FXR激动剂在制备治疗脂肪肉瘤相关疾病药物中的用途的制作方法
本发明属于药物技术领域,具体涉及人法尼酯衍x受体激动剂在抑制脂肪肉瘤方面的新用途。
背景技术:
软组织肉瘤(softtissuesarcoma,sts)是一组源于黏液、纤维、脂肪、平滑肌、滑膜、横纹肌、间皮、血管和淋巴管等结缔组织的恶性肿瘤。软组织肿瘤的发病率在2/10万之内,约占全部恶性肿瘤的1%。好发于青少年和45~55岁中年人。其具有局部复发和晚期远处转移的特征。在美国,估计每年有11,930例sts新病例和4870例死亡。
脂肪肉瘤(lps)是最常见的sts,并大约占成年软组织肉瘤患者中的20%,多发于肢体和腹膜后。根据2002年世界卫生组织(who)的分类标准,脂肪肉瘤分为5个组织亚型,归类为3个生物学类型:①高分化脂肪肉瘤(welldifferentiatedliposarcoma,wdlps)和去分化脂肪肉瘤(dedifferentiatedliposarcoma,ddlps);②黏液细胞型脂肪肉瘤(myxoidliposarcoma,mls)和圆型细胞型脂肪肉瘤(roundcellliposarcoma,rcls);③多形性脂肪肉瘤(plemorphicliposarcoma,pls)。根据具体的亚型和位置,脂肪肉瘤可以在超过80%的病例中复发,其死亡率从1~90%不等,肿瘤的位置以及大小是重要的预后因素。
手术切除是目前主要治疗手段,但往往难以完全切除,术后复发率高,多次复发者复发间期逐渐缩短,手术切除机会也降低。脂肪肉瘤反复复发手术往往导致患者肢体缺失或重要脏器功能的障碍,而且放化疗等对脂肪肉瘤效果都不明显。目前,脂肪肉瘤总的术后5年生存率为40%-60%,患者多数死于肿瘤的局部复发引起的并发症。近年来,虽然关于脂肪肉瘤的分子机制研究取得很大进展,但这并未能显著改善脂肪肉瘤的治疗现状。因此,我们迫切地需要开发新的诊断标记和治疗靶点。
在脂肪肉瘤的多种亚型中,去分化和高分化脂肪肉瘤作为最常见的病理类型,目前临床上却无反应良好的药物,使得不能手术切除或发生复发的患者难以获得良好的治疗。而黏液细胞型的脂肪肉瘤,虽然对多柔吡星联合异环磷酰胺的化疗方案敏感,但该方案对患者具有较大的生理负担,常因多种不良反应发生而被迫停药,影响患者的治疗。
临床上,ddlps比wdlps更具侵略性,转移率约15-20%,并且5年总生存(os)与wdlps的90%相比只有30%,可见ddlps的恶性程度要远高于wdlps。约有10%的初始分化良好的脂肪肉瘤复发时,可表现为去分化区域的特征。组织病理学研究显示,高分化脂肪肉瘤移行为去分化脂肪肉瘤之后,肿瘤细胞恶性程度升高并且伴随着细胞分化能力的减弱或丧失,那么通过促进去分化脂肪肉瘤细胞重新分化,是否有可能降低肿瘤的恶性程度,并抑制肿瘤的某些生物学特性呢?发明人随即将诱导去分化脂肪肉瘤脂分化以改善脂肪肉瘤的恶性程度确定为研究的新思路。
