用于治疗他汀类药物治疗的副作用的白三烯‑介导的活性的抑制剂的制作方法
本发明涉及用于缓解或减轻他汀类药物治疗的副作用的方法。特别地,本发明涉及包含LTC4介导的活性的抑制剂的组合物和药剂盒,用于减轻他汀类药物治疗的至少一种不良副作用。
发明背景
内质网(ER)容易受到广泛范围的生理学信号以及毒性剂的应激,通常导致错误折叠的ER客户蛋白(client proteins)的积聚。ER应激已与多种疾病有关,在这些疾病中它导致细胞死亡。位于ER膜的三个ER应激传感器蛋白,IRE1α、PERK和ATF6,介导一个进化上保守排列的信号传导通路,称为未折叠蛋白反应(UPR)。最初UPR的目的是通过降低ER中错误折叠蛋白的过载应对应激。在过大的应激下,所述UPR传感器触发细胞死亡。鉴定了几种应激触发的细胞死亡机制,但是ER中错误折叠蛋白积聚的毒性基础和涉及的机制尚未完全理解。
在应激触发的细胞死亡中的一个关键角色是C/EBP-同源蛋白CHOP (DDIT3、GADD153),其被所有三个ER应激传感器诱导。CHOP被表明通过下调Bcl2蛋白并将Bax转运至线粒体以触发细胞凋亡。应激触发的TRAF2-ASK1-JNK通路也通过抑制Bcl2蛋白和激活Bim、BAX和BAC触发细胞凋亡。然而,在某些细胞类型和应激条件下,尽管缺乏Bcl-2抑制和ASK-1或Bax/Bak激活,但细胞死亡发生,表明另外的死亡-触发途径的存在。CHOP也通过氧化应激触发细胞死亡,因此,引发细胞凋亡和非细胞凋亡性细胞死亡两种机制。
使用秀丽线虫(C. elegans)的研究证实ERO1,其在ER中生成H2O2作为蛋白质二硫键形成的副产物,是在ER应激下的ROS生产者。然而,在小鼠细胞中就不是这种情况,因为在编码所有ER巯基氧化酶ERO1α、ERO1β和PRDX4的基因中合并的丧失功能的突变导致增加而不是减少H2O2的生产。因此,ER应激触发氧化应激的机制仍不清楚。为鉴定一种替代机制,研究了其它的ER氧化还原酶机制例如结构上相关的酶微粒体谷胱甘肽S-转移酶1 (MGST1)和MGST2。MGST1作为一种促生存因子被广泛研究,通过直接解毒和通过下游保护以避免氧化应激而赋予对细胞毒性药物的抗性。相反,MGST2在氧化应激和ER应激中的作用尚未进行广泛的研究。
MGST2是一种白三烯C4合酶(LTC4S)的同工酶。MGST2主要表达于肥大细胞和一些其它的骨髓细胞中。免疫学提示这样的Fc受体激活通过易位到核膜和细胞溶质磷脂酶A2 (cPLA2)、5-脂氧合酶(5-LO)、5-脂氧合酶激活蛋白(FLAP)和LTC4S的共定位,启动在肥大细胞中的LTC4生物合成。cPLA2从磷脂释放花生四烯酸,5-LO和FLAP将其氧化为LTA4,并且LTC4S缀合LTA4与谷胱甘肽,以形成LTC4。然后LTC4通过转运蛋白MRP1输出到细胞外环境。细胞表面酶将LTC4进一步代谢为更稳定的形式LTD4 (CAS号73836-78-9)和LTE4 (CAS号75715-89-8)。所有三种白三烯结合于两种G-蛋白偶联受体:CysLTR1和CysLTR2。分泌的LTC4及其代谢物触发平滑肌细胞的收缩,从而引起在肺部的支气管收缩和血管收缩,表现为过敏和哮喘的典型的症状。因此,几种LTC4受体拮抗剂(孟鲁司特(CAS号158966-92-8;环戊基3-{2-甲氧基-4-[(邻-甲苯磺酰基)氨基甲酰基]苄基}-1-甲基-1H -吲哚-5-基氨基甲酸酯)、普仑司特(CAS号103177-37-3;N -[4-氧代-2-(1H -四唑-5-基)-4H -苯并吡喃-7-基]-4-(4-苯基丁氧基)苯甲酰胺)、扎鲁司特(CAS号107753-78-6; 环戊基3-{2-甲氧基-4-[(邻-甲苯磺酰基)氨基甲酰基]苄基}-1-甲基-1H -吲哚-5-基氨基甲酸酯),和西鲁司特(CAS号128312-51-6;3-({3-[(E)-2-(4-环丁基-1,3-噻唑-2-基)乙烯基]苯基}氨基甲酰基)-2,2-二乙基丙酸))被开发并被批准为治疗哮喘症状的药物。
尽管LTC4S主要是在肥大细胞中表达并且在过敏和哮喘的情况下已被广泛研究,其同工酶MGST2是广泛表达的,但其生理学作用尚未被广泛研究。
他汀类药物(Statins)是3-羟基-3-甲基戊二酰辅酶A还原酶(HMG-CoA还原酶,HMGCR,UniProt P04035),胆固醇生物合成的限速酶的竞争性抑制剂,其用作高胆固醇血症的主要疗法并预防心血管疾病。他汀类药物对控制胆固醇的治疗作用,包括降低动脉粥样硬化的主要结果,已被很好地确立。为降低高胆固醇水平,或为防止胆固醇水平在危险患者中的增加,患者用一系列的他汀类药物治疗,其包括阿托伐他汀(CAS号134523-00-5)、辛伐他汀(CAS号79902-63-9)、普伐他汀(CAS号81093-37-0)、洛伐他汀(CAS号75330-75-5)和氟伐他汀(CAS号93957-54-1)。西立伐他汀(CAS号145599-86-6)在二十世纪90年代后期投放市场,但由于致死性横纹肌溶解(其中受损的骨骼肌组织分解的一种病症)的报告,已在2001年主动退出全球市场。
此外,他汀类药物具有与内皮功能、胰岛素敏感性和炎症/免疫调节相关的许多多效性的胆固醇-非依赖性作用。也已报道他汀类药物在治疗痴呆症和各种癌症,例如前列腺、皮肤、肺、结肠、膀胱、子宫和肾癌中具有潜在效用。
然而,有许多与他汀类药物治疗有关的潜在的严重副作用,包括肌病,其严重性范围可从肌炎至肌肉损耗(横纹肌溶解)。其它不太严重的不良反应已被报道,包括头痛、关节痛、发热、背痛、腹部绞痛、睡眠障碍、鼻炎、鼻窦炎、咳嗽反射的刺激、头晕和疲劳。对这类药物报道的禁忌症中,最常见的两种是疲劳和/或肌肉疼痛(常常称为"肌痛")。在用他汀类药物治疗期间的不良副作用的风险随着同时给予某些其它的药物,例如环孢菌素、纤维酸衍生物(如吉非贝齐)、烟酸或抗真菌药物而增加。
事实上,估计5-10%的患者由于范围从轻微到中度肌痛(以肌肉无力、疲劳和疼痛为特征),再到威胁生命的横纹肌溶解(其被定义为一种大块的和急性肌肉纤维的破坏,导致肌纤维内容物的释放)的肌病症状而停止他汀类药物的使用。当他汀类药物与其它的药物或与锻炼结合时,肌炎(肌肉炎症)和肌病症状的报告以不同类型的他汀类药物,随着他汀类药物剂量而增加。
已提出各种假设来解释他汀类药物-诱导的肌肉损伤。他汀类药物的作用可以是通过减少胆固醇合成的间接作用或是对不同的肌肉细胞靶的直接作用。他汀类药物肌病的机理基础可能是多因素的且部分归因于他汀类药物对细胞凋亡的调节作用。然而,他汀类药物如何促进细胞凋亡是相当模糊的。在完整的细胞中,他汀类药物被发现触发钙水平的升高,Bax (一种促凋亡蛋白)向线粒体的易位,线粒体通透性转换孔(PTP)开放,并释放细胞色素C,导致细胞凋亡。
除了细胞凋亡外,几个体外和体内研究证实,他汀类药物触发氧化应激和坏死。最重要的是,几种他汀类药物诱导内质网(ER)应激,其特征是诱导蛋白CHOP (CCAAT/-增强子-结合蛋白同源蛋白),其也通过氧化应激触发细胞死亡,引发细胞凋亡和非细胞凋亡性细胞死亡两种机制。他汀类药物的急性应用也已显示触发大量的钙经由兰诺定(ryanodine)受体,从内质网(ER)释放。从ER释放钙是ER应激的特征,ER应激触发氧化应激。总之,他汀类药物-触发的肌病至少部分涉及ER应激触发的氧化应激,导致细胞死亡。
白三烯是一个在白细胞中产生的类花生酸炎性介质的家族,其通过花生四烯酸经酶花生四烯酸5-脂氧合酶(5-脂氧合酶,5-LOX,5-LO,UniProt P 09917)的氧化而生成。顾名思义,白三烯是在白细胞中首次发现的,但此后已在其它的免疫细胞中发现。白三烯使用脂质信号传导以传递信息到产生它们的细胞(自分泌信号传导),或者邻近的细胞(旁分泌信号传导),以调节免疫反应。白三烯产生通常伴有组胺和前列腺素的产生,其也用作炎性介质。它们(特别是,白三烯D4)的作用之一是在细支气管内层的平滑肌中触发收缩;它们的过度生产是哮喘和过敏性鼻炎中的炎症的主要原因。白三烯拮抗剂被用来通过抑制白三烯的生产或活性,治疗这些疾病。
白三烯大致被分为三种类型。LTC4 (CAS号72025-60-6)、LTD4、LTE4 (CAS号75715-89-8)和LTF4 (CAS号83851-42-7)由于在它们的结构中存在氨基酸半胱氨酸,通常被称为“半胱氨酰白三烯”。半胱氨酰白三烯组成过敏性反应的慢反应物质(SRS-A)。LTF4,像LTD4,是LTC4的代谢物,但是不像缺乏谷胱甘肽的谷氨酸残基的LTD4,LTF4缺乏谷胱甘肽的甘氨酸残基。LTB4通过酶LTA4水解酶从LTA4经体内合成。它的主要功能是募集中性粒细胞到组织损伤的区域,虽然它也有助于促进由各种免疫细胞生产的炎性细胞因子。阻断LTB4的行动的药物已显示一些减慢嗜中性粒细胞-介导的疾病的进展的效果。也有人推测存在LTG4,一种LTE4的代谢物,其中半胱氨酰部分已被氧化为α-酮酸(即半胱氨酸已被丙酮酸替代)。
白三烯的受体在药理学上分为三种类型,即BLT受体、CysLT1受体(UniProt Q9Y271),和CysLT2受体(UniProt Q9NS75)。BLT受体特异性地识别LTB4。CysLT1受体和CysLT2受体二者都识别肽白三烯,即白三烯C4 (LTC4)、白三烯D4 (LTD4),和白三烯E4 (LTE4)。
Thompson等(“他汀类药物-相关肌病(Statin-associated myopathy)”, JAMA, 2003, Vol. 289(13):1681-90)进行了文献综述,以提供他汀类药物-相关肌病的临床总结和讨论可能的介导机制。文献综述发现,在他汀类药物临床试验期间,肌肉问题的报告是罕见的,西立伐他汀是最常见涉及的他汀类药物。所述综述进一步指出,他汀类药物如何损害骨骼肌是不清楚的。
