一种淫羊藿苷控释微球、其制备方法及其用途与流程
本发明属于医药卫生领域,涉及一种药物新剂型药物微球的制备方法及其应用,特别涉及一种pH响应的靶向控释微球,其制备方法、药物应用,该药物微球用于预防、缓解和/或治疗结肠类疾病,包括肠易激综合征,溃疡性结肠炎,克隆病,直肠癌等病症。
背景技术:
随着社会的不断发展进步,工作、生活压力增大,作息饮食不规律,长期的精神焦虑,紧张不安等,往往导致免疫失调以及菌群紊乱等健康问题。中医认为溃疡性结肠炎属于泄泻,肠癖等范畴,而现代医学认为其属于自身免疫性疾病或伴有感染,以腹痛,腹泻,粘液便,脓血便等为临床表现,以结肠黏膜的慢性炎症变化及溃疡形成为病理特征。目前关于溃疡性结肠炎的具体病因,病机尚不明确,因此尚缺乏有效的治疗措施。主要采用糖皮质激素类,非甾体抗炎药物或免疫抑制剂对其进行治疗,但效果不佳。
淫羊藿苷是小檗科植物淫羊藿的主要有效成分,具有抗衰老、抗肿瘤、抗结肠炎等药理作用。但是淫羊藿苷水溶性差,自身生物利用度低,这些因素限制了其在临床上的应用。为了保证药物的有效浓度,避免药物的毒副作用,达到有效的治疗剂量,中药现代缓控释制剂应运而生。口服结肠定位释药系统(oral colon specific delivery system,OCDDS)是20世纪90年代发展起来的一种定位给药系统,即指使用适当的方法,避免药物在胃、十二指肠、空肠和回肠前端释放,直接运送到人体回、盲肠部位后释放而发挥局部或全身治疗作用的一种释药系统,具有对结肠局部环境的特异性响应、设计合理以及制备工艺简单等特点。
壳聚糖是甲壳素的脱乙酰产物,是唯一的天然碱性多糖,具有广泛的生物活性而备受医药研究者关注。壳聚糖分子中的D-葡萄糖单元上氨基的正电荷与消化道黏膜上唾液酸残基的负电荷之间的静电引力使得其易于粘附在胃肠道黏膜表面。而且壳聚糖分子中的糖苷键能被结肠酶降解,故可作为结肠定位释药材料的使用。另外海藻酸盐可以实现钠离子与钙离子之间的交换,从而有利于溶胀和海藻酸盐在结肠内的降解。根据多糖类材料可以改善靶向特异性、符合患者的治疗需求以及能够被结肠生理环境降解的特点,选用交联剂对包裹壳聚糖的微球进行交联固化。壳聚糖不同分子链上的胺基与醛基发生交联反应,生成的亚胺基及希夫 碱结构增强了交联壳聚糖在酸性条件下的稳定性,可以降低壳聚糖本身的氢键作用,降低其生物粘附性。戊二醛交联能够实现壳聚糖微球不在胃部滞留,通过胃肠运动到达结肠,从而发挥治疗结肠炎症的作用。
尽管结肠炎发病机理至今仍不清楚,但是炎性细胞因子在介导结肠炎形成的过程中所起的作用是毋庸置疑的。其中促炎因子IL-1β、TNF-α等是公认的介导结肠炎发病的细胞因子,另外,当炎性因子诱导i-NOS表达成功后,前列腺素会促进白细胞转至炎症部位并促使NO的产生,从而引起炎症反应以及COX-II的表达使得机体本身产生炎症以及疼痛感。而且也有研究指出炎症的形成以及COX-II的表达在结肠致癌过程中起着至关重要的作用。
传统结肠疾病给药方式主要有口服和直肠给药两种途径,口服给药由于结肠的特殊生理结构,药物难以到达结肠部位,直肠给药虽然能够使药物在局部释放,但是对于结肠上端疾病并不能有效发挥作用,本发明将淫羊藿苷载入到壳聚糖/海藻酸钙缓控释微球制备的口服结肠定位释药系统用于消化性溃疡的治疗,目前未见其报道。
技术实现要素:
本发明的目的是针对现有治疗结肠疾病的药物存在技术的不足,提供一种pH响应的用于治疗结肠疾病的控释微球及其制备方法,本发明的控释微球具有良好的pH响应性,能够靶向给药,本发明方法制备的结肠靶向控释微球主要用于预防、缓解和/或治疗结肠类疾病,包括肠易激综合征,溃疡性结肠炎,克隆病,直肠癌等相关病症的用途。
为实现上述目的,本发明提供一种用于治疗结肠疾病的控释微球的制备方法,包括如下顺序进行的步骤:
1)配制乳化水相
向海藻酸钠溶液中加入碳酸钙、淫羊藿苷,搅拌,使碳酸钙、淫羊藿苷混悬于海藻酸钠溶液中,制得碳酸钙-淫羊藿苷-海藻酸钠混悬液(即乳化水相);
2)配制乳化油相
将乳化剂加入到液体石蜡中,搅拌,混匀,制成乳化剂-石蜡溶液(即乳化油相);
3)乳化处理
在搅拌状态下,将碳酸钙-淫羊藿苷-海藻酸钠混悬液与乳化剂-石蜡溶液混合,进行乳化处理,形成乳化体系;接着加入酸引发剂,进行酸引发处理,生成淫羊藿苷-海藻酸钙微球;
4)将淫羊藿苷-海藻酸钙微球加入到成膜壳聚糖溶液中,混合均匀,进行复电解质络合反 应,壳聚糖包裹在淫羊藿苷-海藻酸钙微球的表面,制成壳聚糖-淫羊藿苷-海藻酸钙微球;
5)将壳聚糖-淫羊藿苷-海藻酸钙微球与交联剂混合,进行交联反应,即得淫羊藿苷控释微球。