法尼酯衍生物x受体(farnesoidxreceptor,fxr),又称为nr1h4,属于核受体超家族成员。具有典型的核受体结构,即包括氨基端配体非依赖的转录活化域(af-1)、dna结合域(dbd)、铰链区、配体结合域(lbd)和羧基端配体依赖的转录活化域(af-2)等。fxr通过和视黄醛衍生物受体(retinoidxreceptor,rxr)形成异源二聚体,与特定dna反应元件(ir-1)结合,从而调节靶基因的表达。生理状态下fxr调控一系列代谢相关基因的表达,在糖代谢中发挥重要作用。
不同恶性肿瘤具有不同的发病机制和特点。近年来,文献报导了fxr可以在不同肿瘤中表现出不同的作用。在肝癌和睾丸间质瘤中可以抑制肿瘤的生长,而在食管癌和胰腺癌中可以促进肿瘤的生长。说明fxr在不同肿瘤中,依据肿瘤特点不同,可能发挥抑制或促进的作用。fxr在脂肪肉瘤该种恶性肿瘤的作用未有报道及研究。
本发明首次发现fxr受体激动剂诱导多种亚型的脂肪肉瘤细胞分化,抑制脂肪肉瘤的增值、迁移,侵袭。
技术实现要素:
法尼酯x受体(fxr)是一种胆汁酸激活的核激素受体,属于转录调节因子。本发明提供了fxr激动剂在制备治疗脂肪肉瘤相关疾病药物中的用途,本发明发现fxr激动剂能诱导一种或多种亚型的脂肪肉瘤细胞分化,抑制脂肪肉瘤的增殖、迁移、侵袭,从而提供了fxr激动剂在治疗脂肪肉瘤方面的新应用。
本发明所述的fxr激动剂优选自:6-ecdca、way-362450、gw4064、int-767、px-104、cdca、udca,体外实验显示6-ecdca、way-362450、gw4064、int-767、px-104、cdca、udca可抑制多种脂肪肉瘤细胞系的增殖能力、克隆能力和侵袭力,并且具有促进脂肪肉瘤细胞分化的作用,而在pdx脂肪肉瘤小鼠口服fxr激动剂实验中,显示6-ecdca、way-362450、gw4064、int-767、px-104、cdca、udca具有肿瘤抑制效果。
具体而言,本发明的6-ecdca,6-乙基鹅去氧胆酸,又名奥贝胆酸(6-ethylchenodeoxycholicacid,oca),是人初级胆汁酸中鹅去氧胆酸的一种新型衍生物,为法尼酯x受体(farnesoidxreceptor,fxr)的天然配体,其具有式(ⅰ)的结构,其化学式为:分子式c26h44o44,分子量420.63。
本发明的gw4064是一种完全激动剂,1μm浓度时对其它核受体依然没有活性,包括维甲酸受体。因此,gw4064是一种强有效和选择性的非类固醇fxr激动剂,其具有式(ⅱ)的结构,其化学式为:c28h22cl3no4,分子量为542.84。
本发明的way-362450,又称turofexorateisopropyl(xl335),是一种有效的、选择性的fxr激动剂,ec50为4nm,其具有式(ⅲ)的结构,其化学式为:c25h24f2n2o3,分子量为438.47。
本发明的int-767是一种fxr/tgr5双激动剂,其具有式(ⅳ)的结构,其化学式为:c25h43nao6s,分子量为494.66。
本发明的px-104是一种有效的、选择性的fxr激动剂,其具有式(ⅴ)的结构,其化学式为:c29h22cl3no4,分子量为554.85。