因此,对预防或改善与他汀类药物治疗相关的主要副作用的组合物和方法存在未满足的需求。
发明简述
本发明提供用于治疗一种或多种他汀类药物治疗的严重不良副作用的药用组合物、方法和药剂盒。特别地,本发明提供一种药用组合物,其包含有效量的白三烯C4 (LTC4)-介导的活性的至少一种抑制剂,用于减轻他汀类药物治疗的一种或多种不良副作用。
本发明部分基于意外的和令人吃惊的发现,即LTC4受体拮抗剂,特别是半胱氨酰白三烯受体2的拮抗剂,能够减轻HMGCoA (羟基-3-甲基戊二酰辅酶A)还原酶抑制剂(称为他汀类药物)的不良副作用,同时维持其所需的效果。本发明还部分地基于意外的和令人吃惊的发现,即半胱氨酰白三烯受体2 (CysLTR2)的拮抗剂能够减轻他汀类药物在肌细胞培养物中的不良副作用。在某些非-肌细胞性细胞系中,半胱氨酰白三烯受体1 (CysLTR1)的拮抗剂也减少他汀类药物的毒性。然而,鉴于在以下实施例部分中证实的BAY-u9773 (一种CysLTR1和CysLTR2的双特异性拮抗剂)的效果,不希望受任何理论或机制的束缚,CysLTR1的拮抗剂还可以用来保护多种类型的细胞(包括肌细胞),避免他汀类药物的不良作用。
本发明公开一种以前未认识的、由ER应激-触发剂激活的通用信号传导级联,其在启动细胞死亡中起着重要的作用。基于新发现的信号传导级联,本发明提供用于减轻他汀类药物-触发的ER应激的方法,因而治疗他汀类药物治疗的一种或多种不良副作用。
他汀类药物触发坏死,其导致核蛋白HMGB1的释放。HMGB1的释放募集诸如巨噬细胞和粒细胞的免疫细胞,其触发局部炎症和进一步的组织损伤。不与任何理论或机制关联,LTC4抑制剂将弱化由他汀类药物诱导的初始坏死。因此,LTC4抑制剂不用作炎症,特别是肌肉炎症的抑制剂,而是作为例如通过坏死的细胞死亡的抑制剂。因此须理解LTC4抑制剂防止炎症的形成而不是治疗炎症。本发明首次提议,使用LTC4抑制剂作为肌肉损伤或肌肉炎症的预防剂或治疗剂。
根据一个方面,本发明提供包含至少一种他汀类药物,和至少一种选自CysLTR2的拮抗剂,和LTC4生物合成的抑制剂的药物的药用组合物。
在某些实施方案中,他汀类药物选自阿托伐他汀、氟伐他汀、洛伐他汀、匹伐他汀、普伐他汀、罗苏伐他汀,和辛伐他汀,及其任何组合。每一种可能性表示本发明的一个独立的实施方案。
在某些实施方案中,所述药物是CysLTR2的拮抗剂。根据一些实施方案,CysLTR2拮抗剂选自:BAY-cysLT2 (CAS号712313-33-2)、BAY-u9773 (CAS号154978-38-8)、HAMI3379 (CAS号712313-35-4),及其任何组合。每一种可能性表示本发明的一个独立的实施方案。在某些实施方案中,CysLTR2的拮抗剂抑制CysLTR2的活性和半胱氨酰白三烯受体1 (CysLTR1)的活性。在某些实施方案中,所述药物是LTC4生物合成的抑制剂。在某些实施方案中,LTC4生物合成的抑制剂抑制选自微粒体谷胱甘肽S-转移酶2 (MGST2)、细胞溶质磷脂酶A2 (cPLA2)、5-脂氧合酶(5-LO),和5-脂氧合酶激活蛋白(FLAP)的酶的活性。在某些实施方案中,LTC4生物合成的抑制剂是齐留通。在某些实施方案中,5-LO的抑制剂是阿曲留通(CAS号154355-76-7)。在某些实施方案中,FLAP的抑制剂MK-886 (CAS号118414-82-7)。在某些实施方案中,以上描述的药用组合物包含他汀类药物,CysLTR2的拮抗剂,和LTC4生物合成的抑制剂。
在某些实施方案中,以上描述的药用组合物用于预防或减轻由他汀类药物在受试者的肌肉组织中诱导的不良副作用,其中受试者正接受他汀类药物治疗和经历或有风险经历由他汀类药物诱导的不良副作用。
根据某些实施方案,不良副作用是他汀类药物-诱导的肌病。在某些实施方案中,肌病选自肌炎和横纹肌溶解。每一种可能性表示本发明的一个独立的实施方案。
本发明在另一个方面提供一种预防或减轻由他汀类药物在受试者的肌肉组织中诱导的不良副作用的方法,其包括给予受试者至少一种他汀类药物和至少一种选自CysLTR2的拮抗剂和LTC4生物合成的抑制剂的药物的步骤,其中受试者正接受他汀类药物治疗和经历或有风险经历由他汀类药物诱导的不良副作用。
根据某些实施方案,不良副作用是他汀类药物-诱导的肌病。在某些实施方案中,肌病选自肌炎和横纹肌溶解。每一种可能性表示本发明的一个独立的实施方案。
本发明在另一个方面还提供一种药剂盒,其包含含有至少一种他汀类药物的药用组合物,和含有至少一种选自CysLTR2的拮抗剂和LTC4生物合成的抑制剂的药物的药用组合物。
在某些实施方案中,他汀类药物选自阿托伐他汀、氟伐他汀、洛伐他汀、匹伐他汀、普伐他汀、罗苏伐他汀,和辛伐他汀,及其任何组合。每一种可能性表示本发明的一个独立的实施方案。
在某些实施方案中,所述药物是CysLTR2的拮抗剂。根据一些实施方案,CysLTR2拮抗剂选自:BAY-cysLT2 (CAS号712313-33-2)、BAY-u9773 (CAS号154978-38-8)、HAMI3379 (CAS号712313-35-4),及其任何组合。每一种可能性表示本发明的一个独立的实施方案。在某些实施方案中,CysLTR2的拮抗剂抑制CysLTR2的活性和半胱氨酰白三烯受体1 (CysLTR1)的活性。在某些实施方案中,所述药物是LTC4生物合成的抑制剂。在某些实施方案中,LTC4生物合成的抑制剂抑制选自微粒体谷胱甘肽S-转移酶2 (MGST2)、细胞溶质磷脂酶A2 (cPLA2)、5-脂氧合酶(5-LO),和5-脂氧合酶激活蛋白(FLAP)的酶的活性。在某些实施方案中,LTC4生物合成的抑制剂是齐留通。在某些实施方案中,5-LO的抑制剂是阿曲留通(CAS号154355-76-7)。在某些实施方案中,FLAP的抑制剂是MK-886 (CAS号118414-82-7)。在某些实施方案中,以上描述的药剂盒包含含有他汀类药物的药用组合物,包含CysLTR2的拮抗剂的药用组合物,和包含LTC4生物合成的抑制剂的药用组合物。
在某些实施方案中,以上描述的药剂盒还包含给予他汀类药物和所述药物至受试者的用药说明书,所述受试者接受他汀类药物治疗和经历或有风险经历由他汀类药物诱导的不良副作用。在某些实施方案中,以上描述的药剂盒用于预防或减轻由他汀类药物在受试者的肌肉组织中诱导的不良副作用,其中受试者正接受他汀类药物治疗和经历或有风险经历由他汀类药物诱导的不良副作用。在某些实施方案中,不良副作用是他汀类药物-诱导的肌病。在某些实施方案中,肌病选自肌炎和横纹肌溶解。在某些实施方案中,应用包括在给予他汀类药物至受试者之前、期间和/或之后给予所述药物。每一种可能性表示本发明的一个独立的实施方案。
本发明在一方面还提供一种药用组合物,其包含用于预防或减轻由他汀类药物在受试者的肌肉组织中诱导的不良副作用的半胱氨酰白三烯受体2 (CysLTR2)的至少一种拮抗剂,其中受试者正接受他汀类药物治疗和经历或有风险经历由他汀类药物诱导的不良副作用。
根据另一个方面,本发明提供一种药用组合物,其包含用于预防或减轻由他汀类药物在受试者的肌肉组织中诱导的不良副作用的LTC4生物合成的至少一种抑制剂,其中受试者正接受他汀类药物治疗和经历或有风险经历由他汀类药物诱导的不良副作用,和其中LTC4生物合成的至少一种抑制剂抑制选自微粒体谷胱甘肽S-转移酶2 (MGST2)、细胞溶质磷脂酶A2 (cPLA2)、5-脂氧合酶(5-LO),和5-脂氧合酶激活蛋白(FLAP)的酶的活性。
根据某些实施方案,不良副作用是他汀类药物-诱导的肌病。在某些实施方案中,肌病选自肌炎和横纹肌溶解。每一种可能性表示本发明的一个独立的实施方案。
在某些实施方案中,他汀类药物选自阿托伐他汀、氟伐他汀、洛伐他汀、匹伐他汀(CAS号147511-69-1)、普伐他汀、罗苏伐他汀(CAS号287714-41-4),和辛伐他汀,及其任何组合。每一种可能性表示本发明的一个独立的实施方案。
在某些实施方案中,CysLTR2的拮抗剂抑制CysLTR2的活性和半胱氨酰白三烯受体1 (CysLTR1)的活性。根据一些实施方案,CysLTR2拮抗剂选自:BAY-cysLT2 (CAS号712313-33-2)、BAY-u9773 (CAS号154978-38-8)、HAMI3379 (CAS号712313-35-4),及其任何组合。每一种可能性表示本发明的一个独立的实施方案。
在某些实施方案中,描述的药用组合物包含他汀类药物、CysLTR2的拮抗剂和LTC4生物合成的抑制剂,所述抑制剂抑制选自MGST2、cPLA2、5-LO和FLAP的酶的活性。每一种可能性表示本发明的一个独立的实施方案。在某些实施方案中,5-LO的抑制剂是齐留通。在某些实施方案中,5-LO的抑制剂是阿曲留通。在某些实施方案中,FLAP的抑制剂MK-886。
在某些实施方案中,上述药用组合物包含CysLTR2的拮抗剂和LTC4生物合成的抑制剂,所述抑制剂抑制选自微粒体谷胱甘肽S-转移酶2 (MGST2)、细胞溶质磷脂酶A2 (cPLA2)、5-脂氧合酶(5-LO),和5-脂氧合酶激活蛋白(FLAP)的酶的活性。
根据一些实施方案,以上描述的药用组合物还包含有效量的他汀类药物,其选自阿托伐他汀、西立伐他汀、氟伐他汀、洛伐他汀、美伐他汀、匹伐他汀、普伐他汀、罗苏伐他汀、辛伐他汀,及其任何组合。每一种可能性表示本发明的一个独立的实施方案。
根据另外的实施方案,他汀类药物使用的剂量在类似于当单独给予用于它们的批准的适应症时批准的药物的剂量的范围内。
本发明在另一个方面提供一种预防或减轻由他汀类药物在受试者的肌肉组织中诱导的不良副作用的方法,其包括给予CysLTR2的至少一种拮抗剂至受试者的步骤,其中受试者正接受他汀类药物治疗和经历或有风险经历由他汀类药物诱导的不良副作用。
