胺溶液。
步骤(3)中,所述的金属有机框架材料复合光子晶体(mof-pc)薄层为各种mof材料沉积包裹在sio2胶粒表面形成的复合光子晶体薄层;优选地,mof材料为hkust-1、zif-8与mil-100。
本发明制备的mof-pc薄层是仅含有一种mof材料的mof-pc薄层。
步骤(3)中,所述的金属离子溶液为含有生成金属有机框架材料所需的金属离子的溶液,为cu(ch3coo)2·h2o乙醇溶液、zn(no3)2·6h2o甲醇溶液、fecl3·6h2o乙醇溶液等;同理,所述的配体溶液为含有生成该金属有机框架材料所需的配体的溶液,为均苯三甲酸乙醇溶液、2-甲基咪唑甲醇溶液等。
具体地,制备hkust-1复合光子晶体薄层时为cu(ch3coo)2·h2o乙醇溶液(50mm)与均苯三甲酸乙醇溶液(5mm);制备zif-8复合光子晶体薄层时为zn(no3)2·6h2o甲醇溶液(2mm)与2-甲基咪唑甲醇溶液(5mm);制备mil-100复合光子晶体薄层时为fecl3·6h2o乙醇溶液(20mm)与均苯三甲酸乙醇溶液(20mm)。
步骤(3)中,所述的层层液相生长法为一种原位反应制备复合结构的化学方法,即sio2光子晶体薄层先浸润在金属离子溶液中、取出后浸润在配体溶液中反应形成第一层mof层,将上述薄层再次先后浸润在金属离子溶液及配体溶液中形成第二层mof层,如法炮制在液相环境中最终形成mof复合光子晶体薄层。
所述每一层mof生长为sio2光子晶体先后浸泡在金属离子与配体溶液中10-60分钟;优选地,为30分钟。
所述mof生长的反应温度为20-100℃;优选地,为40℃。
所述mof生长过程中sio2光子晶体薄层与金属离子或配体溶液的体积比为1:(1000-5000);优选地为1:2000。
所述mof生长为1-10个生长周期;优选地,为7个生长周期。
本发明还提出了由上述方法制备得到的金属有机框架材料复合光子晶体(mof-pc)薄层。
本发明还提出了所述金属有机框架材料复合光子晶体(mof-pc)薄层在高效薄层色谱分离中的应用。
本发明还提出了一种基于mof-pc的薄层色谱分离方法,具体包括如下步骤:
(1)将含有待分析物的溶液点样在上述制备的mof-pc薄层板上,形成与mof-pc薄层相比具有不同结构色的样品点。
(2)将mof-pc薄层板转移至展开缸中,等待有机溶剂将样品展开,直至展开剂前沿到达预设位置。
(3)从展开缸中取出mof-pc薄层板,拍摄数码照片并采集空间分辨反射光谱。利用样品点与mof-pc薄层板的结构色差异及反射光谱信号差异,确定待分析物中各组分的色谱参数,参考各组分与固定相的相互作用强弱,确定分离后样品点的归属。
本发明所述mof-pc薄层也可称mof-pc薄层板,在步骤(1)中将mof-pc薄层作为薄层色谱固定相,因而称为mof-pc薄层板。
步骤(1)优选用毛细管将含有待分析物的溶液点样在mof-pc薄层板上。
步骤(1)中,所述含有待分析物的溶液,指含有体积分数为30%-60%的邻甲酚、间甲酚的甲醇溶液、含有体积分数为30%-60%的邻甲酚、间甲酚的乙醇溶液、含有体积分数为30%-60%的邻甲酚、间甲酚的水溶液等;优选地,为含有体积分数为50%的邻甲酚与间甲酚的甲醇溶液。
步骤(2)中,所述有机溶剂为石油醚、苯、四氢呋喃、乙酸乙酯、甲醇等常用的薄层色谱展开剂;优选地,为石油醚与乙酸乙酯的混合溶剂。
步骤(3)中,所述色谱参数包括比移值、塔板数、选择性因子、分离度等。