其中,步骤1)中所述碳酸钙的重量与海藻酸钠溶液的体积之比为0.2-2:100,优选为0.5-1.5:100,进一步优选为1:100,即每100ml海藻酸钠溶液中加入0.2-2g纳米级碳酸钙或每1L海藻酸钠溶液中加入2-20g碳酸钙;所述淫羊藿苷的重量与海藻酸钠溶液的体积之比为1-3:100,优选为1.5-2.5:100,进一步优选为2.25:100,即每100ml海藻酸钠溶液中加入1-3g淫羊藿苷或每1L海藻酸钠溶液中加入10-30g淫羊藿苷。
特别是,所述碳酸钙选择纳米级碳酸钙。
其中,所述海藻酸钠溶液的浓度为0.5-2%(w/v),优选为1-2%,进一步优选为1.5%。
特别是,所述海藻酸钠溶液按照如下步骤制备而成:将海藻酸钠加入到纯水中,混合,加热并保持在45±5℃条件下,搅拌,使海藻酸钠充分溶解,即得,其中,海藻酸钠溶液中海藻酸钠的质量与水的体积之比为0.5-2:100,即海藻酸钠溶液的浓度为0.5%-2%(w/v)。
其中,步骤2)中所述乳化剂选择司盘80或/和吐温80。
特别是,所述乳化剂与液体石蜡的体积之比为0.5-2:100,优选为0.5-1.5:100,进一步优选为1.0:100。
其中,步骤3)中所述乳化处理温度为35-45℃,优选为37℃;乳化处理时间为30-60min,优选为40-50min。
特别是,所述碳酸钙-淫羊藿苷-海藻酸钠混悬液与乳化剂-石蜡溶液的体积之比为1:4-6,优选为1:5。
尤其是,所述搅拌速率为200-500rpm,优选为300rpm。
其中,所述酸引发剂选择冰醋酸、盐酸、硫酸、磷酸,优选为冰醋酸。
特别是,所述酸引发剂为冰醋酸与液体石蜡的混合溶液,其中冰醋酸与酸引发剂的体积之比为1:8-12,优选为1:10。
尤其是,所述酸引发剂中冰醋酸的体积与碳酸钙-淫羊藿苷-海藻酸钠混悬液中碳酸钙的重量之比为5:1-3,优选为5:1,即碳酸钙-淫羊藿苷-海藻酸钠混悬液中每含1-3g碳酸钙则添加的酸引发剂中的冰醋酸5ml,或者碳酸钙-淫羊藿苷-海藻酸钠混悬液中每含1-3kg碳酸钙则添加的酸引发剂中的冰醋酸5L。
其中,所述酸引发处理温度35-45℃,优选为37℃;酸引发处理时间为10-20min,优 选为15min。
特别是,还包括对酸引发处理后的混合体系进行静置处理,混合体系上下分层,取下层沉淀,即得所述的淫羊藿苷-海藻酸钙微球。
尤其是,采用体积比浓度为0.5-2%(v/v)的Tween80水溶液清洗沉淀,以去除多余的液体石蜡;然后再用纯水洗涤,收集淫羊藿苷-海藻酸钙微球。
其中,步骤4)中所述成膜壳聚糖溶液为壳聚糖-醋酸溶液。
特别是,所述壳聚糖-醋酸溶液中壳聚糖的浓度为0.5%-2%(w/v),优选为1%。
尤其是,所述成膜壳聚糖溶液按照如下步骤配制而成:将壳聚糖加入到冰醋酸水溶液中,混匀后,加热升温并保持在45±5℃条件下,搅拌,使壳聚糖充分溶解,待其冷却后调节壳聚糖-醋酸溶液的pH为5-6,备用.
其中,所述壳聚糖-醋酸溶液中壳聚糖的质量与冰醋酸水溶液的体积之比为0.5-2:100,即壳聚糖-醋酸溶液中壳聚糖的浓度为0.5%-2%(w/v),优选为1:100。
特别是,所述冰醋酸水溶液体积百分比浓度为0.5-2%(v/v),优选为1%。
尤其是,调节壳聚糖-醋酸溶液的pH为5.5。
其中,所述复电解质络合反应的时间≥20min,优选为20-40min。
特别是,淫羊藿苷-海藻酸钙微球与膜壳聚糖溶液的体积之比为1:5-15,优选为1:8。
尤其是,还包括将复电解质络合反应后的反应体系进行离心处理,然后对离心沉淀物进行水清洗。
其中,步骤5)中所述交联剂选择戊二醛水溶液、PBS缓冲液中的一种,优选为戊二醛水溶液。
特别是,所述戊二醛水溶液的质量百分比浓度为0.25-1%,优选为0.5%。
尤其是,所述壳聚糖-淫羊藿苷-海藻酸钙微球与交联剂的体积之比为1:5-20,优选为1:10。
其中,所述交联反应的温度<10℃,优选为4-10℃,进一步优选为4-8℃,更进一步优选为4℃;交联反应时间为1.5-3h,优选为2h。
特别是,还包括加入交联终止剂,终止所述的交联反应。
其中,所述交联终止剂选择甘氨酸溶液。
特别是,所述甘氨酸溶液的摩尔浓度为0.5-2mol/L,优选为1mol/L。
尤其是,所述加入的甘氨酸溶液与戊二醛溶液的体积之比为2-4:1,优选为2:1。
特别是,还包括对交联反应终止后的混合体系进行离心处理,接着对离心沉淀物用纯水进行清洗,然后进行干燥,即得。
本发明另一方面提供一种按照上述方法制备而成的淫羊藿苷控释微球。
本发明再一方面提供一种按照上述方法制备而成的淫羊藿苷控释微球在制备治疗结肠疾病中的应用,在制备治疗结肠疾病的药物或保健品中的应用。
本发明制备的淫羊藿苷生物控释微球具有以下优点:
1、本发明的淫羊藿苷生物控释微球为pH响应控释微球,具有良好的pH响应性,在胃部低pH环境下不发生明显的降解,药物释放量较低,在结肠较高pH环境下粘附在结肠表面持续释放药物,能够促进药物释放,并延长药物的结肠滞留时间,增加药物吸收,改善口服用药治疗结肠部位疾病的局部生物利用度。体外释药研究结果表明,淫羊藿苷靶向缓控释微球模拟胃液中释放2h后累计释放率仅10%左右,而模拟结肠液中累计释放率可达到65%。
2、本发明方法制备的淫羊藿苷生物控释微球的核心为淫羊藿苷海藻酸钙微球,外壳为壳聚糖,同时具有理想的pH响应控释效果和淫羊藿苷结肠靶向控释药效作用,用于治疗结肠疾病,治疗肠易激综合征,溃疡性结肠炎,克隆病,直肠癌等病症。
本发明方法制备的淫羊藿苷靶向缓控释微球能够显著改善TNBS/乙醇诱导溃疡性结肠炎,能够有效降低结肠部位炎性因子IL-1β、IL-6的分泌,降低引起炎症反应的i-NOS、COX-II蛋白表达水平。
3、本发明的淫羊藿苷pH响应生物控释微球药物负载于微球内部核心,药物载药量高,药物的外层包裹壳聚糖,能够固定药物,从而减少药物在体内的运输过程中的损失。
4、本发明的淫羊藿苷pH响应生物控释微球的靶向给药效果明显,微球在结肠部位长时间停留,提高治疗效果。体内研究表明微球能够在结肠停留超过12h,更有利于药物的释放和发挥治疗作用。
5、本发明制备的淫羊藿苷靶向缓控释微球外面为壳聚糖,为pH敏感性阳离子型水凝胶材料,具有良好的生物粘附性、生物相容性和生物可降解性,对机体无毒性,且具有抗菌消炎、促进伤口愈合作用等。
6、本发明方法工艺条件容易控制,制备的产品质量可控,适宜工业化大生产。
附图说明
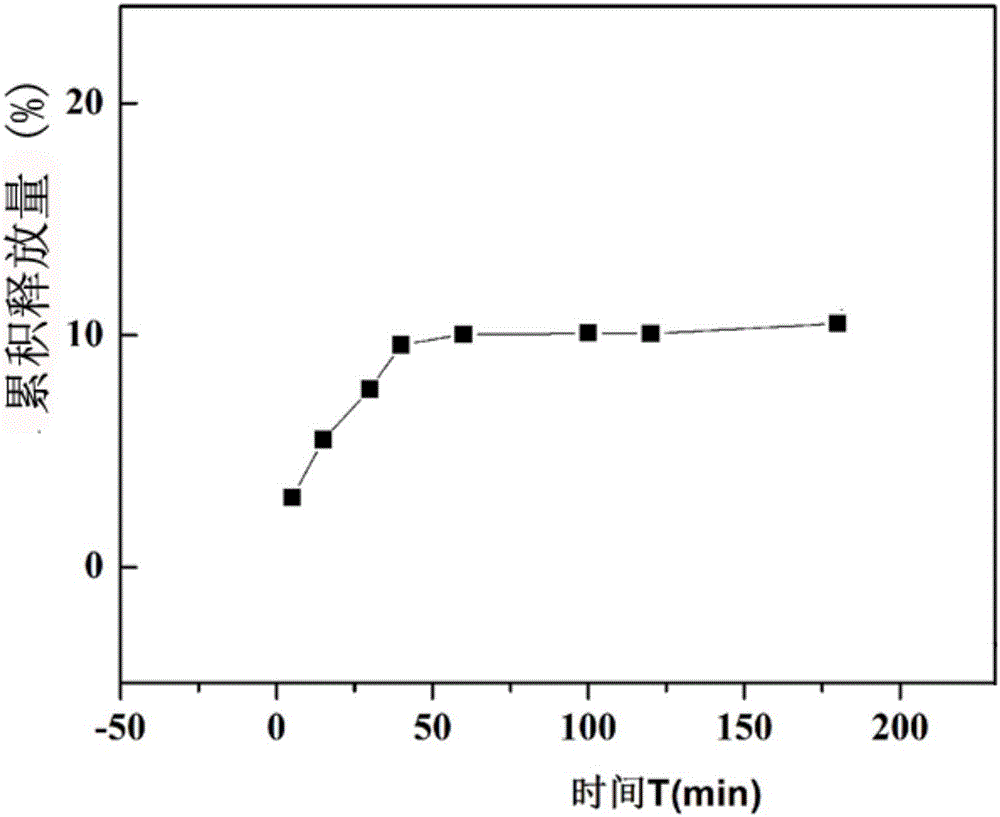
图1淫羊藿苷壳聚糖/海藻酸钙控释微球在模拟胃液中(pH=1.2)的累计释放率图。
图2淫羊藿苷壳聚糖/海藻酸钙控释微球在模拟小肠液和结肠液(pH=6.8、pH=7.4)中的累积释放率图。
图2A为淫羊藿苷/壳聚糖/海藻酸钙微球在模拟小肠液(pH=6.8)中的累积释放率图。
图3 FITC荧光标记微球小鼠消化道内的分布结果图,从左至右分别是胃、十二指肠、空肠和结肠。
图4淫羊藿苷壳聚糖/海藻酸钙控释微球对溃疡性结肠炎模型大鼠结肠组织IL-1β含量的影响结果图。
图5淫羊藿苷壳聚糖/海藻酸钙控释微球对溃疡性结肠炎模型大鼠结肠组织TNF-α含量的影响结果图。
具体实施方式
下面结合具体实施例,进一步阐述本发明。但这些实施例仅限于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体实验条件的实验方法,通常按照常规条件,或按照厂商所建议的条件。
以下通过试验例来进一步阐述本发明所述淫羊藿苷靶向缓控释微球用于结肠炎的治疗作用,这些试验例包括了本发明淫羊藿苷靶向缓控释微球的制备和药物释放试验及本发明淫羊藿苷靶向缓控释微球的药效学试验。
本发明实施例中使用的试剂如下:
实施例1淫羊藿/苷壳聚糖/海藻酸钙微球的制备
1、配制溶液
1-1)配制海藻酸钠溶液
将精确称量的海藻酸钠加入到纯水中,混合,加热并保持在45±5℃条件下,搅拌,使海藻酸钠充分溶解,制得海藻酸钠溶液,待其降至室温(20-25℃左右)后于4℃冰箱保存,使用前加热至45℃,备用,其中,海藻酸钠溶液中海藻酸钠的质量与水的体积之比为0.5:100,即海藻酸钠溶液的浓度为1.0%(w/v);
1-2)配制成膜溶液(即壳聚糖-醋酸溶液)
将精确称量的壳聚糖加入到0.5%的冰醋酸(v/v,体积百分比浓度)水溶液中,混匀后,加热升温并保持在45±5℃条件下,搅拌,使壳聚糖充分溶解,制得壳聚糖-醋酸溶液(即成膜溶液),待其降至室温(20-25℃左右)后于4℃冰箱保存,使用前调节壳聚糖-醋酸溶液的pH为5.5,备用,其中,壳聚糖-醋酸溶液中壳聚糖的质量与冰醋酸水溶液的体积之比为0.5:100,即壳聚糖-醋酸溶液中壳聚糖的浓度为0.5%(w/v);
冰醋酸水溶液的体积百分比浓度除了0.5%之外,其他浓度0.5-2.0%均适用于本发明;本发明使用前的壳聚糖-醋酸溶液的pH值以5.5为例进行说明,其他pH值5-6均适用于本发明。
1-3)配制酸引发剂
将精确量取的冰乙酸加入到液体石蜡中,搅拌,混合均匀,备用,其中冰乙酸与酸引发剂的体积之比为1:8-12,优选为1:10。
本发明实施例中酸引发剂中冰乙酸与液体石蜡的体积之比以1:10为例进行说明,其他冰乙酸与液体石蜡的体积之比为1:8-12均适用于本发明。
2、制备淫羊藿苷-海藻酸钙微球
2-1)配制水相
用注射器移取步骤1)制备的海藻酸钠溶液(20ml)置于烧杯,电磁搅拌,在温度保持为45±5℃条件下加入纳米级碳酸钙(0.1g),搅拌均匀后加入淫羊藿苷(0.3g),搅拌均匀,使得纳米级碳酸钙及淫羊藿苷混悬于海藻酸钠溶液中,制得碳酸钙-淫羊藿苷-海藻酸钠溶液(即水相,20ml);其中:碳酸钙的重量与海藻酸钠溶液的体积之比为0.5:100;淫羊藿苷的重量与海藻酸钠溶液的体积之比为1.5:100。