本发明在又一个方面提供一种预防或减轻由他汀类药物在受试者的肌肉组织中诱导的不良副作用的方法,其包括给予受试者LTC4生物合成的至少一种抑制剂的步骤,其中受试者正接受他汀类药物治疗和经历或有风险经历由他汀类药物诱导的不良副作用,和其中LTC4生物合成的抑制剂抑制选自MGST2、cPLA2、5-LO和FLAP的酶的活性。
在某些实施方案中,不良副作用是他汀类药物-诱导的肌病。在某些实施方案中,不良副作用是他汀类药物-诱导的肌病。
在以上描述的方法的某些实施方案中,白三烯介导的活性的抑制剂在用他汀类药物治疗之前,与用他汀类药物治疗同时,或与用他汀类药物治疗一起给予用他汀类药物治疗的受试者。在以上描述的方法的某些实施方案中,白三烯介导的活性的抑制剂在用他汀类药物治疗之后给予用他汀类药物治疗的受试者。每一种可能性表示本发明的一个独立的实施方案。
在一些实施方案中,本发明提供用于治疗他汀类药物治疗的一种或多种副作用的方法,其包括给予经受他汀类药物治疗的受试者有效量的白三烯介导的活性的抑制剂。根据一些实施方案,白三烯介导的活性的抑制剂可单独或与有效量的他汀类药物联合给予。
给予受试者的任何合适的途径可用于本发明的组合物。优选的给药方式将取决于要治疗的具体适应症并且对本领域技术人员而言将是显而易见的。
根据一些实施方案,本发明的药用组合物可作为一种用于口服给予的丸剂提供。
构成治疗的组分的组合可同时地(作为分开的剂型或作为一种单一的组合物),顺序地,或者经合适的时间间隔分开给予。每一种可能性表示本发明的一个独立的实施方案。当组分作为分开的剂型,即不作为合并的组合物给予时,各组分可以相同的形式或不同的形式,如口服、经鼻、胃肠外或经皮给予。当化合物同时、顺序或分开给予时,组分可作为分开的剂型提供。
当组分作为分开的剂型,即不作为紧密的组合物给予时,各组分可以相同的形式或不同的形式,如口服、经鼻、胃肠外、直肠、阴道或经皮给予。当化合物同时、顺序或分开给予时,组分可作为分开的剂型提供。任选地,组合的组分可以药剂盒形式提供,其中药剂盒优选地以适合于分开给予各组分的隔室形式存在。
或者,当组合的组分同时给予时,它们可作为含有两种或更多种组分的单一的组合物提供或可以药剂盒形式提供,其中药剂盒被分割成用于同时给予各组分的隔室。
当白三烯介导的活性的抑制剂的药用组合物和治疗性他汀类药物作为分开的剂型给予时,各自可用一种或多种药学上可接受的载体配制在一起,以形成组合物。当治疗的组分作为单一组合物给予时,组合物也可任选地包含一种或多种药学上可接受的载体。
药用组合物的制剂是本领域技术人员熟知的。这样的组合物可含有任何合适的载体例如,稀释剂或赋形剂,其在与组合物中的其它成分相容且对受试者无害的意义上说是药学上可接受的。
合适的载体包括所有常规溶剂、油、分散媒介、填充剂、固体载体、包衣料、抗真菌剂和抗菌剂、皮肤渗透剂(合适时)、表面活性剂、等渗剂和吸收剂等。应该理解,本发明的组合物也可包括其它的补充生理活性剂。
本发明的化合物和/或药用组合物可通过本领域已知的任何合适的方法给予。例如,以非限制的方式,它可以经局部、胃肠外、口服、鼻内、静脉内、肌内、皮下,或通过其它的合适的方式给予。每一种可能性表示本发明的一个独立的实施方案。在优选的实施方案中,本发明的化合物和/或组合物可适合配制为口服给予(虽然在适宜的情况下,其它形式也可考虑)并可以选自以下的离散的单位形式配制:离散的单位例如胶囊、袋装粉末剂、颗粒剂和片剂。每一种可能性表示本发明的一个独立的实施方案。根据另一个实施方案,本发明的化合物和/或组合物可以选自粉末剂、颗粒剂、溶液剂、水性或非-水性液体中的混悬剂、油剂、糊剂、水包油液体乳剂和油包水液体乳剂的形式配制。每一种可能性表示本发明的一个独立的实施方案。在一优选的实施方案中,药用组合物被配制为用于口服给予的含有他汀类药物和白三烯介导的活性的抑制剂二者的单一丸剂。
片剂可任选地与一种或多种辅助成分一起经压制或模塑成型制得。压制片剂可通过在合适的机器中压制呈自由流动形式例如粉末或颗粒,任选地与粘合剂(如惰性稀释剂、防腐剂、崩解剂(如羟基乙酸淀粉钠、交联聚乙烯吡咯烷酮、交联羧甲基纤维素钠)、表面活性剂或分散剂混合的活性成分来制备。模塑成型片剂可通过在合适的机器中模压用惰性液体稀释剂润湿的粉末状化合物的混合物制得。片剂可任选地被包衣或刻痕并可使用例如,不同比例的羟丙基甲基纤维素配制,以提供其中活性成分的缓慢或控制释放,以提供所需的释放特性。片剂可任选地用肠溶衣提供,以提供在并非胃的肠道部分释放。化合物也可以硬或软明胶胶囊的形式存在。应该理解,除了上文特别提及的活性成分,考虑到所提及组合物的类型,本发明的组合物还可包含本领域常规的其它试剂或添加剂,例如,适合于口服给予的那些可包括这样的其它试剂如粘合剂、甜味剂、增稠剂、矫味剂、崩解剂、包衣料、防腐剂、润滑剂和/或时间延迟剂。合适的甜味剂包括蔗糖、乳糖、葡萄糖、阿斯巴甜和糖精。合适的崩解剂包括玉米淀粉、甲基纤维素、聚乙烯吡咯烷酮、黄原胶、膨润土、藻酸或琼脂。每一种可能性表示本发明的一个独立的实施方案。
根据一些实施方案,使用与本发明的药用组合物的联合疗法将提供与所述他汀类药物在作为单一疗法给予时类似的胆固醇降低作用,同时减轻其副作用。
根据一些实施方案,以上描述的药用组合物还包含药学上可接受的载体。
本发明的其它目的、特征和优势将从以下的描述和附图而变得清楚。
附图简述
图1.ER应激触发的细胞死亡机制的原理性描述。
图2A-2B.ER应激调节MGST2和5-LO的表达。(2A) 用衣霉素(1 μg/ml, 24小时)处理人WISH上皮细胞和人HaCaT前-角质细胞后,微粒体谷胱甘肽S转移酶(MGST2)和裂解的胱天蛋白酶3 (Casp. 3)表达的动力学,如通过免疫印迹法所测定的。(2B) 在用布雷菲尔德菌素A (0.66 μg/ml, 24小时)处理人WISH上皮细胞和用衣霉素(1 μg/ml, 24小时)处理小鼠B16细胞后,5-脂氧合酶(5-LO)表达的动力学,如通过免疫印迹法所测定的。
图3. 用布雷菲尔德菌素A (0.66 µg/ml, 24小时)处理人WISH细胞,和用衣霉素(1 μg/ml, 24小时)处理小鼠B16细胞后,CysLTR1和CysLTR2表达的动力学,如通过免疫印迹法所测定的。
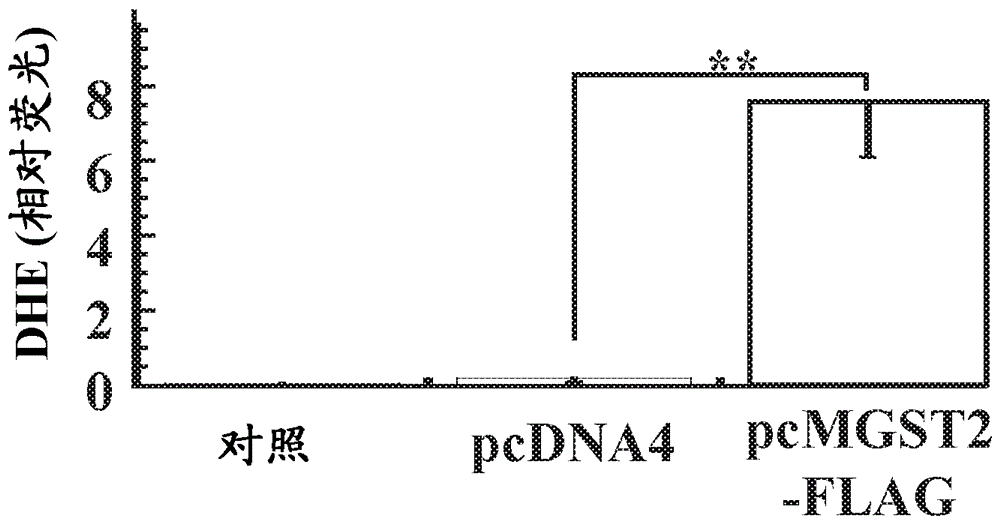
图4A-4E.ER应激触发ROS积聚由MGST2-衍生的LTC4介导。(4A)小鼠B16细胞用对照siRNA或MGST2 siRNA转染,然后用衣霉素处理并用超氧化物阴离子指示剂二氢乙啶(dihydroethidium) (DHE)染色。如通过ImageJ程序确定的表示相对DHE荧光强度的定量。N=3, ***p< 0.001。(4B)小鼠B16细胞用对照siRNA或MGST2 siRNA转染,然后用衣霉素处理并用超氧化物阴离子指示剂二氢乙啶(DHE)染色。如通过qRT-PCR在重复样本上测定的小鼠MGST2 mRNA的沉默效率被示出。N=3, ***p< 0.001。(4C) 人HaCaT前-角质细胞用布雷菲尔德菌素A (0.33 μg/ml, 24小时),伴有或没有MRP1抑制剂reversan (20 µM)或LTC4拮抗剂普仑司特、BAY-cysLT2或BAY-u9773处理。然后用ROS指示剂DCFH-DA对细胞染色。提供相对DCF荧光强度的定量分析。所有抑制剂(除了reversan)显著抑制ROS积聚。N=3, * p< 0.05。(4D) 在用对照pcDNA4载体或者pcMGST2-FLAG转染后,用抗FLAG标签进行HEK 293T细胞提取物的免疫印迹。(4E) HEK 293T细胞经模拟-转染(对照),用pcDNA4转染或者用pcMGST2-FLAG转染,然后用DHE染色。提供相对DHE荧光强度的定量分析,N=4, **p < 0.01。
图5.ER应激触发的ROS积聚由LTC4介导。小鼠B16细胞用衣霉素(1 μg/ml, 24小时),伴有或没有BAY-u9773 (1 µM)或孟鲁司特(5 µM)处理。24小时后,细胞用DCF-DA (10 μM, 40 min)染色。提供了相对DCF荧光强度的定量分析。DCF的平均强度被归一化为使用Photoshop的直方图功能的细胞核染色。数据为测量三个视场的平均±SD。(**p < 0.01)。
图6A-6C.ER应激触发的NADPH氧化酶4 (NOX4)表达,朝向核的易位和DNA氧化通过MGST2-LTC4途径介导。(6A) 在BAY-u9773、MK571或孟鲁司特的存在或不存在下,用媒介(对照)或布雷菲尔德菌素A (0.66 μg/ml, 24小时)处理人WISH细胞。NOX4水平通过免疫印迹评估。(6B) 在孟鲁司特、BAY-u9773或BAY-cysLT2的存在或不存在下,用媒介(对照)或衣霉素(0.5 µg/ml, 24小时)处理小鼠B16细胞。NOX4水平通过免疫印迹评估。(6C) 在普仑司特或BAY-u9773的存在或不存在下,用媒介(对照)或布雷菲尔德菌素A (48小时)处理人HaCaT前-角质细胞,然后用抗8-OHdG免疫染色。8-OHdG荧光强度的定量分析被示出。N= 3 *p <0.05, ****p <0.001。