步骤(3)中,所述采集空间分辨反射光谱,是将光谱仪光纤探头固定在薄层色谱板上方,以1-4cm/min的恒定速率沿展开的方向拉动薄层板,使光谱仪能够连续记录沿展开方向的反射光谱变化;优选地,拉动薄层板的速率为2cm/min。
所述光谱仪记录反射光谱为每隔0.5-2秒记录一个反射峰信号;优选地,为每隔1秒记录。
步骤(3)中,所述空间分辨反射光谱指以x轴表示展开距离,y轴表示反射波长,色彩表示反射强度的三维反射光谱。通过谱图中反射带的凸起位置,精确地确定物质的比移值。
在一个具体实施方式中,所述mof-pc薄层及基于mof-pc的薄层色谱分离方法,具体包括如下步骤:
(1)将含有二氧化硅(sio2)胶粒的过饱和溶液涂布于玻璃基底上组装形成一层均匀的sio2液态光子晶体薄层。加热液膜,挥发溶剂使其干燥固化,形成固态sio2光子晶体薄层。高温煅烧该薄层,进一步增强薄层的机械强度。
(2)通过浸泡处理,恢复上述光子晶体薄层中sio2胶粒的表面羟基。通过室温化学反应,对胶粒表面先后进行氨基修饰和羧基修饰,最终获得表面富含羧基的sio2光子晶体薄层。
(3)将表面富含羧基的sio2光子晶体薄层交替浸润在含有金属离子的溶液及配体溶液中,通过层层液相生长法在sio2胶粒表面原位形成mof层,制得具有结构色的金属有机框架材料复合光子晶体(mof-pc)薄层。
(4)用毛细管将含有待分析物的溶液点样在步骤(3)制备的mof-pc薄层板上,形成与mof-pc薄层相比具有不同结构色的样品点。
(5)当样品点的结构色不再变化时,将mof-pc薄层板转移至盛有特定有机溶剂的展开缸中,密闭容器数分钟,等待有机溶剂将样品展开,直至展开剂前沿到达预设位置。
(6)打开展开缸,将mof-pc薄层板取出,使用数码相机快速记录图像信息,同时使用光谱仪沿展开方向采集空间分辨的反射光谱。利用样品点与mof-pc薄层板的结构色差异及反射光谱信号差异,确定待分析物中各组分的比移值(rf)等色谱参数;参考各组分与固定相的相互作用强弱,最终确定样品点的归属。
与现有技术相比,本发明的有益效果是:本发明提出了一种基于金属有机框架材料复合光子晶体薄层板的高效薄层色谱分离技术,可同步实现物质的高效分离和结构色检出;该技术为本发明首次提出,至目前为止未见有任何报道。与传统的硅胶薄层色谱相比,这种新技术采用高比表面积、特定微孔结构和均匀结构色的mof光子晶体薄层作为固定相,强化了物质的吸附脱附过程,提高了对待分析物的选择性,从而显著提高分离效果,成功分离传统薄层色谱无法分离的结构相近物质。与此同时,这种新色谱技术在分离后样品点检出时,无需紫外辐照显影,无需硫酸、碘染色显影,直接通过样品与mof光子晶体色谱板的结构色差异即可准确识别,为样品检出提供了绿色、便捷、安全的新途径。因此,该技术在未来基础科研与精细化学品的生产中具有广阔的应用前景。
附图说明
图1为基于mof-pc的高效薄层色谱用于甲酚异构体分离的示意图。
图2中图a、d、g、j分别为hkust-1复合sio2光子晶体薄层的实物照片、扫描电镜图、反射光谱及x射线衍射图谱;
图b、e、h、k分别为zif-8复合sio2光子晶体薄层的实物照片、扫描电镜图、反射光谱及x射线衍射图谱;
图c、f、i、l分别为mil-100(fe)复合sio2光子晶体薄层的实物照片、扫描电镜图、反射光谱及x射线衍射图谱。
图3为无样品mof-pc薄层(a)、点样后mof-pc薄层(b)的典型空间分辨反射光谱。
图4为邻甲酚(a-c)、间甲酚(d-f)在hkust-1、zif-8与mil-100复合光子晶体薄层板上的迁移。