本发明的cdca(鹅去氧胆酸)是一种有效的、选择性的fxr激动剂,其具有式(ⅵ)的结构,其化学式为:c24h40o4,分子量为392.58。
本发明的udca(熊去氧胆酸)是一种有效的、选择性的fxr激动剂,其具有式(ⅵ)的结构,其化学式为:c24h40o4,分子量为392.58。
基于以上发现,本发明提供法尼酯x受体激动剂,包括6-ecdca、way-362450、gw4064、int-767、px-104、cdca、udca:式(ⅰ)、(ⅱ)、(ⅲ)、(ⅳ)、(ⅴ)、(ⅵ)和(ⅶ)的化合物治疗脂肪肉瘤方面的新应用,其优点为体外实验显示可抑制多种脂肪肉瘤细胞系的增殖能力、迁移能力和侵袭能力,并且具有促进脂肪肉瘤细胞分化的作用,小鼠口服法尼酯x受体激动剂6-ecdca、way-362450、gw4064、int-767、px-104、cdca、udca显示法尼酯x受体激动剂具有脂肪肉瘤肿瘤抑制效果。
在一个实施方式中,本发明提供式(ⅰ)、(ⅱ)、(ⅲ)、(ⅳ)、(ⅴ)、(ⅵ)和(ⅶ)的化合物抑制多种脂肪肉瘤细胞系的增殖能力、迁移能力和侵袭能力。
在一个实施方式中,本发明提供式(ⅰ)的化合物诱导脂肪肉瘤细胞脂肪源性分化,促进细胞脂肪堆积。
在一个实施方式中,本发明提供式(ⅰ)、(ⅱ)、(ⅲ)、(ⅳ)、(ⅴ)、(ⅵ)和(ⅶ)的化合物在小鼠脂肪肉瘤pdx模型中显著可以抑制脂肪肉瘤增长速度。
在一个实施方式中,所述药物还包括任何活性或非活性的成分。在一个实施方式中,所述药物还包含药学上可接受的赋形剂。
在一个实施方式中,本发明药物可与任何活性或非活性的成分联合制备或联合给药。
在一个实施方式中,本发明药物制备成适合口服、静脉内、腹膜内、局部、透皮、经眼、经鼻、局部、吸入、皮下、肌内、口含、舌下或直肠给药的剂型。在一个实施方式中,本发明药物剂型包括但不限于片剂、胶囊、丸剂、颗粒制剂、注射液。
附图说明
图1是6-ecdca抑制脂肪肉瘤细胞的迁移,左侧6-ecdca是实验组,右侧nc为对照组。
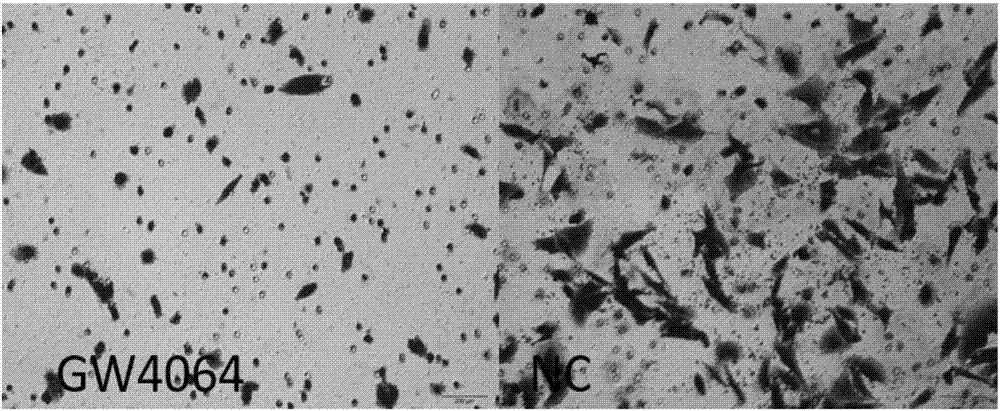
图2是gw4064抑制脂肪肉瘤细胞的迁移,左侧gw4064是实验组,右侧nc为对照组。
图3是way-362450抑制脂肪肉瘤细胞的迁移,左侧way-362450是实验组,右侧nc为对照组。
图4是int-767抑制脂肪肉瘤细胞的迁移,左侧int-767是实验组,右侧nc为对照组。
图5是px-104抑制脂肪肉瘤细胞的迁移,左侧px-104是实验组,右侧nc为对照组。
图6是cdca抑制脂肪肉瘤细胞的迁移,左侧cdca是实验组,右侧nc为对照组。
图7是udca抑制脂肪肉瘤细胞的迁移,左侧udca是实验组,右侧nc为对照组。
图8是6-ecdca诱导脂肪肉瘤细胞分化,6孔板内细胞生长到约50%~60%时,分组分别滴加激动剂6-ecdca以及阴性对照组(只加dmso),进行油红染色。6-ecdca处理组油红染色率高,表明6-ecdca处理脂肪肉瘤细胞发生了分化。
图9是6-ecdca、way-362450、gw4064、int-767、px-104、cdca、udca,在去分化脂肪肉瘤小鼠pdx模型中,都显示了显著的肿瘤抑制效果。