2-2)配制油相
用注射器移取表面活性剂Span 80(0.5ml)加入到液体石蜡(100ml)中,于37±5℃水浴条件下,搅拌,使其充分混匀,制得Span-石蜡溶液(油相)备用,其中表面活性剂Span 80与液体石蜡的体积之比为0.5:100;
本发明实施例中以表面活性剂Span80为例进行说明,其他表面活性剂均适用于本发明,例如Tween80等。
2-3)酸引发处理
在37±1℃水浴、搅拌速度为300rpm(200-500rpm也适用)的条件下,将水相加入到油相中进行乳化处理,搅拌使水相与油相充分混匀,形成乳化体系;
乳化处理45min后立即向乳化体系中加入酸引发剂液体石蜡-醋酸混合液(5mL,含冰乙酸0.5ml),进行酸引发处理,形成淫羊藿苷-海藻酸钙微球,其中,酸引发剂中冰乙酸与液体石蜡的体积之比为1:10;酸引发剂中冰乙酸的体积与水相中碳酸钙的重量之比为5:1(v/w);
酸引发处理15min后停止搅拌,然后静止放置,混合体系上下分层(即静置分层)静置30min后,取下层淫羊藿苷-海藻酸钙微球沉淀,用体积比0.5%的Tween80水溶液清洗3次沉淀,以去除多余的液体石蜡,接着用纯水洗涤,直至清洗干净,收集淫羊藿苷-海藻酸钙微球。
本发明实施例中水相与油相的体积之比以1:5为例进行说明,其他水相与油相的体积之比为1:4-6均适用于本发明;酸引发剂中冰乙酸与液体石蜡的体积之比以1:10为例进行说明,其他配比如1:9-11均适用于本发明
本发明将冰乙酸与与液体石蜡混合制成的混合液作为酸引发剂,可以将引发剂酸与乳化体系混合均匀,冰醋酸迅速均匀地分散到乳化体系中,避免局部酸浓度过高或过低,同时加入的冰醋酸与纳米碳酸钙发生化学反应,使纳米碳酸钙中钙以离子的状态游离出来,加入的酸引发剂液体石蜡-醋酸混合液在乳化体系中形成一个缓冲溶液体系,使钙离子能够缓慢、持续的释放,释放的钙离子与乳化体系微乳内的海藻酸钠发生反应,形成内部交联,生成海藻酸钙均匀分布于形成的微球,从而有利于形成粒径均匀、结构紧密的微米级微球。
本发明乳化处理形成淫羊藿苷-海藻酸钙微球过程中,采用冰乙酸引发纳米级碳酸钙释放钙离子,不仅使得本发明制备工艺条件反应温和,可控性强,有利于形成粒径均匀的微米级微球;而且由于在乳化过程中由于纳米碳酸钙均匀分布于微乳内部,由醋酸引发释放钙离子与海藻酸分子形成凝胶,这样形成的凝胶属于内部交联,使得形成的凝胶微球结构更为紧密,有良好的药物控释性能;而其他制备微球通常采用在微乳液形成之后,再添加氯化钙或其它钙离子溶液固化形成凝胶,由于微乳液的水相液滴直径在100微米甚至更大,微乳液外部最先接触到钙离子,迅速固化,形成了一个比较致密的凝胶“壳层”,不利于钙离子向内部迁移,而影响固化效率,使得形成的微球内部未能很好固化,而是一个微囊,这样会造成药物释放 过快或者微囊解体突释药物,不利于结肠靶向给药。
3、壳聚糖-淫羊藿苷-海藻酸钙微球的制备
将精确量取步骤2)制备的淫羊藿苷-海藻酸钙微球加入到成膜溶液壳聚糖-醋酸溶液(pH5.5)中,在搅拌状态下使壳聚糖与海藻酸钠发生复电解质络合反应,壳聚糖包裹在淫羊藿苷-海藻酸钙微球的表面,磁力搅拌40min后,使壳聚糖充分与海藻酸钠发生复电解质络合反应,包裹在淫羊藿苷海藻酸钙微球的表面。反应完毕后离心收集微球,用纯水清洗微球表面,除去未反应的壳聚糖溶液,将微球收集,获得络合反应后的壳聚糖-淫羊藿苷-海藻酸钙微球,其中,淫羊藿苷-海藻酸钙微球与成膜溶液的体积比为1:5;所述复电解质络合反应时间为40min。
4、淫羊藿苷/壳聚糖/海藻酸钙控释微球的制备
4-1)将壳聚糖-淫羊藿苷-海藻酸钙微球加入到交联剂溶液(质量百分比浓度为0.25%的戊二醛水溶液)中,搅拌、混合均匀,在4℃条件下,于滚轴上振摇,戊二醛与壳聚糖-淫羊藿苷-海藻酸钙微球发生交联反应,其中,壳聚糖-淫羊藿苷-海藻酸钙微球与戊二醛溶液的体积之比为1:5;
本发明实施例中交联反应温度以4℃为例进行说明,低温条件下能够避免戊二醛与壳聚糖发生剧烈反应,使得交联反应速度更加缓慢平稳,形成的微球表面厚度更加均匀一致,其他温度低于10℃(例如4-8℃)条件下均适用于本发明。
4-2)交联反应时间为1.5h后,向反应体系中加入交联终止剂摩尔浓度为0.5mol/L的甘氨酸溶液去除未反应的戊二醛,其中,甘氨酸溶液的用量与戊二醛溶液体积比为2:1;
4-3)将上述溶液通过离心机离心得到湿态的淫羊藿苷控释微球,然后用纯水清洗微球表面,再次离心,反复3次,最后将得到的淫羊藿苷控释微球冷冻干燥24h备用。
实施例2
1、配制溶液