图5为邻甲酚与间甲酚的混合物在mil-100复合光子晶体薄层板上的分离(a)与识别(b)。
图6为邻甲酚、间甲酚及邻甲酚与间甲酚的混合物在商业硅胶薄层板上的分离。
图7为甲酚异构体混合物在不同色谱分离中,固定相呈现出的理论塔板数、选择性因子以及甲酚异构体的实际分离度,图(a)为样品的理论塔板数;图(b)为固定相对待分析物的选择性因子及实际分离度。
具体实施方式
结合以下具体实施例和附图,对本发明作进一步的详细说明。实施本发明的过程、条件、实验方法等,除以下专门提及的内容之外,均为本领域的普遍知识和公知常识,本发明没有特别限制内容。
实施例1.制备sio2光子晶体薄层
采用
实施例2.制备金属有机框架材料复合光子晶体(mof-pc)薄层
首先,将本发明实施例1煅烧后的sio2光子晶体薄层浸泡在食人鱼溶液中12小时恢复表面羟基。随后将羟基化的sio2光子晶体薄层浸泡在体积分数0.06%的氨丙基三乙氧基硅烷(aptes)乙醇溶液中,室温反应12小时,取出用乙醇洗涤干燥,得到表面氨基修饰的光子晶体薄层。最后,将氨基修饰的光子晶体薄层浸泡在浓度为0.005g/ml的丁二酸酐二甲基甲酰胺溶液中,室温反应12小时,取出后用乙醇洗涤并干燥,得到表面富含羧基的光子晶体薄层。
将羧基修饰的光子晶体薄层交替浸润在金属离子和配体溶液中,诱导mof材料在胶粒表面原位生长,制备得到金属有机框架材料复合光子晶体(mof-pc)薄层。以制备mil-100-pc薄层为例,将羧基修饰的光子晶体薄层浸入浓度为20mm的fecl3·6h2o乙醇溶液,在40℃下反应1小时,取出后乙醇冲洗并烘干。随后将其浸润在浓度为20mm的均苯三甲酸乙醇溶液,同样在40℃下反应1小时,取出后乙醇冲洗并烘干。通过上述表面吸附及原位反应,胶粒表面生成了非常薄的一层mil-100。重复上述浸润操作若干次,就能在胶粒表面生长出一层一层的mof结构,最终得到厚度可控且mil-100均匀包覆的光子晶体薄层。运用相同的层层生长技术,以浓度为50mm的cu(ch3coo)2·h2o乙醇溶液和浓度为5mm的均苯三甲酸乙醇溶液为原料,可以制备hkust-1复合光子晶体薄层;以浓度为2mm的zn(no3)2·6h2o甲醇溶液和浓度为5mm的2-甲基咪唑甲醇溶液为原料,可以制备zif-8复合光子晶体薄层。图2中的照片及反射光谱表明,制得的三种mof-pc薄层具有均匀的结构色和光子晶体的特征反射信号。扫描电镜图和x射线衍射图表明,胶粒表面粗糙,确实生长出一定厚度的mof层;而胶粒排列仍呈现密堆积有序结构,说明mof材料的层层生长并未破坏胶体的有序排列,解释了复合光子晶体薄层的结构色与反射信号。此外,氮气吸附脱附实验和x射线衍射图证明这三种mof-pc薄层还具有高比表面积及均匀的微孔结构,为吸脱附增强和选择性吸附奠定了基础。
实施例3.运用空间分辨反射光谱确定光子晶体薄层板上样品点的位置
化学物质样品点在本发明实施例2所述的mof-pc薄层板上的位置可以用肉眼直接观察,也可以运用空间分辨的反射光谱(srrs)来精准确定。在实际操作中,通常将光谱仪光纤探头固定在薄层色谱板上方,以2cm/min的恒定速率沿样品展开的反方向拉动薄层板,使光谱仪能够连续记录mof-pc薄层板沿样品展开方向的光谱变化。将一系列光谱数据绘制成以比移值(rf)值为x轴,反射波长为y轴,反射强度以颜色表示的三维反射光谱,就可以直观的显示出mof-pc薄层上反射信号/结构色的变化,从而确定样品的位置。如图3所示,无加载化学物质的mil-100-pc薄层板上各个位置的反射信号完全一致,因此沿展开方向收集的srrs图谱表现为一条由反射峰组成的水平反射带。当样品点出现在扫描路径上时,样品点导致的反射峰红移会造成反射带的凸起,且凸起的位置、宽度与薄层板上样品点的位置、拖尾展宽完全吻合,故通过srrs图谱可以更加精准地确定物质的rf值。