图7A-7G.MGST2-LTC4途径介导ER应激-触发的细胞死亡。(7A)小鼠B16细胞用对照siRNA (siControl)或MGST2-特异性siRNA (siMGST2)转染,用媒介或衣霉素(1 μg/ml, 24小时) (Tm)处理,然后用结晶紫染色。细胞的相对成活力被示出。N=3 ***p< 0.0001。(7B)小鼠B16细胞用对照siRNA (siControl)或MGST2-特异性siRNA (siMGST2)转染,用媒介或衣霉素(Tm)处理,然后用结晶紫染色。如通过qRT-PCR在重复样本上测定的小鼠MGST2 mRNA的沉默效率被示出。N=3, ***p<0.001。(7C) HEK 293T细胞被模拟转染,用pcDNA4或用pcMGST2-FLAG转染,用媒介(对照)或布雷菲尔德菌素A (0.66 μg/ml, 24小时)处理,然后用结晶紫染色。细胞的相对成活力被示出。N=3 ****p< 0.0001。(7D) 在BAY-u9773 (80 nM)的存在或不存在下,用媒介(对照)或布雷菲尔德菌素A (BfA, 1.33 µg/ml, 48小时)处理人HaCaT前-角质细胞。然后用结晶紫染色各板。细胞的相对成活力被示出。N=4 ***p< 0.0001。(7E) 人WISH细胞用媒介(对照)或者衣霉素(1 µg/ml, 48小时)在BAY-u9773 (80 nM)的存在或不存在下处理,然后用中性红染色。细胞的相对成活力被示出。N=4 ***p< 0.0001。(7F) 人HaCaT前-角质细胞用媒介(对照)或MG262 (0.05 µM)在齐留通(10 μM, 24小时)的存在或不存在下处理。然后用结晶紫染色各板。细胞的相对成活力被示出。N=4, ***p< 0.001。(7G) 人WISH细胞用媒介(对照)或布雷菲尔德菌素A (0.66 μg/ml, 24小时)在普仑司特(10 μM)的存在或不存在下处理。然后用结晶紫染色各板。细胞的相对成活力被示出。N=3, ***p< 0.001。
图8A-8C. MGST2-LTC4途径的抑制剂减少ER应激-触发的细胞死亡。(8A) 人WISH细胞用媒介(对照)或布雷菲尔德菌素A (0.66 μg/ml, 48小时)在BAY-cysLT2的存在或不存在下处理。然后用结晶紫染色各板。细胞的相对成活力被示出。N=4, ***p< 0.001。(8B)小鼠B16细胞用媒介(对照)或衣霉素(1 μg/ml, 24小时) (Tm)、毒胡萝卜素(50 nM, 24小时) (Tg)或布雷菲尔德菌素A (BfA 1.3 µM, 24小时)在MK571的存在或不存在下处理24 h。用结晶紫染色各板。细胞的相对成活力被示出。N=4, ***p< 0.001。(8C) 释放至用媒介(对照)或布雷菲尔德菌素A (1.3 µg/ml, 24小时)在MK571的存在或不存在下处理的B16细胞的培养基的坏死标记物HMGB1的免疫印迹。丽春红(Ponceau)染色用作加载对照。
图9A-9G.MGST2缺乏减弱ER应激触发的体内死亡。(9A) 通过来自WT和纯合MGST2-缺乏(KO) 129svEvBrd小鼠的尾端的DNA的PCR获得的PCR产物的琼脂糖凝胶电泳。杂合ES细胞(ES)和阴性PCR对照的DNA也示出。(9B) WT和第2代MGST2-缺乏小鼠胚胎的成纤维细胞用媒介(对照)或衣霉素(1 μg/ml, 24小时)处理。然后用结晶紫染色各板。如通过中性红染色所测定的,用媒介(对照)或衣霉素处理的WT和第2代MGST2-缺乏小鼠胚胎的相对成活力被示出。(9C) 如通过在WT和在MGST2缺乏MEFs中的裂解的胱天蛋白酶3 (Casp. 3)的免疫印迹所测定的布雷菲尔德菌素A诱导的细胞凋亡。(9D) WT和第2代MGST2-缺乏小鼠胚胎的成纤维细胞用媒介(对照)或衣霉素(2 µg/ml, 24小时)或布雷菲尔德菌素A (0.25 μg/ml, 24小时)处理,然后用DCFH-DA染色。DCF荧光强度的定量分析被示出。N=3 *** p < 0.001。(9E) 衣霉素(1 mg/kg)在时间=0经ip给予WT和MGST2-缺乏小鼠(5/组)一次。切除肾并用苏木精-曙红染色切片。对肾近端小管的损害的定量分析被示出。使用Photoshop的套索工具(lasso tool)选择含有近端小管的整个肾面积的图像并使用ImageJ程序对空泡计数。N=5, ***p < 0.03。(9F) 在时间=0经ip给予衣霉素(2.5 mg/kg)一次的WT和MGST2-缺乏小鼠(20/组)的存活。对显示出严重发病率的小鼠实施安乐死以减少痛苦。(9G) 在时间=0经ip给予衣霉素(2.5 mg/kg)一次的和或者给予媒介(对照)或者普仑司特(1 mg/kg/天,3天)的WT小鼠(4/组)的存活。对显示出严重发病率的小鼠实施安乐死以减少痛苦。
图10. 普仑司特减少辛伐他汀触发的细胞死亡。用指定浓度的辛伐他汀在普仑司特(10 μM, 48小时)的存在(虚线)或不存在(连续线)下处理的人WISH细胞的存活。然后用结晶紫染色各板并确定细胞的相对成活力。
图11A-11B. BAY-cysLT2和BAY-u9773而不是普仑司特减少辛伐他汀-触发的分化的C2C12小鼠肌细胞的细胞死亡。(11A) 在小鼠C2C12细胞分化为肌细胞后,在媒介、普仑司特(10 µM)、BAY-cysLT2 (10 µM)、BAY-u9773 (1 µM)或甲羟戊酸(71.4 µM)的存在下,用10 uM 辛伐他汀处理5天的存活。然后用结晶紫染色各板并测定相对细胞成活力。(11B) 染色强度的定量被示出。
图12A-12B. 齐留通而不是孟鲁司特减少辛伐他汀-触发的分化的C2C12小鼠肌细胞的死亡。(12A) 使C2C12无限增殖化小鼠成肌细胞(15,000细胞/100 ul DMEM)接种24,然后分化3天(由补充有1xITS培养基的无血清培养基启动),然后用20 µg/ml辛伐他汀(伴有或没有孟鲁司特(2 µM)、齐留通(10 µM)或甲羟戊酸(71.4 µM))处理。4天后,细胞用结晶紫染色并在光镜下拍照。(12B) 染色强度的定量被示出。
发明详述
本发明提供用于治疗他汀类药物治疗的一种或多种不良副作用的方法、药用组合物和药剂盒。特别地,本发明提供一种药用组合物,其包含有效量的用于减轻他汀类药物治疗的一种或多种不良副作用的白三烯介导的活性的至少一种抑制剂。
本发明的发明人已揭示先前未识别的、由ER应激-触发剂激活的通用信号传导级联,其在启动细胞死亡中起着重要的作用。基于新发现的信号传导级联,本发明提供减轻他汀类药物-触发的ER应激的方法,因而治疗他汀类药物治疗的一种或多种不良副作用。
为有利于理解本发明,许多术语和短语在下文定义。要理解这些术语和短语是为了描述的目的并不是限制目的,以致本说明书的术语或措辞是由熟练技术人员根据本文提出的教导和指导,结合本领域普通技术人员的知识来解释。
本发明首次揭示先前未识别的、由ER应激-触发他汀类药物激活的通用信号传导级联,其在接受他汀类药物的患者中启动细胞死亡方面起着重要的作用。本发明提供通过抑制所述信号传导级联,减少他汀类药物-触发的ER应激的方法。此外,本发明公开由ER应激-触发剂,以及由他汀类药物激活的、作为一种先前未识别的信号传导级联的MGST2-LTC4。
本发明部分基于以下意外的发现:(a) 由特异性试剂例如衣霉素、毒胡萝卜素和布雷菲尔德菌素A引发的ER应激,至少部分通过白三烯C4 (LTC4)的生成触发细胞死亡;(b) 这种LTC4通过酶MGST2生成,并且ER应激通过其共易位(co-translocation)至核被膜且其与cPLA2、5LO和FLAP共定位在一起激活MGST2;(c) ER应激也触发两个LTC4受体(CysLT1和cysLT2)的易位至核被膜,并且它们与LTC4的合成机器共定位,从而能使LTC4的胞内分泌行动局部化;(d) LTC4结合于其内在化的受体激活NADPH氧化酶4,导致ROS积聚,并且由于ER应激,LTC4是氧化应激的主要触发剂;和(e) 由ER应激-激活的MGST2-LTC4途径介导的ROS积聚导致DNA损伤以及随后的细胞死亡(图1)。在对ER应激的反应(通常称为“未折叠蛋白反应” (UPR))中,LTC4生物合成机器及其受体易位和在核被膜共定位。结果NOX4被激活,生成ROS,其造成氧化性DNA损伤和随后的细胞死亡。因此,本发明公开了一种由ER应激激活的主要的死亡-触发途径。
本发明还公开了LTC4受体拮抗剂例如孟鲁司特和普仑司特减少他汀类药物例如辛伐他汀以及阿托伐他汀、西立伐他汀、氟伐他汀、洛伐他汀、美伐他汀(CAS号73573-88-3)、匹伐他汀、普伐他汀和罗苏伐他汀的毒性。本发明还公开了LTC4受体拮抗剂保护野生型小鼠避免ER应激触发的发病。几种LTC4受体拮抗剂(孟鲁司特、普仑司特等)被开发并被批准为治疗哮喘综合征的药物。所有批准为药物的LTC4受体拮抗剂是CysLT1受体的选择性抑制剂。选择性抑制剂也被开发用于CysLT2,第二种LTC4受体,但到目前为止,它们还没有经受临床的开发。早先的研究证实CysLTR1和CysLTR2,两种LTC4受体,在不同类型的组织中并非同等表达的。因此,本发明也提供用于缓解他汀类药物的一些主要不良副作用的Cys LT2受体拮抗剂以及cysLT1和CysLT2受体拮抗剂的组合。本发明也公开非-选择性LTC4受体拮抗剂在缓解他汀类药物的一些不良副作用中的用途。本发明还提供用于缓解他汀类药物的一些不良副作用的白三烯生物合成的抑制剂。
根据一个方面,本发明提供包含至少一种他汀类药物,和至少一种选自CysLTR2的拮抗剂,和LTC4生物合成的抑制剂的药物的药用组合物。
本发明在另一个方面提供一种预防或减轻由他汀类药物在受试者的肌肉组织中诱导的不良副作用的方法,其包括给予受试者至少一种他汀类药物和至少一种选自CysLTR2的拮抗剂和LTC4生物合成的抑制剂的药物的步骤,其中受试者正接受他汀类药物治疗和经历或有风险经历由他汀类药物诱导的不良副作用。
本发明在另一个方面还提供一种药剂盒,其包含含有至少一种他汀类药物的药用组合物,和包含至少一种选自CysLTR2的拮抗剂和LTC4生物合成的抑制剂的药物的药用组合物。
