双亲性羟丙基-β-环糊精纳米药物载体及其载药纳米粒子的制备方法与流程
本发明属于纳米药物载体及载药纳米粒子的合成技术领域,具体涉及一种双亲性羟丙基-β-环糊精纳米药物载体及其载药纳米粒子的制备方法。
背景技术:
环糊精(Cyclodextrins,CD)首次由Villiers在1891年作为淀粉降解产品分离出来,1904年由Schardinger表征为环状低聚糖,因此也称为Schardinger糊精,1938年Freudenberg等证明环糊精的葡萄糖单元以1,4糖苷键链接,并在之后根据葡萄糖单元组成将其分类为α-CD、β-CD和γ-CD,1953年获得CD在药物组方的应用专利,提出CD可以提高药物溶解度和稳定性。由于CD的杯状结构内部相对非极性,能提供一个疏水性环境,因此可以作为“主体”将“客体”分子包合在内部,从而大大提高疏水性药物的溶解性。环糊精对人体酶解不敏感,可以原形从肾排除,但细菌和真菌可促使其降解,可在胃肠道系统被代谢。而小鼠通过注射α-CD、β-CD及γ-CD的LD50分别为1.0g/kg、0.79g/kg及4.0g/kg。在同样条件下,α-CD、β-CD及γ-CD水中溶解度分别为13%、2%和26%。而当浓度足够大时,β-CD因其溶解度低,且足够浓度能溶解红血球,因此无法直接注射用药,而对其进行功能化研究则可克服该问题。羟丙基-β-CD已经被美国FDA所批准应用,水中溶解度可达到60%(w/w)以上,因此是将疏水性药物制备成以水为溶媒的液体制剂的理想载体。
环糊精尽管具有包合性能和可溶解难溶性药物的性能,但却不具有将药物靶向到病变部位的能力。而且由于天然环糊精外部亲水基团多,使其无法与脂质生物膜接近,因此,功能化的环糊精使其既能够维持其安全性也能够保持其包合能力,目前成为学者研究的热点。将环糊精包裹在脂质体可因为脂质体的特性而将携带的药物靶向到病变部位,同时环糊精也可以延长药物在脂质体内的保留时间。
双亲性聚合物能自组装成胶束体系,影响传统双亲性共聚物胶束作为药物传递载体的一大缺陷就是载药量和包封率低及初始释药太快,进而限制了其在药物传递体系的应用。双亲性的PEG-PCL多嵌段共聚物作为载体材料,其中亲脂性的PCL片段充当疏水性药物的储库,PEG的亲水性则可形成称之为“stealth”结构,避免颗粒被网状内皮系统的非特异性吸收,从而延长药物在体内的循环时间。而将羟丙基-β-环糊精与双亲性mPEG-PCL共聚物反应可得到新的双亲性高分子,并在适宜条件下形成载药纳米粒子。
技术实现要素:
本发明解决的技术问题是提供了一种能够有效实现对水难溶/不溶性药物进行包裹的双亲性羟丙基-β-环糊精纳米药物载体的制备方法。
本发明解决的另一个技术问题是提供了一种双亲性羟丙基-β-环糊精载药纳米粒子的制备方法。
本发明为解决上述技术问题采用如下技术方案,双亲性羟丙基-β-环糊精纳米药物载体的制备方法,其特征在于具体步骤为:
(1)丙烯酰化mPEG-b-PCL(AC-mPEG-b-PCL)的合成,将mPEG-b-PCL溶于二氯甲烷中,在冰浴条件下加入三乙胺形成溶液A,将丙烯酰氯溶于二氯甲烷中形成溶液B,将溶液B滴加到溶液A中反应1h后升至室温继续反应2h,用0.45μm的微孔滤膜过滤,滤液滴入冰乙醚,沉淀,过滤,冷冻干燥得到丙烯酰化mPEG-b-PCL;
(2)丙烯酰化羟丙基-β-CD(AC-HP-β-CD)的合成,将HP-β-CD溶于二氯甲烷并在冰浴条件下加入三乙胺,反应20min得到溶液A,将丙烯酰氯溶于DMF中形成溶液B,将溶液B滴加到溶液A中,在冰浴条件下搅拌反应1h后升至室温继续反应2h,用0.45μm微孔滤膜过滤,滤液滴入冰乙醚,沉淀,过滤,冷冻干燥得到丙烯酰化羟丙基-β-CD;
(3)双亲性羟丙基-β-环糊精纳米药物载体mPEG-b-PCL-co-AC-HP-β-CD的合成,将步骤(1)得到的丙烯酰化mPEG-b-PCL和步骤(2)得到的丙烯酰化羟丙基-β-CD溶于二甲基亚砜作为油相,将过硫酸铵和亚硫酸氢钠溶于水中作为水相,将水相加入到油相中于37℃反应12h,透析24h,冷冻干燥得到mPEG-b-PCL-co-AC-HP-β-CD。
进一步优选,步骤(1)中各原料的用量分别为mPEG-b-PCL3.0g,三乙胺0.71mL,丙烯酰氯0.41mL。
进一步优选,步骤(2)中各原料的用量分别为HP-β-CD 1.5g,三乙胺0.14mL,丙烯酰氯0.2mL。
进一步优选,步骤(3)中各原料的用量分别为丙烯酰化mPEG-b-PCL0.10g,丙烯酰化羟丙基-β-CD 0.02g,过硫酸铵0.032g,亚硫酸氢钠0.032g。