图10是6-ecdca、way-362450、gw4064、int-767、px-104、cdca、udca,在粘液型脂肪肉瘤小鼠pdx模型中,都显示了显著的肿瘤抑制效果。
图11是6-ecdca口服处理的实验裸小鼠,进行油红染色发现其肿瘤发生了较对照组更明显的分化,细胞脂肪堆积增加。
具体实施方式
在描述本发明材料和方法之前,应该理解本发明不限于所述的具体方法、方案、材料和试剂,因为其可以不同。还应理解本文所用的术语仅仅出于描述具体实施方式的目的,而并非旨在限制仅由随后提交的非临时申请所限定的本发明范围。
除非另有定义,否则本文所用的所有技术和科学术语具有与本发明所属领域的普通技术人员通常理解的相同的意义。虽然可采用与本文所述那些相似或等效的任何方法和材料来实施或测试本发明,但目前描述的是优选的方法和材料。本文具体提及的所有出版物和专利通过引用纳入本文以用于所有目的,包括描述和公开出版物中报道的可与本发明联用的化学物质、仪器、统计学分析和方法。本说明书中引用的所有参考文献应视作表明本领域的技术水平。本文所有内容均不应理解为承认本发明本能凭借在先发明而先于这些公开内容。
i.概述
如本文所用,术语“包含”、“包括”和其等同形式包括“含有”以及“由……组成”的含义,例如“包含”x的组合物可仅由x组成或可含有其它物质,例如x+y。
除非上下文中另有明确指示,否则本文和所附权利要求中使用的单数形式“一个”、“一种”和“该”包括复数指示物。同样,术语“一个”(或“一种”)、“一种或多种”和“至少一个”在本文可互换使用。
如本文所用,术语“脂肪肉瘤”是成人最常见的软组织肉瘤,也可见于青少年和儿童。脂肪肉瘤通常体积较大,一般为深在性、无痛性、逐渐长大的肿物,最常发生于下肢(如腘窝和大腿内侧)、腹膜后、肾周、肠系膜区以及肩部。在不同部位的发生率主要取决于该肿瘤的亚型,包括非典型性脂肪瘤性肿瘤/高分化脂肪肉瘤,去分化脂肪肉瘤,黏液样脂肪肉瘤,多形性脂肪肉瘤,混合型脂肪肉瘤。
如本文所用,“pdx”是指来源于患者原代肿瘤组织的肿瘤移植模型(patient-derivedxenograft模型,pdx模型)。是当前临床前药效的评估以及生物标志物的鉴定的研发手段。与使用细胞系建立的肿瘤模型相比,pdx模型减少了体外培养步骤,保持临床肿瘤细胞的形态和分子生物学特征,因而用该模型做出的药效结果将更为准确,与临床试验结果更为接近。
如本文所用,术语“核受体超家族”是一组配体(包括固醇类激素、维生素d、蜕化素、9-顺式和全部反式视黄酸、甲状腺激素、脂肪酸、氧化甾醇、前列腺素j2、白三烯b4、法呢醇代谢产物等)激活的转录因子家族,通过在信号分子与转录应答间建立联系,调控着细胞的生长和分化。在人类,核受体家族包含48个成员,例如ppar、fxr、lxr、vdr、rxr等。近年来,核受体家族在代谢性疾病领域受到广泛的关注,已有研究证明,它们与糖尿病、脂肪肝等疾病的发生发展密切相关,也被称为代谢性核受体。