本发明在一个方面还提供一种药用组合物,其包含用于预防或减轻由他汀类药物在受试者的肌肉组织中诱导的不良副作用的半胱氨酰白三烯受体2 (CysLTR2)的至少一种拮抗剂,其中受试者正接受他汀类药物治疗和经历或有风险经历由他汀类药物诱导的不良副作用。
根据另一个方面,本发明提供一种药用组合物,其包含用于预防或减轻由他汀类药物在受试者的肌肉组织中诱导的不良副作用的LTC4生物合成的至少一种抑制剂,其中受试者正接受他汀类药物治疗和经历或有风险经历由他汀类药物诱导的不良副作用,和其中LTC4生物合成的至少一种抑制剂抑制选自微粒体谷胱甘肽S-转移酶2 (MGST2)、细胞溶质磷脂酶A2 (cPLA2)、5-脂氧合酶(5-LO),和5-脂氧合酶激活蛋白(FLAP)的酶的活性。
本发明在另一个方面提供一种预防或减轻由他汀类药物在受试者的肌肉组织中诱导的不良副作用的方法,其包括给予CysLTR2的至少一种拮抗剂至受试者的步骤,其中受试者正接受他汀类药物治疗和经历或有风险经历由他汀类药物诱导的不良副作用。
本发明在又一个方面提供一种预防或减轻由他汀类药物在受试者的肌肉组织中诱导的不良副作用的方法,其包括给予受试者LTC4生物合成的至少一种抑制剂的步骤,其中受试者正接受他汀类药物治疗和经历或有风险经历由他汀类药物诱导的不良副作用,和其中LTC4生物合成的抑制剂抑制选自MGST2、cPLA2、5-LO和FLAP的酶的活性。
胆固醇治疗领域是日益增长和不断变化的,因为广泛的研究和资源致力于克服这种病症。因此,正如上面所阐述的,任何类型的影响人体胆固醇水平的分子均被认为是根据本发明的“他汀类药物”。如本文所用的术语“他汀类药物”指用来通过抑制酶HMG-CoA还原酶降低人的胆固醇水平的任何药剂、分子或药物。一般来说,减少或消除酶HMG-CoA还原酶的酶活性的任何分子均被本发明认为是“他汀类药物”。特别是,减弱从3-羟基-3-甲基-戊二酰-CoA (HMG-CoA)转化为甲羟戊酸的HMG-CoA还原酶的任何分子均被本发明认为是“他汀类药物”。
如本文所用的术语“药用组合物”指包含至少一种生物活性剂,和至少一种药学上可接受的载体的任何组合物。生物活性分子的非-限制性实例是白三烯受体的拮抗剂,例如受体CysLTR1和CysLTR2的拮抗剂,和LTC4生物合成的抑制剂,例如酶MGST2、cPLA2、5-LO和FLAP的抑制剂。如本文所用的术语“药剂”指具有生物学活性的任何分子。
如本文所用的,术语"药学上可接受的载体"意指药学上可接受的物质、组合物或媒介,例如液体或固体填充剂、稀释剂、赋形剂、溶剂或封囊物质,涉及携带或转运受试者体内的本发明的化合物或携带或转运本发明的化合物至受试者,以致它能执行其预定的功能。载体在与制剂中的其它成分相容且对患者无害的意义上说必须是"可接受的"。
如本文所用的术语“白三烯C4 (LTC4)受体的拮抗剂”、“CysLTR1的拮抗剂”、“CysLTR1拮抗剂”、“CysLTR2的拮抗剂”,和“CysLTR2拮抗剂”指能够阻断、抑制、减轻或干扰所指定的白三烯C4受体的活性或功能的任何药剂。这些术语还指能够阻断、抑制、减轻或干扰白三烯C4受体的表达的任何药剂。白三烯C4受体拮抗剂的非-限制性实例是孟鲁司特、扎鲁司特和普仑司特。
如本文所用的,术语“白三烯介导的活性的抑制剂”指白三烯受体拮抗剂、白三烯生物合成抑制剂或其组合。根据本发明的发现,白三烯受体拮抗剂和白三烯生物合成抑制剂二者都能够抑制白三烯介导的活性,因此可作为替代或相互组合使用。如本文所用的,术语“LTC4介导的活性的抑制剂”指LTC4受体拮抗剂、LTC4生物合成抑制剂或其组合。如本文所用的,术语“白三烯”指选自以下的白三烯:LTC4、LTD4、LTE4及其任何组合。如本文所用的,术语"受体拮抗剂"指一种受体的配体,其在结合于受体时发挥对该受体活性的完全或部分抑制作用,例如,LTC4受体拮抗剂引起LTC4受体的抑制作用。如本文所用的,术语"受体的配体"指特异性地结合受体并从而引起受体的激活或者抑制作用的化合物。如本文所用的,“白三烯受体拮抗剂”是能够特异性地结合于白三烯受体,和能够完全或部分地抑制即灭活所述受体的任何化合物。因此白三烯受体拮抗剂是一种通过白三烯受体的结合和灭活而发挥其主要作用的化合物。术语"LTC4受体拮抗剂"、"白三烯受体拮抗剂"、"CysLT1受体拮抗剂",和"CysLT2受体拮抗剂"在此意欲覆盖任何药学上可接受的盐、酯、溶剂合物,或水合物,其在给予接受者时能够提供(直接地或间接地)如本文描述的拮抗剂。盐的制备可通过本领域已知的方法进行。根据一些实施方案,白三烯介导的活性的抑制剂选自:白三烯C4(LTC4)介导的活性的抑制剂、白三烯D4 (LTD4)介导的活性的抑制剂、白三烯E4 (LTE4)介导的活性的抑制剂,及其任何组合。每一种可能性表示本发明的一个独立的实施方案。
术语“预防”一般指在接受他汀类药物的受试者中消除或延缓与他汀类药物治疗有关的急性和/或慢性不良副作用的最初发生。预防可以是完全的,例如,完全没有对组织或细胞的损伤。预防也可以是部分的,以致由他汀类药物诱导的对组织或细胞的损伤小于在没有LTC4受体拮抗剂和/或LTC4生物合成抑制剂的作用时将出现的损伤。
术语“减轻”一般指在接受他汀类药物的受试者中减轻与他汀类药物治疗有关的急性和/或慢性不良副作用的总体严重性和/或加速与他汀类药物治疗有关的急性和/或慢性不良副作用的消散。
如本文可互换使用的术语"治疗"和"缓解损伤"包括减少、缓解,或改善与他汀类药物的毒性相关的或由汀类药物的毒性诱导的至少一种症状。如本文所用的术语"治疗"也包括预防性(如防止)、姑息性和治愈性治疗。
如本文所用的术语“肌病”指任何一种已知的肌肉疾病,其中肌肉纤维由于许多原因中的任何一种而不起作用,导致肌肉无力。肌肉抽搐、僵硬和痉挛也可能与肌病相关。肌肉疾病按性质可分类为神经肌肉的或肌肉骨骼的。一些病症,例如肌炎,可被考虑同时为神经肌肉的和肌肉骨骼的。肌病是遗传的或后天获得的。在某些实施方案中,术语“肌病”指药物-诱导的肌病,例如在国际疾病分类(International Classification of Diseases) (ICD)-10-CM诊断代码G72.0中列出的。
如本文所用的术语“肌炎”一般指肌肉的炎症。受试者血液中肌酸激酶(一种坏死的标记物)的升高通常是肌炎的指示,因为肌炎往往作为细胞坏死的结果发生。肌炎的类型包括,但不限于骨化性肌炎、特发性炎性肌病、皮肌炎、青少年皮肌炎、多发性肌炎,包涵体肌炎,和脓性肌炎。在某些实施方案中,肌炎选自骨化性肌炎和特发性炎性肌病。每一种可能性表示本发明的一个独立的实施方案。
如本文所用的术语“横纹肌溶解”指其中损伤的骨骼肌肉组织迅速分解的一种病症。损伤的肌肉细胞的分解产物被释放进入血流中,这些中的一些,例如蛋白肌红蛋白,对肾脏有害并可能导致肾衰竭。症状的严重性(可包括肌肉疼痛、呕吐、意识错乱)取决于肌肉损伤的程度和肾衰竭是否发展。诊断通常是进行血液检验和尿分析。
术语“受试者”或“有需要的受试者”在此可互换使用并指经受他汀类药物治疗并罹患所述不良他汀类药物治疗副作用的受试者。根据一些实施方案,受试者是人。在一些实施方案中,受试者是哺乳动物。
在某些实施方案中,受试者患有高水平的血液低密度脂蛋白(LDL)胆固醇。在某些实施方案中,受试者患有高胆固醇血症。如本文所用的术语“高胆固醇血症”、“血胆甾醇过多”和“血脂障碍”指在血液中存在高水平的胆固醇。高胆固醇血症是"高脂血症" (血液中的脂质水平升高)和"高脂蛋白血症" (血液中的脂蛋白水平升高)的一种形式。
在某些实施方案中,接受他汀类药物治疗的受试者正接受高剂量的他汀类药物。在某些实施方案中,短语“接受高剂量的他汀类药物”意思是接受每日至少20、至少30、至少40、至少50、至少60、至少80,或至少80 mg的他汀类药物。
术语“包含”、"含有"、"有"、“包括”、"含"、"包纳"、“具有”意指"包括但不限于"。术语“由…组成”意指“包括且限于”。如本文所用的,单数形式"一"、"一个"和"该" 包括复数指代,除非文中另外清楚地指明。例如,术语"一个化合物"或"至少一个化合物"可包括多个化合物,包括其混合物。
如本文所用的术语“抑制受体的活性”指能够预防、阻断或减少受体的信号-转导级联或生物学活性的任何药剂。例如,半胱氨酰白三烯激活CysLTR1和CysLTR2、G蛋白-偶联受体,以在非-造血细胞中启动包括MGST2的激活的信号转导级联。
如本文所用的术语“白三烯生物合成抑制剂”指选自以下的抑制剂:微粒体谷胱甘肽S-转移酶2 (MGST2)抑制剂、细胞溶质磷脂酶A2 (cPLA2)抑制剂、5-脂氧合酶(5-LO)抑制剂、5-脂氧合酶激活蛋白(FLAP)抑制剂及其组合。如本文所用的术语“LTC4生物合成的抑制剂”指能够预防、阻断或减少LTC4的合成的任何药剂。如在背景部分所详细描述的,cPLA2从磷脂释放花生四烯酸,5-LO和FLAP将其氧化为反应性中间体LTA4和LTC4S缀合LTA4与谷胱甘肽,以形成LTC4。这些酶的任何一种的任何抑制剂被考虑为LTC4生物合成的抑制剂。如本文所用的术语“抑制酶的活性”指能够预防、阻断或减少酶的活性的任何药剂。
如本文所用的术语"治疗有效量"指药物以单一或多个剂量给予受试者时,在提供治疗益处给受试者和/或在预防由他汀类药物造成的不良副作用中是有效的量。在一个实施方案中,治疗益处是抑制或改善这样的不良副作用的症状。如本文所用的术语"治疗有效量"也指他汀类药物和/或白三烯介导的活性的抑制剂以单一或多个剂量给予受试者时,在提供给有需要的受试者治疗益处,例如胆固醇降低作用,同时减轻不良他汀类药物治疗副作用中是有效的量。
如本文所用的,术语"给予"指使哺乳动物细胞与本发明的化合物或组合物接触。用来实施本发明以疗效性治疗由他汀类药物引起的病症或归因于他汀类药物的病症的组合物的有效量根据他汀类药物,他汀类药物治疗的方案,给予方式,患者的年龄、体重和一般健康而变化。最终,主治医师将决定适宜的量和剂量方案。这样的量被称为有效量。