进一步优选,所述的双亲性羟丙基-β-环糊精纳米药物载体mPEG-b-PCL-co-AC-HP-β-CD中PEG的平均相对分子质量为1000-10000,PCL的平均相对分子质量为1000-20000。
进一步优选,所述的双亲性羟丙基-β-环糊精纳米药物载体mPEG-b-PCL-co-AC-HP-β-CD为mPEG2000-b-PCL4000-co-AC-HP-β-CD。
本发明所述的双亲性羟丙基-β-环糊精载药纳米粒子的制备方法,其特征在于具体步骤为:将上述制得的双亲性羟丙基-β-环糊精纳米药物载体mPEG-b-PCL-co-AC-HP-β-CD与难溶/不溶药物溶于四氢呋喃中形成溶液A;将泊洛沙姆溶于水中形成溶液B;将溶液A滴加到溶液B中挥干,冷冻干燥得到载药纳米粒子冻干粉。
进一步优选,所述的药物为姜黄素,双亲性羟丙基-β-环糊精纳米药物载体mPEG-b-PCL-co-AC-HP-β-CD、姜黄素与泊洛沙姆的质量比为1:0.05:0.3。
本发明与现有技术相比具有以下有益效果:本发明制得的双亲性羟丙基-β-环糊精纳米药物载体能够有效实现对水难溶性药物进行包裹,实现双载药位点。作为新聚合物的一部分,mPEG-b-PCL具有双亲性,可以自组装成为纳米粒子,并且其亲油基团可以与水难溶/不溶性药物亲和并载药,同时环糊精本身可以提高水难溶/不溶性药物的亲水性,因此具有两个载药位点,本发明设计合理,步骤简单,用于水难溶/不溶性药物载药具理想释药效果。
附图说明
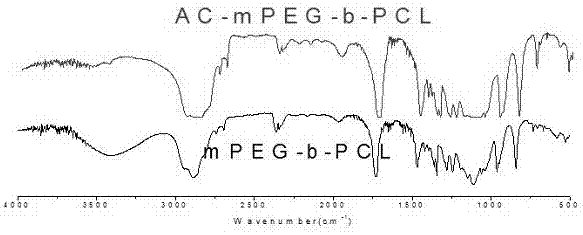
图1是实施例1中丙烯酰化前后的mPEG2000-b-PCL4000的红外光谱图;
图2是实施例1中丙烯酰化前后的mPEG2000-b-PCL4000的H1NMR图;
图3是实施例1中丙烯酰化前后羟丙基-β-CD的红外光谱图;
图4是实施例1中丙烯酰化前后羟丙基-β-CD的H1NMR图;
图5是实施例1中mPEG2000-b-PCL4000-co-AC-HP-β-CD的H1NMR图;
图6是实施例1中双亲性羟丙基-β-环糊精载药纳米粒子的透射电镜图;
图7是姜黄素载药纳米粒子在pH=7.4的缓冲溶液中的释放曲线。
具体实施方式
以下通过实施例对本发明的上述内容做进一步详细说明,但不应该将此理解为本发明上述主题的范围仅限于以下的实施例,凡基于本发明上述内容实现的技术均属于本发明的范围。
本发明合成过程的反应方程式为:
实施例1
(1)丙烯酰化mPEG-b-PCL(AC-mPEG-b-PCL)的合成,将3.0g mPEG2000-b-PCL4000溶于5mL二氯甲烷中,在冰浴条件下加入0.71mL三乙胺形成溶液A,将0.41mL丙烯酰氯溶于1mL二氯甲烷中形成溶液B,将溶液B滴加到溶液A中反应1h后升至室温继续反应2h,用0.45μm的微孔滤膜过滤,滤液滴入冰乙醚(体积比为1:10),沉淀,过滤,冷冻干燥得到丙烯酰化mPEG2000-b-PCL4000;
(2)丙烯酰化羟丙基-β-CD(AC-HP-β-CD)的合成,将1.5g HP-β-CD溶于2mL二氯甲烷并在冰浴条件下加入0.14mL三乙胺,反应20min得到溶液A,将0.2mL丙烯酰氯溶于0.5mLDMF中形成溶液B,将溶液B滴加到溶液A中,在冰浴条件下搅拌反应1h后升至室温继续反应2h,用0.45μm微孔滤膜过滤,滤液滴入冰乙醚(体积比为1:10),沉淀,过滤,冷冻干燥得到丙烯酰化羟丙基-β-CD;
(3)双亲性羟丙基-β-环糊精纳米药物载体mPEG-b-PCL-co-AC-HP-β-CD的合成,将0.10g步骤(1)得到的丙烯酰化mPEG2000-b-PCL4000和0.02g步骤(2)得到的丙烯酰化羟丙基-β-CD溶于2mL二甲基亚砜作为油相,将0.032g过硫酸铵和0.032g亚硫酸氢钠溶于2mL水中作为水相,将水相加入到油相中于37℃反应12h,透析24h,冷冻干燥得到mPEG2000-b-PCL4000-co-AC-HP-β-CD。
(4)双亲性羟丙基-β-环糊精载药纳米粒子的合成,将0.1g步骤(3)制得的双亲性羟丙基-β-环糊精纳米药物载体mPEG-b-PCL-co-AC-HP-β-CD与0.005g姜黄素溶于5mL四氢呋喃中形成溶液A;将0.03g泊洛沙姆溶于100mL水中形成溶液B;将溶液A滴加到溶液B中挥干,冷冻干燥得到载药纳米粒子冻干粉。
以上实施例描述了本发明的基本原理、主要特征及优点,本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明原理的范围下,本发明还会有各种变化和改进,这些变化和改进均落入本发明保护的范围内。
- 还没有人留言评论。精彩留言会获得点赞!