实施例4.单一组分甲酚分子在金属有机框架材料复合光子晶体薄层板上的迁移
为了验证不同化学物质在本发明制备的mof复合光子晶体薄层板上具有差异性的迁移行为,首先进行单一组分邻甲酚或间甲酚在hkust-1、zif-8与mil-100复合光子晶体薄层板上的展开研究。在所有六组实验中,展开剂为体积比5:1的石油醚与乙酸乙酯混合溶剂,将其加入展开缸中直至深度约1厘米,静置几分钟后确保溶剂蒸汽达到饱和。用毛细管在mof复合光子晶体薄层板上点样,使其直径约2毫米,将点样后的薄层板转移至展开缸中展开。待展开剂达到预设位置,取出薄层板,使用数码相机记录图像,同时使用光谱仪沿展开方向采集空间分辨的反射光谱。
图4中的数码照片表明,展开后的邻甲酚与间甲酚样品点与周围mof复合光子晶体薄层形成明显的结构色差异,因而在薄层板上留下可肉眼辨识的痕迹。图4中的空间分辨反射光谱图则更加清晰的表明,邻甲酚与间甲酚在三种mof复合光子晶体薄层上均展现出不同的迁移距离,其中间甲酚在hkust-1-pc、zif-8-pc、mil-100-pc薄层上的比移值分别为0.37、0.40、0.27,而邻甲酚的比移值分别为0.54、0.53、0.67,与肉眼观察的结果一致。通过对比发现,两种甲酚异构体在mil-100复合光子晶体薄层上的迁移差异最大,且样品点相对集中,拖尾现象不明显,有望在相同条件下实现甲酚异构体混合物的分离。
实施例5.甲酚异构体混合物在mil-100复合光子晶体薄层板上的高效分离
为了验证基于mof复合光子晶体的薄层色谱技术具有高效的分离能力,选用在传统薄层色谱上难以分离的甲酚异构体混合物作为研究对象,采用本发明实施例2中制备的mil-100复合光子晶体薄层作为固定相,采用本发明实施例4中的条件进行展开,研究异构体混合物的实际分离效果。在典型实验中,展开剂为体积比5:1的石油醚与乙酸乙酯混合溶剂,将其加入展开缸中直至深度约1厘米,静置几分钟后确保溶剂蒸汽达到饱和。将体积比为1:1的邻甲酚与间甲酚混合物用毛细管点样在mof复合光子晶体薄层板上,使样品点直径约2毫米,将点样后的薄层板转移至展开缸中展开,并使用数码相机记录整个展开过程。待展开剂达到预设位置,取出薄层板,使用光谱仪沿展开方向采集空间分辨的反射光谱。
图5中的一系列数码照片表明,在展开过程中,橙黄色的mil-100复合光子晶体因溶剂的浸润而呈现出亮绿色;将展开后的薄层板从展开缸中取出,mil-100复合光子晶体又因溶剂的挥发而快速恢复至橙黄色。此时,由于甲酚分子与溶剂的部分残留,载有样品点的区域呈现绿色,因此可通过肉眼直接观察并确定邻甲酚与间甲酚的位置。图5中的空间分辨反射光谱图与数码照片的结果完全一致,充分证明甲酚异构体可以通过mil-100复合光子晶体薄层来有效分离。
mil-100复合光子晶体薄层的高效分离性能主要来源于两个方面。一是高比表面积增强了待分析物在薄层板上的吸附脱附过程。测试表明mil-100比表面积为1857m2/g,高于hkust-1和zif-8的表面积(1365m2/g,1711m2/g),也远高于商用sio2硅胶颗粒的比表面积。二是mil-100具有最适合甲酚异构体分子扩散及吸脱附的孔窗尺寸和孔道结构,使其具有良好的选择性。mil-100中存在两种取向不同的交错孔道,它们的孔窗尺寸分别为0.86nm和0.58nm,与甲酚分子大小较为匹配,使得mof复合光子晶体薄层对不同的甲酚分子产生了选择性吸附,进而提高了分离效果。