1-1)配制海藻酸钠溶液
将精确称量的海藻酸钠加入到纯水中,混合,加热并保持在45±5℃条件下,搅拌使海藻酸钠充分溶解,制得海藻酸钠溶液,待其降至室温后于4℃冰箱保存,使用前加热至45℃,备用,其中,海藻酸钠溶液中海藻酸钠的质量与水的体积之比为1.5%;
1-2)配制壳聚糖-醋酸溶液(成膜溶液)
将精确称量的壳聚糖加入到1%的冰醋酸(v/v,体积百分比浓度)水溶液中,混匀后,加 热升温并保持在45±5℃条件下,搅拌,使壳聚糖充分溶解,制得壳聚糖-醋酸溶液(成膜溶液),待其降至室温后于4℃冰箱保存,使用前调节壳聚糖-醋酸溶液的pH为5.5,备用,其中,壳聚糖-醋酸溶液中壳聚糖的质量与冰醋酸水溶液的体积之比为1:100,即壳聚糖-醋酸溶液中壳聚糖的浓度为1%;
冰醋酸水溶液的体积百分比浓度除了1%之外,其他浓度0.5-2.0%均适用于本发明;本发明使用前的壳聚糖-醋酸溶液的pH值除了5.5之外,其他pH值5-6均适用于本发明。
1-3)配制酸引发剂
将精确量取的冰乙酸加入到液体石蜡中,搅拌,混合均匀,备用,其中冰乙酸与酸引发剂的体积之比为1:10。
2、制备淫羊藿苷-海藻酸钙微球
2-1)配制水相
用注射器移取步骤1)制备的海藻酸钠溶液(20ml)置于烧杯,电磁搅拌,在温度保持为45±5℃条件下加入纳米级碳酸钙(0.2g),搅拌均匀后加入淫羊藿苷(0.45g),搅拌均匀,使得纳米级碳酸钙及淫羊藿苷混悬于海藻酸钠溶液溶液中,制得碳酸钙-淫羊藿苷-海藻酸钠溶液(即水相,20ml);其中:碳酸钙的重量与海藻酸钠溶液的体积之比为1:100;淫羊藿苷的重量与海藻酸钠溶液的体积之比为2.25:100。
2-2)配制油相
用注射器移取表面活性剂Span 80(1ml)加入到液体石蜡(100ml)中,于37±5℃水浴条件下,机械搅拌,使其充分混匀,制得Span-石蜡溶液(油相)备用,其中表面活性剂Span80与液体石蜡的体积之比为1:100;
2-3)酸引发处理
在37±1℃水浴、搅拌速度为300rpm的条件下,将水相加入到油相中进行乳化处理,搅拌使水相与油相充分混匀,形成乳化体系;
乳化处理40min后立即向乳化体系中加入酸引发剂液体石蜡-醋酸混合液(5mL,含冰乙酸0.5ml),进行酸引发处理,形成淫羊藿苷-海藻酸钙微球,其中,酸引发剂中冰乙酸的体积与水相中碳酸钙的重量之比为5:2(v/w);
酸引发处理15min后停止搅拌,然后静止放置,混合体系上下分层(即静置分层)静置30min后,取下层淫羊藿苷-海藻酸钙微球沉淀,用体积比1%的Tween80水溶液清洗3次沉淀,以去除多余的液体石蜡,接着用纯水洗涤,直至清洗干净,收集淫羊藿苷-海藻酸钙微球。3、壳聚糖-淫羊藿苷-海藻酸钙微球的制备
将精确量取步骤2)制备的淫羊藿苷-海藻酸钙微球加入到成膜溶液壳聚糖-醋酸溶液(pH5.5)中,在搅拌状态下使壳聚糖与海藻酸钠发生复电解质络合反应,壳聚糖包裹在淫羊藿苷-海藻酸钙微球的表面,磁力搅拌30min后,使壳聚糖充分与海藻酸钠发生复电解质络合反应,包裹在淫羊藿苷海藻酸钙微球的表面。反应完毕后离心收集微球,用纯水清洗微球表面,除去未反应的壳聚糖溶液,将微球收集,获得络合反应后的壳聚糖-淫羊藿苷-海藻酸钙微球,其中,淫羊藿苷-海藻酸钙微球与成膜溶液的体积比为1:8;所述复电解质络合反应时间为30min;
4、淫羊藿苷/壳聚糖/海藻酸钙控制微球的制备
4-1)将壳聚糖-淫羊藿苷-海藻酸钙微球加入到交联剂溶液(质量百分比浓度为0.5%的戊二醛溶液)中,搅拌、混合均匀,在4℃条件下,于滚轴上振摇,戊二醛与包裹壳聚糖的淫羊藿苷-海藻酸钙微球发生交联反应,其中,壳聚糖-淫羊藿苷-海藻酸钙微球与戊二醛溶液的体积之比为1:10;
4-2)交联反应时间为2h后,向反应体系中加入甘氨酸溶液(摩尔浓度为1mol/L)去除未反应的戊二醛,其中,甘氨酸溶液的用量与戊二醛溶液体积比为2:1;
4-3)将上述溶液通过离心机离心得到湿态的淫羊藿苷控释微球,然后用纯水清洗微球表面,再次离心,反复3次,最后将得到的淫羊藿苷控释微球冷冻干燥24h备用。
实施例3
1、配制溶液
1-1)配制海藻酸钠溶液
将精确称量的海藻酸钠加入到纯水中,混合,加热并保持在45±5℃条件下,搅拌,使海藻酸钠充分溶解,制得海藻酸钠溶液,待其降至室温后于4℃冰箱保存,使用前加热至45℃,备用,其中,海藻酸钠溶液中海藻酸钠的质量与水的体积之比为2%;
1-2)配制壳聚糖-醋酸溶液(成膜溶液)
将精确称量的壳聚糖加入到2%的冰醋酸(v/v,体积百分比浓度)水溶液中,混匀后,加热升温并保持在45±5℃条件下,搅拌,使壳聚糖充分溶解,制得壳聚糖-醋酸溶液(即成膜溶液),待其降至室温后于4℃冰箱保存,使用前调节壳聚糖-醋酸溶液的pH为5.5,备用,其中,壳聚糖-醋酸溶液中壳聚糖的质量与冰醋酸水溶液的体积之比为2%;
