一种慢病毒包装系统辅助质粒的超螺旋结构纯度的检测方法与流程
1.本发明涉及药物分析领域,具体涉及一种利用高效液相色谱法检测质粒dna超螺旋结构纯度的方法。
背景技术:
2.质粒(plasmid)广泛存在于生物界,从细菌、放线菌、丝状真菌、大型真菌、酵母到植物,甚至人类机体中都含有。从分子组成看,有dna质粒,也有rna质粒;从分子构型看,有线型质粒、也有环状质粒,其表型也多种多样。细菌质粒是基因工程中最常用的载体。质粒(plasmid)是细菌或细胞染色质以外的,能自主复制的,与细菌或细胞共生的遗传成分,是染色质外的双链共价闭合环形dna,可自然形成超螺旋结构。
3.质粒dna是一类能在细菌染色体外自主复制的环形双链dna分子。不同类型的质粒dna分子量大小不同,大的可达105kda,小的则仅为103kda。质粒dna有三种不同的构型:
4.①
共价闭合环形dna所呈现的超螺旋结构(sc构型),在电泳图谱中,其处于凝胶的前面;
5.②
一条链保持完整的环形结构,另一条链上有一个至数个缺口的、开环的dna所呈现的开环构型(oc构型),其走在凝胶的后面;
6.③
双键断裂的dna呈现的线性结构(l构型)其走在凝胶的中间部位。
7.由于超螺旋质粒dna转染效率高,可大幅减少用药剂量,所以在dna疫苗、基因治疗和细胞治疗中使用的质粒dna为超螺旋构型。但在质粒纯化过程中会造成质粒dna超螺旋结构的破坏,形成多种不同构型的质粒dna,从而影响质粒包装效率,因而对质粒dna超螺旋含量做出准确的定量分析就很有必要。
8.现有的检测超螺旋dna含量技术有琼脂糖凝胶电泳法和毛细管凝胶电泳法,但均存在检测方法上的不足,如毛细管凝胶电泳法需要繁琐的样品前处理过程,且不适合在质粒dna生产的每一步中使用;琼脂糖凝胶电泳法在od
260
波长下检测范围太窄,并且样品稀释过程中很容易造成误差,准确度很低,也不适合在质粒dna生产的每一步中使用。更重要地,上述两种方法均无法定量地检测超螺旋dna的含量。
技术实现要素:
9.针对现有技术中存在的缺点与不足,本发明的目的是提供一种高效液相色谱法检测一种质粒超螺旋结构纯度的方法,使用waters e2695型高效液相色谱仪对一种慢病毒包装系统辅助质粒bz2k进行高效液相色谱分析检测,通过采用外标法即可快速计算出样品中超螺旋结构所占比例。
10.本发明的一个方面,提供一种质粒超螺旋纯度的高效液相色谱检测方法,该方法包括如下步骤:
11.(1)使用waters e2695型高效液相色谱仪和赛分proteomix sax强阴离子交换色谱柱
12.(2)检测条件的选择:
13.①
检测波长为260nm;
14.②
流速为0.5ml/min;
15.③
柱温为35℃;
16.④
上样量20μl;
17.⑤
流动相a为20mmol/l tris ph8.0溶液;
18.⑥
流动相b为20mmol/l tris 1mol/l nacl ph8.0溶液;
19.⑦
洗脱梯度改变量为1%。
20.(3)质粒各构型工作标准品的制备:采用单酶切的方式获得线性结构工作标准品;采用回收液相色谱图中的杂峰和主峰的方式获得开环结构和超螺旋结构工作标准品,并结合琼脂糖凝胶电泳的方法和高效液相色谱的方法再次对获得的待定开环结构和超螺旋结构工作标准品进行验证确认;
21.(4)利用确认的超螺旋结构工作标准品,结合优化后的色谱检测条件对供试品进行超螺旋结构纯度的检测。
22.综上所述,本发明的有益效果至少包括如下:
23.1.摸索获得一套色谱纯化参数,在该套参数下获得的超螺旋结构质粒纯度可高达99%以上;
24.2.制备出一种质粒超螺旋结构工作标准品,采用外标标曲法即可准确测定质粒中超螺旋结构纯度,该分析方法简便、快速、灵敏度高,比常规的凝胶电泳法有更高的分辨率。
附图说明
25.附图用来提供对本发明的进一步理解,并且构成说明书的一部分,与本发明的实施例一起用于解释本发明,并不构成对本发明的限制。
26.图1为bz2k质粒结构图;
27.图2为tskgel dna
‑
npr色谱柱bz2k质粒高效液相色谱图;
28.图3优化实验中洗脱梯度以0.8%改变时bz2k质粒高效液相色谱图;
29.图4优化实验中洗脱梯度以1%改变时bz2k质粒高效液相色谱图;
30.图5 bz2k质粒优选参数下的高效液相色谱图;
31.图6质粒各构型的琼脂糖凝胶电泳图;
32.图7 bz2k质粒超螺旋标准品色谱图;
33.图8 bz2k质粒中加入开环结构色谱图;
34.图9 bz2k质粒中加入超螺旋结构、线性结构色谱图;
35.图10 bz2k质粒超螺旋结构工作标准品纯度的凝胶电泳检测结果;
36.图11空白进样色谱图;
37.图12系统适用性验证结果;
38.图13标准曲线。
具体实施方式