实施例6.商业硅胶薄层色谱与mof复合光子晶体薄层色谱在甲酚异构体分离中的效果对比
为了确定商业硅胶薄层色谱对甲酚异构体的分离效果,分别进行邻甲酚、间甲酚、甲酚异构体混合物在商业硅胶薄层板上的展开研究。在实验中,分别采用体积比1:0、7:1、6:1、5:1、3:1、1:1的石油醚与乙酸乙酯混合溶剂作为展开剂,将其加入展开缸中直至深度约1厘米,静置几分钟后确保溶剂蒸汽达到饱和。用毛细管在商业硅胶薄层板上点样,使其直径约2毫米,将点样后的薄层板转移至展开缸中展开。待展开剂达到预设位置,取出薄层板,将其置于紫外灯辐照下,使用数码相机记录样品点位置。如图6所示,在各种展开剂条件下,邻甲酚与间甲酚展开后的样品点十分接近;因此当两者混合物展开时,邻甲酚与间甲酚样品点有明显重叠,形成一个严重拖尾的样品点,这就使得甲酚异构体混合物始终难以完全分离。
如图7所示,为了研究不同薄层色谱板的分离效果,选用5:1石油醚与乙酸乙酯展开剂在各种薄层板上的展开结果,包括理论塔板数、选择性因子和分离度等进行比较。从理论塔板数来看,本发明制备的mof复合光子晶体薄层板和商业硅胶色谱板的塔板数均为100左右,远低于气相色谱及液相色谱的层板数,这主要是因为待分析物在薄层色谱板上的扩散距离较短,仅2至4厘米。从选择性因子来比较,本发明所述的mil-100-pc薄层对邻甲酚与间甲酚的选择性因子(2.51)高于hkust-1及zif-8复合光子晶体薄层的选择性因子(1.3-1.5),更远远高于商品化薄层色谱板的选择性因子(1.11)。因此,本发明制备的mof复合光子晶体薄层具有高选择性因子,有利于两者的分离。在实际分离中,甲酚异构体在商品化硅胶薄层色谱中的分离度仅为0.33,无法实现有效分离;这是因为甲酚在商品化硅胶薄层上的理论塔板数和其对甲酚异构体的选择性因子均较低。当采用本发明所述的mil-100复合光子晶体薄层作为固定相时,甲酚异构体的分离度达到1.96。这是因为,mil-100复合光子晶体薄层对甲酚异构体具有高选择性,有效弥补了薄层色谱塔板数较低的缺点,在较短的展开距离内实现了高效分离。
实施例7.气相色谱与mof复合光子晶体薄层色谱在甲酚异构体分离中的效果对比
将甲酚异构体混合物在气相色谱中进行分离,在色谱图中可以观察到两个完全分离的色谱峰,经计算其分离度为1.54,证明气相色谱可以对甲酚异构体实现有效分离。如图7所示,甲酚分子在气相色谱柱中的理论塔板数高达20000,远远高于其在商品化硅胶薄层板、mof复合光子晶体薄层板中的塔板数。而气相色谱柱对甲酚异构体的选择性因子仅为1.05,与商品化硅胶薄层板的选择因子相近,均远低于mof复合光子晶体薄层板的选择因子。由此可见,商业硅胶薄层色谱因为较低的层板数和较低的选择因子而无法分离甲酚异构体;气相色谱通过极高的层板数,弥补了较低的选择因子,实现了甲酚异构体的分离;mof复合光子晶体薄层色谱则通过较高的选择性因子,弥补了较低的层板数,同样可以分离甲酚异构体,甚至实现了比气相色谱更高的分离度。
本发明的保护内容不局限于以上实施例。在不背离发明构思的精神和范围下,本领域技术人员能够想到的变化和优点都被包括在本发明中,并且以所附的权利要求书为保护范围。
- 还没有人留言评论。精彩留言会获得点赞!