冰醋酸水溶液的体积百分比浓度除了2%之外,其他浓度0.5-2.0%均适用于本发明;本发明使用前的壳聚糖-醋酸溶液的pH值除了5.5之外,其他pH值5-6均适用于本发明。
2、制备淫羊藿苷-海藻酸钙微球
2-1)配制水相
用注射器移取步骤1)制备的海藻酸钠溶液(20ml)置于烧杯,电磁搅拌,在温度保持为45±5℃条件下加入纳米级碳酸钙(0.3g),搅拌均匀后加入淫羊藿苷(0.5g),搅拌均匀,使得纳米级碳酸钙及淫羊藿苷混悬于海藻酸钠溶液溶液中,制得碳酸钙-淫羊藿苷-海藻酸钠溶液(即水相,20ml);其中:碳酸钙的重量与海藻酸钠溶液的体积之比为1.5:100;淫羊藿苷的重量与海藻酸钠溶液的体积之比为2.5:100。
2-2)配制油相
用于注射器移取表面活性剂Span 80(1.5ml)加入到液体石蜡(100ml)中,于37±5℃水浴条件下,搅拌,使其充分混匀,制得Span-石蜡溶液(油相)备用,其中表面活性剂Span80与液体石蜡的体积之比为1.5:100;
2-3)酸引发处理
在37±1℃水浴、搅拌速度为300rpm的条件下,将水相加入到油相中进行乳化处理,搅拌使水相与油相充分混匀,形成乳化体系;
乳化处理50min后立即向乳化体系中加入酸引发剂液体石蜡-醋酸混合液(5mL,含冰乙酸0.5ml),进行酸引发处理,形成淫羊藿苷-海藻酸钙微球,其中,酸引发剂中液体石蜡与冰醋酸的体积之比为10:1;酸引发剂中冰乙酸的体积与水相中碳酸钙的重量之比为5:3(v/w);
酸引发处理20min后停止搅拌,然后静止放置,混合体系上下分层(即静置分层)静置30min后,取下层淫羊藿苷-海藻酸钙微球沉淀,用体积比1.5%的Tween80水溶液清洗3次沉淀,以去除多余的液体石蜡,接着用纯水洗涤,直至清洗干净,收集淫羊藿苷-海藻酸钙微球。
3、壳聚糖-淫羊藿苷-海藻酸钙微球的制备
将精确量取步骤2)制备的淫羊藿苷-海藻酸钙微球加入到成膜溶液壳聚糖-醋酸溶液(pH5.5)中,在搅拌状态下使壳聚糖与海藻酸钠发生复电解质络合反应,壳聚糖包裹在淫羊藿苷-海藻酸钙微球的表面,磁力搅拌20min后,使壳聚糖充分与海藻酸钠发生复电解质络合反应,包裹在淫羊藿苷海藻酸钙微球的表面。反应完毕后离心收集微球,用纯水清洗微球表面,除去未反应的壳聚糖溶液,将微球收集,获得络合反应后的壳聚糖-淫羊藿苷-海藻酸钙微球,其中,淫羊藿苷-海藻酸钙微球与成膜溶液的体积比为1:15;所述复电解质络合反应时间为20min。
4、淫羊藿苷/壳聚糖/海藻酸钙控制微球的制备
4-1)将壳聚糖-淫羊藿苷-海藻酸钙微球加入到交联剂溶液(质量百分比浓度为1%的戊二醛水溶液)中,搅拌、混合均匀,在8℃条件下,于滚轴上振摇,戊二醛与壳聚糖-淫羊藿苷-海藻酸钙微球发生交联反应,其中,壳聚糖-淫羊藿苷-海藻酸钙微球与戊二醛溶液的体积之比为1:20;
4-2)交联反应时间为3h后,向反应体系中加入甘氨酸溶液(摩尔浓度为2mol/L)去除未反应的戊二醛,其中,甘氨酸溶液的用量与成膜溶液体积比为1:1;
4-3)将上述溶液通过离心机离心得到湿态的淫羊藿苷控释微球,然后用纯水清洗微球表面,再次离心,反复3次,最后将得到的淫羊藿苷控释微球冷冻干燥24h备用。
实施例4淫羊藿苷/壳聚糖/海藻酸钙控释微球体外释放药物实验
根据2015版《中国药典》一部附录,“释放度”测定的第一种方法“转篮法”测定,分别以模拟胃液(2h、pH=1.2);模拟小肠液(3h、pH=6.8);模拟结肠液(24h、pH=7.4)为溶出介质,温度为37±0.5℃,转速为50r/min。
分别精密称取实施例2制备的淫羊藿苷缓控释微球各2g,共8份,投入8个干燥的转篮内,模拟胃液4份;模拟小肠液、模拟结肠液4份,调节转篮转速50rpm,待其平稳后,将转篮降入恒温的含有200mL溶出介质(模拟胃液、模拟小肠液)的溶出杯中,自样品接触溶出介质起立即计时,于不同时间点分别取样1mL,其中模拟胃液取样时间点为5,10,15,20,25,30,40,50,60,80,100,120min;模拟小肠液取样时间点为5,10,15,20,25,30,40,50,60,80,100,120,150,180min;在本发明制备的淫羊藿苷缓控释微球在模拟小肠液中释放药物3h后将装有淫羊藿苷控释微球的转篮从模拟小肠液转移到模拟结肠液中,进行本发明制备的淫羊藿苷缓控释微球在模拟结肠液中的药物释放试验,模拟结肠液的取样时间点为:200,260,320,400,600,720,1440,2880min,每次取样后同时补加相同体积的模拟液,将取得样品经0.45μm微孔滤膜过滤后采用高效液相色普法(HPLC)测定释放液中淫羊藿苷的含量。