术语“有效量”和“有效的量”在此可互换使用,指包含他汀类药物和/或白三烯介导的活性的抑制剂的组合物在单一或多个剂量给予时有效减轻不良他汀类药物治疗副作用的量和/或剂量。例如,以非-限制性方式,为用本发明的组合物治疗他汀类药物治疗的副作用,他汀类药物的有效量是所述他汀类药物当作为一种单一疗法给予时导致可测量的或可检测的胆固醇降低作用的量。根据一些实施方案,白三烯受体拮抗剂的有效量是导致可测量的或可检测的减轻、阻断、抑制或预防他汀类药物治疗的不良作用的量。
如本文所用的术语"方法"指完成给定任务的方式、方法、技术和程序,包括,但不限于,化学、药理学、生物学、生物化学和医学领域的专业人员已知的,或者由化学、药学、生物学、生物化学和医学领域的专业人员从已知的方式、方法、技术和程序容易地开发的那些方式、方法、技术和程序。
如本文所用的短语“预防或减轻”和“减轻或预防"意指受试者的细胞、组织或器官中的病症或疾病通过在病症或疾病,或其症状出现之前、期间或之后给予药剂被预防或严重性减轻。因此,在一些实施方案中,短语“预防或减轻”和“减轻或预防"意指“预防”。在一些实施方案中,短语“预防或减轻”和“减轻或预防"意指“减轻”。
在某些实施方案中,他汀类药物选自阿托伐他汀、氟伐他汀、洛伐他汀、匹伐他汀、普伐他汀、罗苏伐他汀和辛伐他汀。每一种可能性表示本发明的一个独立的实施方案。
根据一些实施方案,CysLTR2拮抗剂选自:BAY-cysLT2 (CAS号712313-33-2)、BAY-u9773 (CAS号154978-38-8)、HAMI3379 (CAS号712313-35-4),及其任何组合。每一种可能性表示本发明的一个独立的实施方案。
在某些实施方案中,CysLTR2的拮抗剂是双特异性的并且还抑制CysLTR1的活性。在某些实施方案中,以上描述的药用组合物包含LTC4受体拮抗剂的混合物或多个LTC4受体拮抗剂,包含CysLTR1的拮抗剂和CysLTR2的拮抗剂。
除了干扰或阻断LTC4及其受体CysLTR1和CysLTR2之间的相互作用,LTC4的生物学活性可通过消除受试者体内的LTC4生物合成而减至最小。在某些实施方案中,以上描述的药用组合物包含CysLTR2的拮抗剂。在某些实施方案中,以上描述的药用组合物包含LTC4生物合成的抑制剂,其抑制选自微粒体谷胱甘肽S-转移酶2 (MGST2)、细胞溶质磷脂酶A2 (cPLA2)、5-脂氧合酶(5-LO),和5-脂氧合酶激活蛋白(FLAP)的酶的活性。每一种可能性表示本发明的一个独立的实施方案。在某些实施方案中,LTC4生物合成的抑制剂抑制MGST2的活性。在某些实施方案中,LTC4生物合成的抑制剂抑制cPLA2的活性。在某些实施方案中,LTC4生物合成的抑制剂抑制5-LO的活性。在某些实施方案中,5-LO的抑制剂是齐留通。在某些实施方案中,5-LO的抑制剂是阿曲留通。在某些实施方案中,LTC4生物合成的抑制剂抑制FLAP的活性。在某些实施方案中,FLAP的抑制剂是MK-886。
在某些实施方案中,以上描述的药用组合物包含他汀类药物、CysLTR2的拮抗剂,和LTC4生物合成的抑制剂,所述抑制剂抑制选自微粒体谷胱甘肽S-转移酶2 (MGST2)、细胞溶质磷脂酶A2 (cPLA2)、5-脂氧合酶(5-LO),和5-脂氧合酶激活蛋白(FLAP)的酶的活性。
在某些实施方案中,以上描述的药用组合物被用于预防或减轻由他汀类药物在受试者的肌肉组织中诱导的不良副作用,其中受试者正接受他汀类药物治疗和经历或有风险经历由他汀类药物诱导的不良副作用。
根据某些实施方案,不良副作用是他汀类药物-诱导的肌病。在某些实施方案中,肌病选自肌炎和横纹肌溶解。每一种可能性表示本发明的一个独立的实施方案。
在某些实施方案中,受试者正接受至少每日20 mg的阿托伐他汀或每日40-80 mg的阿托伐他汀。在某些实施方案中,受试者正接受每日0.8 mg的西立伐他汀。在某些实施方案中,受试者正接受每日超过80 mg的氟伐他汀。在某些实施方案中,受试者正接受每日超过80 mg的洛伐他汀。在某些实施方案中,受试者正接受至少每日4 mg的匹伐他汀。在某些实施方案中,受试者正接受每日超过40 mg的普伐他汀。在某些实施方案中,受试者正接受至少每日10 mg的罗苏伐他汀或每日20-40 mg的罗苏伐他汀。在某些实施方案中,受试者正接受至少每日40 mg的辛伐他汀或每日80 mg的辛伐他汀。每一种可能性表示本发明的一个独立的实施方案。
患有高水平的胆固醇的患者往往用他汀类药物,或他汀类药物的组合治疗。因为这些他汀类药物是全身给予的,它们往往部分地通过与肌肉细胞相互作用而造成不良副作用。在某些实施方案中,他汀类药物治疗高水平的胆固醇。在某些实施方案中,他汀类药物诱导不良副作用。
在某些实施方案中,以上描述的药剂盒包含含有他汀类药物的药用组合物,包含CysLTR2的拮抗剂的药用组合物,和包含LTC4生物合成的抑制剂的药用组合物。在某些实施方案中,以上描述的药剂盒还包含给予他汀类药物和所述药剂至受试者的用药说明书,所述受试者接受他汀类药物治疗和经历或有风险经历由他汀类药物诱导的不良副作用。在某些实施方案中,以上描述的药剂盒用于预防或减轻由他汀类药物在受试者的肌肉组织中诱导的不良副作用,其中受试者正接受他汀类药物治疗和经历或有风险经历由他汀类药物诱导的不良副作用。在某些实施方案中,应用包括在给予他汀类药物至受试者之前、期间和/或之后给予所述药剂。每一种可能性表示本发明的一个独立的实施方案。
根据一些实施方案,本发明的药用组合物还包含有效量的选自以下的他汀类药物:阿托伐他汀、西立伐他汀、氟伐他汀、洛伐他汀、美伐他汀、匹伐他汀、普伐他汀、罗苏伐他汀、辛伐他汀,及其任何组合。每一种可能性表示本发明的一个独立的实施方案。
根据一些实施方案,使用通过本发明的药用组合物的组合疗法将提供与他汀类药物在作为单一疗法给予时类似的或更好的胆固醇降低作用,同时减轻其副作用。
根据另外的实施方案,对他汀类药物使用的剂量在类似于当分开给药用于它们被批准的适应症时被批准的药物的剂量的范围内。
他汀类药物治疗是一类胆固醇治疗。为防止或减轻由他汀类药物造成的不良副作用,例如肌病,必须理解以上描述的药用组合物(包含LTC4受体的拮抗剂,LTC4生物合成的抑制剂,或其任何组合),可在用他汀类药物治疗之前,与用他汀类药物治疗同时,与用他汀类药物治疗一起,用他汀类药物治疗之后,和以其任何组合,给予用他汀类药物治疗的受试者。每一种可能性表示本发明的一个独立的实施方案。
在某些实施方案中,受试者先前未用他汀类药物治疗。在其它的某些实施方案中,受试者先前已用功能类似于他汀类药物的他汀类药物治疗。在某些实施方案中,受试者先前已用他汀类药物治疗。在某些这样的实施方案中,受试者先前已用他汀类药物治疗,并且已知获得由他汀类药物造成的不良副作用。
给予受试者的任何合适的途径可用于本发明的组合物。优选的给药方式将取决于要治疗的具体适应症并且对本领域技术人员而言将是显而易见的。
根据一些实施方案,本发明的药用组合物可作为一种丸剂、片剂或胶囊提供用于口服给予。
根据一些实施方案,本发明的药用组合物可作为一种粉末剂、溶液剂或混悬剂提供,用于静脉内给予。
在某些实施方案中,由本发明提供的药用组合物被配制用于他汀类药物的延迟释放。关于本文公开的制剂的术语"延迟释放"意指所述制剂并不立即释放活性成分至环境(如血液、胃、肠、结肠)中,而是在预定量的时间内释放活性成分。因此,相对恒定或可以预见的变化量的活性剂可以在指定的时间段内递送。措辞例如"延长作用"、"重复作用"、"持续释放"、"缓慢释放"和"控制释放"也已被用来描述这样的制剂或剂型。延缓释放可因此被描述为一种与常规即释剂型比较,允许至少减少给药频率一半的剂型或制剂。如本文所用的,术语"活性成分"意指他汀类药物、CysLTR2的拮抗剂和/或LTC4生物合成的抑制剂。
在某些实施方案中,由本发明提供的药用组合物包含10、20、30、40、50、60、70或80 mg他汀类药物。
构成治疗的组分的组合可同时地(作为离散的剂型或作为单一组合物)、顺序地,或者经一个合适的时间间隔分开给予。每一种可能性表示本发明的一个独立的实施方案。当组分作为分开的剂型,即不作为合并的组合物给予时,各组分可以相同的形式或不同的形式,如口服、经鼻、胃肠外,或经皮给予。当化合物同时、顺序或分开给予时,各组分可作为分开的剂型提供。
在某些实施方案中,本发明提供一种选自阿托伐他汀、西立伐他汀、氟伐他汀、洛伐他汀、美伐他汀、匹伐他汀、普伐他汀、罗苏伐他汀、辛伐他汀的他汀类药物或任何其它的胆固醇生物合成抑制剂,与选自批准的药物例如齐留通和实验药物例如BAY-cysLT2、MK-886、阿曲留通等的LTC4活性抑制剂一起的组合疗法。组合疗法的剂量将在与批准的药物在分开给予时的剂量类似的范围内。组合疗法可通过任何批准的给予形式给予并优选作为口服给予的含有他汀类药物和MGST2-LTC4途径的抑制剂二者的单一丸剂提供。
在一个方面,LTC4抑制剂是能够抑制选自以下的多肽的表达的RNA干扰剂:LTC4、MGST2、cPLA2、5-LO、FLAP、CysLTR2及其任何组合。每一种可能性表示本发明的一个独立的实施方案。
根据本发明,“RNA干扰剂”或"RNAi药物"是经基因改造为能够在靶细胞内转录双链RNA的双链RNA或者DNA构建体,所述双链RNA包含与选自以下的多肽的核苷酸序列至少70%互补的RNA链:LTC4、MGST2、cPLA2、5-LO、FLAP、CysLTR2或其同源物或片段或部分。
根据进一步的实施方案,本发明提供与选自LTC4、MGST2、cPLA2、5-LO、FLAP、CysLTR2或其同源物或片段的多肽的核苷酸序列互补的反义寡核苷酸序列,其中反义寡核苷酸序列能够与所述多肽或其同源物或片段特异性地杂交,从而抑制LTC4的表达。