如本文所用,术语“过氧化物酶体增殖物激活受体(ppar)”是核激素受体家族中的配体激活受体,在不同的物种中已经发现了它的3种亚型,控制许多细胞内的代谢过程,属于配体诱导核受体(ligand-induciblenuclearreceptors)。ppars与配体结合激活后,与视黄醇类x受体(rxr)形成异二聚体,形成的pparγ/rxr异二聚体与靶基因启动子上游的ppar反应元件(ppre)结合,最终调节靶基因的转录。其配体为脂溶性分子,受体与配体结合后,主要通过调节靶基因的表达产生生物效应。ppars与各自的配体结合后引起本身的构象的变化,促进或抑制靶基因的表达。
如本文所用,术语“激动剂”指的是促进核受体激动表达的药剂,也叫诱导剂。本发明所述激动剂包括了配体的诱导剂以及受体天然配体的衍生物。本发明所述6-ecdca,是法尼酯衍生物x受体天然配体鹅脱氧胆酸的衍生物。6-ecdca是fxr配体中激动效率较高的激动剂。
如本文所用,术语“抑制”指药物或化合物在脂肪肉瘤细胞系的体外实验或者pdx的体内实验中,使得脂肪肉瘤细胞的增长速度低于对照组,或者使得脂肪肉瘤细胞在细胞迁移实验、克隆形成实验或者增殖实验中的功能低于对照组。
如本文所用,术语“去分化”指高分化型脂肪肉瘤移行为去分化为去分化脂肪肉瘤的过程,伴随着细胞分化能力的减弱或丧失和肿瘤细胞侵袭力的增强,发生去分化之后的脂肪肉瘤细胞被认为具有更差的远期生存率和预后。
本发明涉及将化合物以治疗有效量给予患者。化合物可单独给予或作为药学上可接受的组合物的一部分来给予。此外,化合物或组合物可例如通过推注一次性全部给予、例如通过一系列片剂多次给予、或例如通过使用透皮递送在一段时间内基本上均匀地递送。另外,化合物的剂量可随时间而变化。可使用即释型制剂、控释型制剂或其组合来给予化合物。
本发明的药物组合物可以单一单位剂量或以多种单一单位剂量制备、包装或大批出售。如本文中所用,“单位剂量”为包含预定量的活性成分的药物组合物的个别量。活性成分的量通常等于将给予患者的活性成分的剂量或此剂量的适当比率,例如,此剂量的二分之一或三分之一。
在一个实施方式中,本发明的化合物单独给药。在另一个实施方式中,本发明化合物与其它活性或非活性的成分联合给药,在另一个实施方式中,本发明化合物与其它活性或非活性的成分联合制备。
本发明的药物组合物中的活性成分、药学上可接受的载体和任何附加成分的相对量将根据所治疗的人的种类、身材和病症而变化,并且还根据所述组合物的给予途径而变化。举例而言,所述组合物可包含0.1%-100%(w/w)的活性成分。或者本发明药物组合物包含3μm–20μm奥贝胆酸,优选3μm、5μm、10μm、20μm奥贝胆酸。在一个实施方式中,本发明药物组合物包含100μm鹅去氧胆酸。
如本文中所用,“给药”包括将本发明的化合物引入身体,优选引入全身循环的任何方式。本发明的化合物可以通过任何有效途径来给药,包括但不限于口服、静脉内、腹膜内、局部、透皮、经眼、经鼻、局部、吸入、皮下、肌内、口含、舌下、直肠给药等。