39.下面将结合实施例对本发明的方案进行解释。本领域技术人员将会理解,下面的
实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体技术或条件的,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
40.实施例1:检测条件的筛选和优化
41.bz2k质粒是慢病毒包装过程中的必需辅助质粒之一,其结构如图1所示。使用waters e2695型高效液相色谱仪和tskgel dna
‑
npr色谱柱对bz2k质粒各个结构进行分离。
42.实验1:
43.检测波长 260nm;
44.流速 0.5ml/min;
45.柱温 35℃;
46.上样量 20μl(2μg);
47.流动相a:20mmol/l tris ph8.8溶液;
48.流动相b:20mmol/l tris 1mol/l nacl ph8.8溶液;
49.洗脱梯度 1%(表1)。
50.表1
51.时间(分钟)流动相a%流动相b%0505055050451090505050
52.结果如图2所示,发现tskgel dna
‑
npr色谱柱的色谱图中bz2k质粒的不同组份(超螺旋结构质粒和开环结构质粒)分离效果不够理想,分离度仅有1.9。基于该效果不佳,我们更换另一款色谱柱proteomix sax强阴离子交换色谱,并且对流动相ph、柱温、洗脱梯度进行优化筛选。
53.实验2:对流动相ph的筛选
54.将流动相a和b的ph值调整为7.5、8.0、8.5、9.0四组;其余条件同实验1,结果如下:
55.表2
[0056][0057]
结果显示流动相a和b的ph值优选为8.0。
[0058]
实验3:对柱温的筛选
[0059]
将柱温调整为25℃、30℃、35℃、40℃三组;其余条件同实验1,结果如下:
[0060]
表3
[0061]
柱温分组主峰分离度usp拖尾usp理论塔板数25℃1.780600e+0001.311389e+0001.238183e+00430℃1.820771e+0011.106584e+0003.235684e+00335℃1.951852e+0001.021085e+0003.951557e+00340℃1.965713e+0001.251282e+0012.542138e+004
[0062]
结果显示柱温调整为30
‑
35℃为较优选择。
[0063]
实验4:对洗脱梯度的筛选
[0064]
将洗脱梯度设置为0.8%、1.0%两组,其余条件同实验1。结果如图3
‑
4所示,当洗脱梯度改变量为0.8%时,峰型出现较为严重的前置(主峰拖尾因子为0.85),提示1.0%的洗脱梯度优于0.8%的洗脱梯度。
[0065]
实验5:确定最佳检测条件
[0066]
通过一系列单因素实验,确定能够实现分辨超螺旋质粒和开环结构质粒的最优检测条件为:检测波长为260nm;流速为0.5ml/min;柱温为30
‑
35℃;上样量20μl;流动相a为20mmol/l tris ph8.0溶液;流动相b为20mmol/l tris 1mol/l nacl ph8.0溶液;梯度洗脱改变量为1%,梯度洗脱如表4:
[0067]
表4
[0068]
时间(分钟)流动相a%流动相b%050505257525595305050
[0069]
在该检测条件下,bz2k质粒的图谱如图5所示,可以清晰看出bz2k质粒出现主成分峰和小峰,其中主成分峰为超螺旋质粒,而小峰则为开环结构质粒。
[0070]
实施例2:bz2k质粒各构型工作标准品的制备
[0071]
1、bz2k质粒线性结构工作标准品制备
[0072]
取bz2k质粒2μg~5μg,用mlui
‑
hf进行单酶切,将单酶切产物进行醇化,醇化后产物用流动相a溶解至50μl,即为bz2k质粒线性质粒工作标准品溶液。
[0073]
2、bz2k质粒开环结构工作标准品制备
[0074]
利用实施例1中确定的最优色谱检测条件(检测波长为260nm;流速为0.5ml/min;柱温为35℃;上样量20μl;流动相a为20mmol/l tris ph8.0溶液;流动相b为20mmol/l tris 1mol/l nacl ph8.0溶液;梯度洗脱为1%),对bz2k质粒原液进样,对色谱图中杂峰(主峰前约1分钟的杂峰)进行峰回收,将回收的质粒dna进行醇化,纯化产物用流动相a溶解至50μl,即为bz2k质粒开环结构工作标准品。
[0075]
3、bz2k质粒超螺旋结构工作标准品制备
[0076]
利用实施例1中的最终确定的色谱检测条件,对bz2k质粒原液进样,对色谱图中主峰进行峰回收,将回收的质粒dna进行醇化,醇化后产物用流动相a溶解至100μl。
[0077]
采用两种方法对回收到的杂峰(开环结构)和主峰(超螺旋结构)再次进行鉴定:
[0078]
(1)琼脂糖凝胶电泳法:
[0079]
如图6所示,单酶切产物、回收到的杂峰(开环结构)和主峰(超螺旋结构)电泳条带位置分明,与质粒原液电泳条带位置一一对应。
[0080]
(2)hlpc法:
[0081]
将回收的主成分峰按实施例1中摸索的色谱条件再次上样,如图7所示,有一主峰(18.911min)和杂峰(17.918min),杂峰仅是主峰千分之几;在回收的主成分峰中加入回收的杂峰(主峰前约1分钟处),如图8所示,在原色谱图杂峰位置(17.862min)出现明显峰增加;在回收的主成分峰中加入bz2k质粒的单酶切产物,如图9所示,在主峰和杂峰之间,出现明显的色谱峰(18.031min)。
[0082]
用琼脂糖凝胶电泳将bz2k质粒超螺旋结构工作标准品进行纯度检测,如图10所示,取hplc峰(主峰)回收液200ng,进行琼脂糖凝胶电泳,计算其主条带(超螺旋)的纯度为99.23%,大于最小峰纯度的99.0%(当峰分离度为1.5时,峰纯度99.0%),即色谱图主峰应为单一物质,该检测色谱条件能够将bz2k质粒超螺旋结构与其他bz2k质粒组分(开环和线性结构质粒)有效分开。由此可见,上述两种方法均能够证明实施例1中摸索获得的色谱条件获得了纯度很高的超螺旋结构,可以作为bz2k质粒超螺旋结构工作标准品。
[0083]
实施例3:系统适用性溶液制备
[0084]
线性质粒工作粒标准品50μl、开环质粒工作标准品50μl和20μg超螺旋质粒工作标准品,用流动相a溶解至200μl混匀,即得系统适用性溶液。
[0085]
实施例4:标准曲线溶液配制
[0086]
根据bz2k质粒工作标准品溶液的浓度及超螺旋质粒的纯度,取不同体积的bz2k质粒工作标准品,分别用流动相a将其稀释成超螺旋质粒浓度为50ng/μl、100ng/μl、150ng/μl、200ng/μl和250ng/μl的溶液100μl,混匀,即得。
[0087]
实施例5:供试品溶液处理
[0088]
根据测试得到bz2k质粒制品dna浓度,用流动相a将pbz2k质粒制品稀释成浓度为100ng/μl的溶液200μl,即为供试品溶液。
[0089]
实施例6:检测方法
[0090]
1、空白无干扰实验:取流动相a溶液100μl放入进样盘中,连续进样2次,每次20μl,记录色谱图。如图11所示,在4min~30min色谱图,未见色谱峰。
[0091]
2、系统适用性实验:取系统适用性溶液200μl放入进样盘中,按照拟定好的色谱条件(检测波长为260nm;流速为0.5ml/min;柱温为35℃;上样量20μl;流动相a为20mmol/l tris ph8.0溶液;流动相b为20mmol/l tris 1mol/l nacl ph8.0溶液;梯度洗脱为1%),连续进样6次,每次进样20μl,记录色谱图,出系统适用性报告,计算主峰分离度、理论塔板数、拖尾和rsd(峰面积和出峰时间)(表4)。如图12所示,主峰同杂质峰完全分开,质粒开环结构和质粒线性结构的峰高约达到主峰峰高的十分之一左右。
[0092]
表4系统适用性溶液的分离情况
[0093][0094]
3、标准曲线的绘制(外标标准曲线法):取标准曲线溶液放入进样盘中,按照拟定好的色谱条件,依次进样,每个样品进样2次,每次进样20μl,利用峰面积(两次的平均值)和
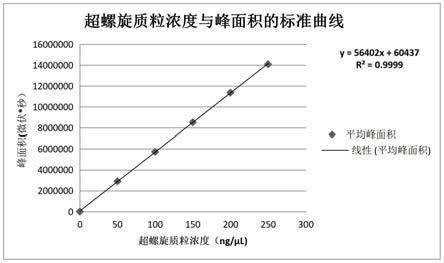
超螺旋质粒的浓度绘制标准曲线和线性回归方程式,如图13所示。
[0095]
4、供试品溶液测试:取供试品溶液100μl放入进样盘中,按照拟定好的色谱条件,进行进样,每个供试品进样2次,每次进样20μl,记录色谱图。
[0096]
5、计算方法:纯度c%=cx/c
测
×
100%。cx为供试品超螺旋质粒的浓度;c
测
为超微量紫外分光光度法(q5000)测得质粒浓度。
[0097]
实施例7:实验结果与分析
[0098]
通过色谱图及实验数据分析(表5
‑
表6),在该检测方法条件下bz2k质粒超螺旋结构质粒在上样量浓度为49.94ng/μl~249.71ng/μl之间有良好的线性关系,质粒中超螺旋与其他两种结构质粒完全有效的分离开来,通过外标法即可快速准确计算出其超螺旋质粒的浓度,进而算出其超螺旋质粒纯度,为84.71%。
[0099]
表5
[0100][0101]
表6
[0102][0103]
尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为对本发明的限制,本领域的普通技术人员在本发明的范围内可以对上述实施例进行变化、修改、替换和变型。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1