贯穿本申请,本发明的各种实施方案可以一定的范围格式呈现。应该理解,在范围格式内的描述仅仅是为了方便和简洁,而不应视为对本发明范围的硬性限制。因此,一个范围的描述应视为已特别地公开所有可能的子范围以及在该范围内的各个数值。例如,范围例如从1-6的描述应视为已特别地公开子范围例如从1-3、从1-4、从1-5、从2-4、从2-6、从3-6等,以及在该范围内各个数值。例如,1、2、3、4、5和6。这适用于无论怎样的范围宽度。
要理解某些本发明的某些特征,为了简洁起见,其在独立的实施方案的文中描述,也可在单一实施方案中组合提供。相反,本发明的各个特征,为了简洁起见,其在单一实施方案的文中描述,也可分开或以任何合适的亚组合或在合适时以任何其它描述的本发明实施方案提供。在各个实施方案的上下文中描述的某些特征不被视为那些实施方案的必要特征,除非实施方案在没有这些要素时是不起作用的。
如上所述的和如在下文权利要求部分要求的本发明的各个实施方案和方面在下面的实施例中找到实验支持。
特定实施方案的前文描述将充分揭示本发明的一般性质,以至于对于各种应用,其它实施方案可通过运用现有的知识容易地修改和/或改编所述特定实施方案而无需过度的实验且不背离通用概念,并且因此,这样的改编和修改应当和意欲在本公开的实施方案的等价物的意义和范围内来理解。要理解本文所用的措辞或术语是为了描述而不是限制的目的。执行各种公开的功能的方法、材料和步骤可采取多个可供选择的形式而不脱离本发明。
提出以下实施例,以更全面地说明本发明的一些实施方案。然而,它们决不应视为限制本发明的广泛范围。本领域技术人员可容易地设计本文公开的原理的许多变化和修改,而不背离本发明的范围。
实施例
实施例1 - ER应激调节MGST2和5-LO的表达
为确定ER应激对人WISH上皮细胞和人未分化的HaCaT角质细胞中的MGST2水平的影响,在用衣霉素,一种蛋白糖基化抑制剂(1 μg/ml, 24小时)处理人WISH上皮细胞和人HaCaT前-角质细胞后,微粒体谷胱甘肽S转移酶(MGST2)和裂解的胱天蛋白酶3 (Casp. 3)表达的动力学通过免疫印迹测定。在图2A中所示的结果证实用衣霉素处理这些细胞导致ER应激,推测是由于蛋白的错误折叠所致。值得注意的是,在两种细胞类型中,MGST2在早期(未折叠蛋白反应(UPR)的保护期)被下调,并在晚期(死亡促进期)上调,伴有裂解的胱天蛋白酶3的升高。为评估MGST2及其产物LTC4在ER应激触发的细胞死亡中的作用,进行免疫印迹试验用于测定在用布雷菲尔德菌素A,一种蛋白从内质网(ER)转运至高尔基氏体的抑制剂(0.66 μg/ml, 24小时)处理人WISH上皮细胞,和用衣霉素(1 μg/ml, 24小时)处理小鼠B16细胞后5-脂氧合酶(5-LO)表达的动力学。在图2B中所示的结果证实5-LO,白三烯生物合成的限速酶也在对ER应激的反应中表现出倒钟形的表达模式。因此,LTC4生物合成的两个关键组分受ER应激的严格调节。它们在早期(UPR的促生存期)衰减,并在晚期(死亡促进期)上调。
实施例2 - ER应激触发MGST2和5-LO的核易位
为评价ER应激对MGST2、cPLA2、5-LO和FLAP的细胞易位的影响,对未处理的和用布雷菲尔德菌素A (0.66 µg/ml, 24小时)处理的WISH上皮细胞进行免疫染色。结果显示在对照人WISH细胞中,MGST2既存在于ER中,又存在于细胞核中(数据未示出),并且5-LO既存在于胞质中,又存在于细胞核中(数据未示出)。尽管它们在对照细胞中的共同的核位置,5-LO没有与MGST2共同定位于人WISH上皮细胞或者HaCaT前-角质细胞中(数据未示出)。用布雷菲尔德菌素A,一种阻断蛋白从ER通往高尔基氏体的毒素,触发ER应激,导致MGST2和5-LO二者向核的移动,如通过用核被膜标记物核纤层蛋白(lamin)和核标记物Hoechst 33258染色所确定的(数据未示出)。此外,ER应激触发MGST2和5-LO在WISH和HaCaT两种细胞中的共同定位(数据未示出)。
实施例3 - ER应激触发LTC4的整个生物合成机器的核易位
肥大细胞中的Fc受体的激活通过触发其整个生物合成机器向核被膜的易位启动LTC4的生产。因此,为了检查ER应激对用于LTC4的生物合成所需的两种其它的酶的细胞定位的影响,在用布雷菲尔德菌素A (0.66 μg/ml, 24小时)处理后的WISH细胞中,5-LO激活蛋白(FLAP)和5-LO的易位和共同定位,通过免疫染色测定。结果显示在对照细胞中,FLAP被定位于核和ER中,而cPLA2主要在胞质中。尽管它们在对照细胞中的重叠定位,5-LO不与FLAP或cPLA2共同定位(数据未示出)。用布雷菲尔德菌素A触发ER应激造成向核的易位和5-LO与FLAP和cPLA2的共同定位(数据未示出)。因此,ER应激触发向核的易位和一组能够生成LTC4的酶的共同定位。这种ER应激触发的易位事件与肥大细胞的Fc受体激活时见到的类似,除了其由ER应激诱导而不是免疫学提示并且其涉及MGST2而非LTC4S外。
实施例4 - ER应激触发LTC4的基于MGST2的生物合成
为进一步评估基于MGST2的机器的ER应激触发的易位是否导致LTC4的生产,进行EDC-固定的细胞的免疫染色,以确定ER应激的3种诱导剂(它们通过不同的机制起作用)对LTC4生物合成的影响。衣霉素抑制蛋白糖基化,毒胡萝卜素触发钙离子从ER释放到胞质中,而布雷菲尔德菌素A阻断蛋白从ER转运至高尔基氏体。结果证实由这些毒素诱导的ER应激造成LTC4的广泛的生物合成。总而言之,这些结果提示ER应激通过MGST2、5-LO、FLAP和cPLA2的表达、核易位和共同定位,触发MGST2-介导的LTC4生物合成。
实施例5 - ER应激调节LTC4受体的表达和向核的易位
在肥大细胞中,LTC4被分泌并通过结合于其两个胞质膜受体,CysLTR1和CysLTR2对邻近的细胞起作用。然而,先前证实外源性加入LTC4和LTD4触发它们的受体向核被膜的易位(Nielsen, C.K., et al. Cancer Res, 2005. 65(3): p. 732-42)。在用布雷菲尔德菌素A (0.66 µg/ml, 24小时)处理人WISH细胞,和用衣霉素(1 μg/ml, 24小时)处理小鼠B16细胞后,通过使用免疫印迹进行CysLTR1和CysLTR2表达的动力学分析。令人吃惊地,如可从图3中所示结果见到的,在人WISH上皮细胞和小鼠B16细胞中,CysLTR1和CysLTR2在早期(UPR的保护期)下调和恢复,并在UPR的晚期死亡促进期进一步诱导。
此外,结果证实意外的和令人吃惊的发现,即ER应激也触发CysLTR1和CysLTR2向核被膜的易位和它们与5-LO的共同-定位。因此,在ER应激后,LTC4生物合成机器和LTC4受体二者共同定位于相同的细胞隔室,从而促进MGST2-生成的LTC4的胞内分泌行动。
实施例6 - ER应激触发LTC4的基于MGST2的生物合成
ER应激和氧化应激密切相关,因为ER应激触发氧化应激以及反之亦然。因此,用于测定MGST2及其产物LTC4是否参与ER应激触发的ROS积聚的试验被进行。如先前所报告的,由衣霉素触发的ER应激导致ROS的积聚,如通过用超氧化物阴离子指示剂二氢乙啶(DHE)染色所检测的。在MGST2用特异性siRNA有效沉默后(图4B),ER应激触发的ROS积聚的水平显著减少,达到对照细胞的水平(图4A)。MGST2通过使其不仅偶合于LTA4,而且也偶合于其它的底物而消耗GSH。为发现MGST2是否通过耗尽GSH或通过LTC4的生物合成触发ROS积聚,使用几种LTC4受体拮抗剂以及LTC4生物合成或转运的抑制剂。由于白三烯介导哮喘的症状,白三烯生物合成、转运和活性的许多不同的抑制剂是可利用的。Reversan抑制LTC4从细胞输出。孟鲁司特和普仑司特是选择性CysLTR1受体拮抗剂,BAY-cysLT2是选择性CysLTR2拮抗剂和BAY-u9773是CysLTR1和CysLTR2二者的双重抑制剂。由布雷菲尔德菌素A触发ER应激生成ROS,如通过指示剂二氯-二氢-荧光素二乙酸酯(DCFH-DA, 图4C, 5)所测量的。结果令人惊讶和出乎意料地显示,经测试的所有CysLTR1和CysLTR2拮抗剂显著地抑制ER应激触发的ROS积聚(图4C, 5)。
与LTC4拮抗剂对比,MRP1转运蛋白抑制剂reversan (20 µM)在对ER应激的反应中对ROS积聚具有一种小的,但在统计学上无意义的抑制作用(图4C),表明LTC4活性主要在胞内分泌。这一重要发现与先前两个LTC4受体易位至核的发现一致,进一步支持ER应激触发的LTC4行动主要是胞内分泌的主张。MGST2在ROS积聚中的作用通过其在HEK 293T细胞中的过度表达被进一步确立(图4D),这导致在细胞ROS中的显著升高(图4E)。总之,这些结果表明,MGST2-生成的LTC4是ER应激触发的氧化应激的主要指示物。
实施例7 - MGST2-LTC4途径激活核NADPH氧化酶4 (NOX4)和随后的DNA损伤
最近的研究已鉴定作为最重要的细胞ROS生产酶的NADH/NADPH氧化酶(NOX),且ER应激被发现通过上调NOX4同工型触发氧化应激。CysLTR1和CysLTR2属于的G-蛋白偶联受体的几种配体通过受体-介导的NOX的激活引起氧化应激。人WISH细胞和小鼠B16细胞用媒介(对照)或布雷菲尔德菌素A (0.66 μg/ml, 24小时)在BAY-u9773、MK571或孟鲁司特的存在或不存在下处理,NOX4水平通过免疫印迹评估。显著地,LTC4受体拮抗剂大大地抑制在ER应激时NOX4的表达(图6A, 6B)。此外,用媒介(对照)或布雷菲尔德菌素A (0.66 μg/ml, 24小时)处理WISH细胞,固定和免疫染色揭示ER应激触发的NOX4朝向核的易位。因此,NOX4 (在ER应激下的主要的ROS生产者)的表达和激活是LTC4-依赖的。