将淫羊藿苷缓控释微球首先在模拟小肠液中释放180min,然后放入模拟结肠液中继续释放,主要看其在微球到达结肠环境下的释放情况,由于结肠的特殊结构,微球在其中停留的时间较长,因此我们模拟的时候也延长了释放时间,延长到2800min。
淫羊藿苷/壳聚糖/海藻酸钙微球在模拟胃液中释放2h后累计释放率仅有10%(如图1),图2显示微球在模拟小肠及结肠液中的释放情况,结果表明本发明制备的淫羊藿苷控释微球在模拟小肠及结肠液中药物能够大量释放,累计释放率可达到65%,在较高pH值的模拟结 肠液中淫羊藿苷/壳聚糖/海藻酸钙控释微球可以持续释放药物。
图2中虚线左边为模拟小肠液(pH 6.8)的药物释放情况,释药时间从5-180min;虚线的右边表示为模拟结肠液(pH7.4)的药物释放情况,释药时间从200-2880min。
图2A为淫羊藿苷/壳聚糖/海藻酸钙微球在模拟小肠液中释放情况,本发明制备的淫羊藿苷控释微球在释放180min后累计释放率为10.51%。
实施例5淫羊藿/苷壳聚糖/海藻酸钙控释微球体内消化道内分布
取一定体积的按照实施例2步骤2制备的淫羊藿苷-海藻酸钙微球,将淫羊藿苷-海藻酸钙微球加入到6倍的FITC荧光标记的成膜溶液壳聚糖-醋酸溶液(pH5.5)中,磁力搅拌30min,反应完毕后,用70%的甲醇溶液洗涤微球至上清在495nm无吸收,然后用纯水清洗微球表面,得到FITC标记壳聚糖-淫羊藿苷-海藻酸钙微球;其中,所述FITC荧光标记的成膜溶液中FITC荧光标记的壳聚糖的浓度为10mg/mL;
将FITC标记壳聚糖-淫羊藿苷-海藻酸钙微球分散于10倍的0.5%戊二醛交联剂中,4℃条件下置于滚轴上振摇交联反应2h。用1mol/L的甘氨酸溶液洗去多余未反应的戊二醛溶液,最后用纯水清洗微球表面,冷冻干燥24h,制得FITC荧光标记的淫羊藿苷/壳聚糖/海藻酸钙控释微球,备用。
选取健康的雄性ICR小鼠进行试验,体重20±2g,实验前禁食24h,自由饮水;将得到的FITC标记的淫羊藿苷/壳聚糖/海藻酸钙控释微球,用蒸馏水分散均匀,按照6mg/20g(微球的重量/小鼠重量)小鼠灌胃给予FITC标记的淫羊藿苷/壳聚糖/海藻酸钙控释微球,共7组,每组3只小鼠,分别在灌胃后的不同时间点(2、4、6、12、16、24、36h),将实验组小鼠麻醉后分离胃、十二指肠、空肠、结肠四部分,2h为第一组,4h为第二组,6h为第三组,12h为第四组,16h为第五组,24h为第六组,36h为第七组,使用小动物成像仪观察FITC标记交联微球在小鼠胃肠道的分布情况。
FITC荧光标记微球小鼠消化道内的分布结果分析如图3所示。
为了进一步考察本发明的pH响应控释微球在小鼠消化道内的分布情况,给小鼠灌胃FITC标记壳聚糖/海藻酸钙控释微球,在灌胃后的不同时间点(2、4、6、12、16、24、36h)观察微球在胃肠道内的分布情况,以观察控释微球的定位情况。从图3的荧光强度照片可以看出,前12h微球主要聚集在胃部,而自16h开始,微球开始大量集聚于结肠,24h时集聚最多。结果表明本发明制备的结肠定位释药微球能够在给药16-36h内,将淫羊藿苷药物聚集在结肠部位,从而达到靶向性治疗结肠炎症的目的。
实施例6淫羊藿苷/壳聚糖/海藻酸钙控释微球的药效学评价分析
1、TNBS/乙醇诱导结肠炎模型的建立
TNBS/乙醇诱导的大鼠溃疡性结肠炎模型为经典的溃疡性结肠炎模型。选健康雄性SD大鼠,禁食24h后,按0.3mL/100g腹腔注射10%的水合氯醛麻醉,仰卧固定于大鼠实验板上,将灌胃针由肛门轻缓插入深约8cm处,按100mg/kg剂量(即4mL/kg)推入5%TNBS/乙醇溶液。灌入结肠后保持肛门高位5分钟,防止药物流出,随即将大鼠放回笼内,待自然苏醒。2、试验药物
本发明实施例2制备的淫羊藿苷/壳聚糖/海藻酸钙控释微球;
阳性药物:地塞米松(片剂),天津力生制药股份有限公司(0.75mg/片,共100片)
阳性药物:淫羊藿苷提取物(纯度>99%):南京替斯艾么中药研究所
3、分组与给药
将造模成功肠炎大鼠随机分为7组,每组8只,分别为空白组(Control)、模型组(Model)、阳性药组地塞米松(DEX,0.2mg/kg)、低剂量组(Low-dose,30mg淫羊藿苷微球/kg)、中剂量组(Medium-dose,60mg淫羊藿苷微球/kg)、高剂量组(High-dose,90mg淫羊藿苷微球/kg)、淫羊藿苷组(50mg淫羊藿苷/kg)。造模成功24h后开始灌胃给药,空白组灌胃给予生理盐水,模型组灌胃给予生理盐水,阳性药组灌胃给予地塞米松(0.2mg/kg),淫羊藿苷组灌胃给予淫羊藿苷微球,低剂量组(Low-dose,30mg淫羊藿苷微球/kg)、中剂量组(Medium-dose,60mg淫羊藿苷微球/kg)、高剂量组(High-dose,90mg淫羊藿苷微球/kg)、淫羊藿苷组灌胃给以淫羊藿苷(50mg淫羊藿苷/kg),连续给药6天,末次给药24h后将大鼠处死,取材。
大鼠灌胃剂量参照《中药药理研究方法学》,用成人每日给药剂量乘以大鼠换算系数。4、取材及样本处理
取材前大鼠禁食24h,10%水合氯醛经腹腔注射麻醉后仰卧固定。沿腹中线切开腹壁,曝露腹腔组织,小心分离结肠(从回盲部到肛口)。切断结肠,沿肠系膜纵向剖开肠段,冰生理盐水冲洗后,肉眼观察结肠黏膜形态,取完整结肠肠段,用滤纸吸除多余水分,平展于玻璃板上,观察结肠外观,切开结肠肠腔后迅速观察有无血性内容物,检查肠黏膜有无充血水肿、糜烂及溃疡形成,记录结肠黏膜损伤程度并照相,称重,将剩余结肠段分装于样本冻存管中,在液氮中速冻及时转移至-80℃保存待用(取材操作均在冰上进行)。
5、药效学评价结果
5.1结肠黏膜损伤指数(CMDI)
剖取大鼠肛门至盲肠末端的结肠段,清理肠系膜及粘连组织,沿肠系膜纵轴剪开,冰磷酸盐缓冲液反复冲洗肠内残余粪便,干净后平铺于滤纸上以吸去水分,并立即观察大体黏膜损伤程度。
CMDI评分表如表1所示。
表1 CMDI评分表
结肠黏膜损伤指数(CMDI)的测定结果如表2所示。
表2淫羊藿苷/壳聚糖/海藻酸钙控释微球对结肠炎模型大鼠CMDI的影响结果
结肠炎模型大鼠结肠黏膜损伤指数实验结果表明,与空白组比较,模型组结肠粘膜损伤较严重,CMDI值较高,而给药组相应的CMDI值均出现显著性地下调,阳性药物地塞米松也能够下调结肠炎大鼠CMDI值,给药高剂量组要优于阳性药组;阳性药物淫羊藿苷也能够 下调结肠炎大鼠CMDI值,给药高剂量组要优于阳性药组。本发明控释微球与阳性药物淫羊藿苷对照组相比,有效降低了药物的使用剂量,同时能够使药物通过控释微球到达结肠部位,药物能够在结肠部位持续释放,而口服给予淫羊藿苷药物较难到达结肠部位,控释微球不仅能够实现靶向治疗,还提高药物的生物利用度,更有效达到治疗效果。
5.2淫羊藿苷/壳聚糖/海藻酸钙控释微球对溃疡性结肠炎模型大鼠结肠组织中炎性相关因子分泌的影响
将结肠组织样本采用蛋白裂解液进行蛋白提取,用BCA蛋白浓度测定试剂盒(美国Pierce公司)测定蛋白含量;采用ELISA法测定结肠组织样本中IL-1β和TNF-α的分泌;IL-1β的浓度采用Rat IL-1βELISA试剂盒(美国PeproTech公司)进行测定;TNF-α的浓度采用Rat TNF-αELISA试剂盒(美国eBioscience公司)进行测定,具体实验步骤按照试剂盒说明书操作。
淫羊藿苷/壳聚糖/海藻酸钙控释微球对溃疡性结肠炎模型大鼠结肠组织IL-1β含量的影响测定结果如表3、图4所示;淫羊藿苷壳聚糖/海藻酸钙控释微球对溃疡性结肠炎模型大鼠结肠组织TNF-α含量的影响如表3、图5所示。
表3.淫羊藿苷/壳聚糖/海藻酸钙控释微球对溃疡性结肠炎模型大鼠结肠组织IL-1β含量的影响
IL-1β主要介导溃疡性结肠炎初期阶段炎症的产生,可以促使炎性细胞进入结肠内部病变位置。由表2及图4可以看出,与空白组比较,模型组结肠组织中的IL-1β明显升高,具有极显著性差异(P<0.01);阳性药地塞米松对照组和微球给药治疗组均能显著抑制IL-1β的产生(P<0.01或P<0.05),其中载药微球中剂量组在抑制结肠炎大鼠IL-1β的产生上具有极显著性差异(P<0.01)。阳性药物淫羊藿苷对照组和微球给药治疗组均能显著抑制IL-1β的产生(P<0.01 或P<0.05),本发明的控释微球有效降低了药物的使用剂量,同时能够使药物通过控释微球到达结肠部位,药物能够在结肠部位持续释放,而口服给予淫羊藿苷药物较难到达结肠部位,控释微球不仅能够实现靶向治疗,还提高药物的生物利用度,更有效达到治疗效果。
表4.淫羊藿苷壳聚糖/海藻酸钙控释微球对溃疡性结肠炎模型大鼠结肠组织TNF-α含量的影响
TNF-α主要启动炎性因子,可以刺激单核巨噬细胞和中性粒细胞等合成其他炎性细胞因子。由表4和图5可知,与空白组比较,模型组TNF-α含量明显升高,具有极显著性差异(P<0.01);与模型组相比,阳性药物地塞米松对照组和载药微球高、中、低剂量组TNF-α含量均显著降低(P<0.05),载药微球高剂量组抑制效果最明显;阳性药物淫羊藿苷对照组和载药微球高、中、低剂量组TNF-α含量均显著降低(P<0.05),载药微球高剂量组抑制效果最明显。有效降低了药物的使用剂量,同时能够使药物通过控释微球到达结肠部位,药物能够在结肠部位持续释放,而口服给予淫羊藿苷药物较难到达结肠部位,控释微球不仅能够实现靶向治疗,还提高药物的生物利用度,更有效达到治疗效果。
- 还没有人留言评论。精彩留言会获得点赞!