对ER应激的反应中的核ROS的积聚将导致氧化DNA损伤。为测试MGST2-LTC4途径是否涉及ER应激触发DNA损伤,测定氧化DNA衍生物8-羟基-2’脱氧鸟苷(8-OHdG)的形成。用布雷菲尔德菌素A处理HaCaT细胞导致8-OHdG出现在核中,如通过免疫染色所测定的(图6C)。用普仑司特抑制LTC4结合于CysLTR1显著地减少核8-OHdG的形成,使用 BAY-u9773抑制LTC4结合于CysLTR1和CysLTR2二者在减轻DNA损伤中甚至更有效(图6C)。总之,ER应激触发的DNA损伤在很大程度上通过MGST2-LTC4途径介导。
实施例8 - MGST2-LTC4途径介导ER应激触发的细胞死亡
虽然适度水平的ROS促进细胞增殖,但称为“毒性阈值”的水平以上的浓度触发细胞死亡。如果LTC4升高ROS在该阈值之上,则它可以是ER应激触发的细胞死亡机器的一部分,如在由缺氧/再灌注所致心肌细胞死亡的情况下报告的。此外,NOX2的诱导被鉴定为导致ER应激-触发的巨噬细胞的细胞凋亡的机制之一。为确定MGST2-LTC4途径是否介导ER应激触发的细胞死亡,小鼠B16细胞用对照siRNA (siControl)或MGST2-特异性siRNA (siMGST2)转染,用媒介或衣霉素(1 μg/ml, 24小时) (Tm)处理,然后用结晶紫染色。结果证实有效敲减MGST2显著地减少ER应激触发的B16黑素瘤细胞的死亡(图7A, 7B)。在一逆向研究中,MGST2在HEK 293T细胞中的过度表达显著地增加细胞死亡(图7C)。最终结论是,MGST2参与ER应激触发的细胞死亡。
接着,进行了另一个检查MGST2产物LTC4在人HaCaT前-角质细胞的ER应激触发的细胞中的作用的试验。在BAY-u9773 (80 nM)的存在或不存在下,用媒介(对照)或布雷菲尔德菌素A (BfA, 1.33 µg/ml, 48小时)处理HaCaT细胞,然后用结晶紫染色。
结果显示用双重CysLTR1和CysLTR2拮抗剂,BAY-u9773预处理人HaCaT角质细胞,减少由布雷菲尔德菌素A引起的ER应激触发的细胞死亡(图7D)。BAY-u9773也显著地减少衣霉素-诱导的人WISH细胞死亡,如通过中性红分析所测定的(图7E)。由于错误折叠蛋白在ER中的积聚,蛋白酶体抑制剂触发ER应激和随后的细胞死亡。先前已发现5-脂氧合酶抑制剂齐留通在脊柱损伤和脑缺血后减少细胞凋亡性细胞死亡。结果显示齐留通(10 μM)显著地保护HaCaT角化细胞避免由蛋白酶体抑制剂MG262 (50 nM, 24小时, 图7F)触发的死亡。两种LTC4受体CysLTR1和CysLTR2在肥大细胞和在人肠上皮细胞中二聚化。因此可能这些受体之一的选择性抑制作用将足以减轻LTC4活性。确实,普仑司特(10 μM),一种选择性CysLTR1拮抗剂,显著地减少布雷菲尔德菌素A-触发的人WISH细胞的死亡(图7G)。类似地,BAY-cysLT2 (5 µM),一种选择性CysLTR2拮抗剂,同样保护这些细胞以避开布雷菲尔德菌素A (图8A)。
在其作为MRP1抑制剂的活性上面,MK571是有效的选择性CysLTR1拮抗剂。用MK571 (10 µM)预处理鼠B16黑素瘤细胞显著地减少由衣霉素、毒胡萝卜素和布雷菲尔德菌素A触发的细胞死亡(图8B)。这种保护作用与减轻细胞坏死相关,如通过减少HMGB1向培养基中的分泌所证实(图8C)。
总之,本文提供的结果鉴定了一种先前未识别的ER应激触发的信号传导途径,其介导ER应激触发的细胞死亡。
实施例9 - MGST2缺乏减少体内ER应激触发的死亡
纯合子MGST2缺乏小鼠从129svEvBrd小鼠品系ES细胞基因诱捕文库建立。从Mgst2敲除小鼠和它们的WT和杂合同窝出生小鼠的尾尖获得的DNA样本的PCR证实Mgst2的靶向等位基因是无效突变(图9A)。MGST2-缺乏小鼠正常饲养并且看起来与它们的野生型同窝出生小鼠无法区别,在体重和食物摄取方面没有明显的差别。WT和第2代MGST2-缺乏小鼠胚胎的成纤维细胞用媒介(对照)或衣霉素(1 μg/ml, 24小时)处理。然后用结晶紫染色各板。结果显示第2代MGST2-缺乏的鼠胚胎成纤维细胞(MEFS)对由衣霉素引起的ER应激触发的细胞死亡,比WT MEFS具有显著更大的抗性(图9B)。与WT MEFs比较,用DCFH-DA染色显示在对ER应激的反应中几乎完全缺乏ROS积聚(图9C)。
为评价MGST2在体内ER应激的作用,采用急性肾损伤的小鼠模型。在0天用衣霉素(1 mg/kg体重)经ip注射给WT和MGST2-缺乏小鼠。在第4天收集的苏木精-曙红染色的WT肾切片的组织学检查揭示对WT小鼠近端小管的预期的损伤,而MGST2-缺乏小鼠的组织学照片是明显轻微的,如由空泡的数量测定的(图9D, 9E)。为进一步研究MGST2-LTC4途径在ER应激触发的发病率中的作用,更高剂量的衣霉素被注射到小鼠中。MGST2缺乏小鼠对2.5 mg/kg的单一衣霉素剂量具有比WT小鼠显著更大的抗性(图9F)。而且,给予普仑司特伴随2.5 mg/kg衣霉素与单独的衣霉素比较大大地减少WT小鼠的发病率和死亡率(图9G)。
实施例10 - 普仑司特保护人WISH细胞避免辛伐他汀-触发的细胞死亡
为研究MGST2-LTC4途径在他汀类药物-触发的细胞死亡中的作用,在普仑司特(10 µM)或者媒介的存在下,用各种浓度的辛伐他汀或媒介在96孔板中处理人WISH上皮细胞48 h。结果显示普仑司特减少由宽范围(0.5-4 µg/ml)的辛伐他汀浓度触发的细胞死亡(图10)。
实施例11 – BAY-cysLT2和BAY-u9773保护小鼠C2C12肌细胞避免辛伐他汀-触发的细胞死亡
为研究MGST2-LTC4途径在他汀类药物-触发的细胞死亡中的作用,在媒介、BAY-cysLT2 (10 µM)或者BAY-u9773 (1 µM)或者甲羟戊酸纳的存在下,用10 uM辛伐他汀或媒介在96孔板中处理分化的小鼠C2C12肌细胞5天。结果显示 BAY-cysLT以及BAY-u9773减少由辛伐他汀触发的细胞死亡。对比之下,CysLTR1拮抗剂普仑司特不能保护这些细胞免遭辛伐他汀的毒性作用(图11A, 11B)。
实施例12 – 齐留通保护小鼠C2C12肌细胞避免辛伐他汀-触发的细胞死亡
为研究MGST2-LTC4途径在他汀类药物-触发的细胞死亡中的作用,在媒介、齐留通(10 µM)或甲羟戊酸纳的存在下,用20 uM辛伐他汀或媒介在96孔板中处理分化的小鼠C2C12肌细胞4天。结果显示齐留通减少由辛伐他汀触发的细胞死亡。作为对比,CysLTR1拮抗剂孟鲁司特不能保护这些细胞避免辛伐他汀的毒性作用(图12A, 12B)。
实施例13 - 齐留通在减轻他汀类药物治疗副作用中的作用
齐留通对经历他汀类药物治疗(用于治疗高胆固醇血症)和已报道患有不同程度的肌肉疼痛和疲劳的患者的作用被测试。
患者在-1、0、+1、+2和+4周接触或者参与临床。在第一周(WK(-l))的临床中,患者的他汀类药物的详情被记录且注明每日剂量。也使用McGill疼痛问卷表(McGill Pain Questionnaire) (Melzack, R., 1975 "The McGill Pain Questionnaire: Major Properties and Scoring Methods". Pain 1:277-299,其公开内容通过引用以其全文包括在本文中),疼痛评分指数(Pain Rating Index) (PRI)和现时疼痛强度(Present Pain Intensity) (PPI)量表对患者的疼痛评分。在PRI指数中,其包含两个分数,较高的评分表示疼痛的水平增加。在PPI指数中,现时疼痛给出0-5的分数,其中0表示无疼痛和5表示极度疼痛。在WK(-1)也对患者的疲劳使用疲劳冲击量表(Fatigue Impact Scale)进行评分(Fisk, J.D. et al, 1994, "Measuring the functional impact of fatigue: Initial Validation of the Fatigue Impact Scale",Clinical Infectious Disease, 18 (Suppl l):S79-83,其公开内容通过引用以其全文包括在本文中)。
在WK(0)和WK(+4)从患者采集血样以测定肌酸激酶浓度(CK) (单位/L) (作为肌肉创伤的血液测量)和HDL-胆固醇或LDL-胆固醇各自的基线水平和甘油三酯水平。要注意到血肌酸激酶浓度的正常范围是0-200单位/L。
在WK(0)开始并在整个研究中继续,患者被要求服用2400 mg齐留通的每日总剂量,每日口服给予4次(600 mg片)或每日两次1200 mg缓释片剂。
为测试齐留通对患者的作用,确定PRI、PPI和FIS评分的问卷表在+1、+2和+4周后进行。在+1和+2周时,患者通过电话联系并被要求邮寄他们的自我评估问卷表到诊所。
特定实施方案的前文描述将充分揭示本发明的一般性质,以至于对于各种应用,其它实施方案可通过运用现有的知识容易地修改和/或改编所述特定实施方案而无需过度的实验且不背离通用概念,并且因此,这样的改编和修改应当和意欲在本公开实施方案的等价物的意义和范围内来理解。要理解本文所用的措辞或术语是为了描述而不是限制的目的。执行各种公开的功能的方法、材料和步骤可采取多个可供选择的形式,而不脱离本发明。
- 还没有人留言评论。精彩留言会获得点赞!