本发明的药物组合物可以制备成任何适当的剂型,例如片剂、胶囊、丸剂、颗粒制剂、注射液、栓剂、气雾剂、泡腾片、滴剂、滴丸剂、滴眼剂、滴鼻剂、眼用软膏剂、含漱剂、舌下片剂、粘贴片、贴膜剂、溶液剂、洗剂、搽剂、软膏剂、硬膏剂、糊剂等。本发明所述剂型还包括固体或可注射植入物或贮存剂。
适于注射的组合物包含与药学上可接受的载体例如生理学上可接受的无菌水溶液或非水溶液、分散体、悬浮液或乳剂联用的活性成分,或可包含用于在无菌可注射溶液或分散体中复溶的无菌粉末。合适的水性和非水性载体、稀释剂、溶剂或载剂的实例包括水、等渗盐水、乙醇、多元醇(丙二醇、聚乙二醇、甘油等)、其合适的混合物、甘油三酯,包括植物油如橄榄油,或可注射的有机酯如油酸乙酯。可例如通过使用包衣如卵磷脂、如果是分散体则通过保持所需粒度、和/或通过使用表面活性剂来保持适当的流动性。此类制剂可以适于推注给予或连续给予的形式制备、包装或出售。可注射制剂可以单位剂型制备、包装或出售,例如安瓿瓶、包含防腐剂的多剂量容器或用于自动注射或通过医师注射的单次使用装置。
药物组合物还以可注射的无菌水性或油性(乳剂)悬浮液或溶液的形式制备、包装或出售。此悬浮液或溶液可根据本领域已知技术进行配制,并且除了活性成分还可包含附加成分,例如本文所述的分散剂、润湿剂或悬浮剂。例如,可使用胃肠外可接受的无毒稀释剂或溶剂例如水或1,3-丁二醇来制备此类无菌可注射制剂。其它可接受的稀释剂和溶剂包括林格溶液、等渗氯化钠溶液和非挥发性油类,例如合成单甘油酯或二甘油酯。其它有用的胃肠外可接受的制剂包括含活性成分的那些制剂,所述活性成分呈微晶形式、在脂质体制剂中或作为可生物降解聚合物体系的一部分。用于缓释或植入的组合物可包含药学上可接受的聚合或疏水材料,例如乳剂、离子交换树脂、微溶聚合物或微溶盐。
根据本发明的化合物还可包含佐剂,例如防腐剂、润湿剂、乳化剂和/或分散剂,包括例如对羟基苯甲酸脂、氯代丁醇、酚、山梨酸等。还可能需要包括等渗剂,例如糖、氯化钠等。可通过使用能延迟吸收的药剂例如单硬脂酸铝和/或明胶来引起可注射药物组合物的长期吸收。具体而言,其中可用脂质体、胶粒和乳化剂,从而使本发明化合物更易于溶解以便递送。
用于口服给予的固体剂型包括胶囊、片剂、粉末和颗粒。在此类固体剂型中,活性化合物与至少一种常规惰性赋形剂(或载体)混合,所述赋形剂例如柠檬酸钠或磷酸二钙或(a)填料或增量剂,例如淀粉、乳糖、蔗糖、甘露糖醇或硅酸;(b)粘结剂,例如羧甲基纤维素、海藻酸盐、明胶、聚乙烯吡咯烷酮、蔗糖或阿拉伯树胶;(c)湿润剂,例如甘油;(d)崩解剂,例如,琼脂、碳酸钙、马铃薯或木薯淀粉、藻酸、某些复合硅酸盐或碳酸钠;(e)溶液缓聚剂,例如石蜡;(f)吸收加速剂,例如季铵化合物;(g)润湿剂,例如鲸蜡醇或单硬脂酸甘油酯;(h)吸附剂,例如高岭土或膨润土;和/或(i)润滑剂,例如滑石、硬脂酸钙、硬脂酸镁、固体聚乙二醇、月桂基硫酸钠或它们的混合物。在胶囊和片剂的情况中,剂型还可包含缓冲剂。
片剂可例如通过压缩或模制活性成分来制备,其任选地包含一种或多种附加成分。可通过在合适的装置中将活性成分压缩为自由流动形式(如粉末或颗粒制剂)来制备压缩片剂,所述活性成分任选地与粘结剂、润滑剂、赋形剂、表面活性剂和分散剂中的一种或多种混合。可通过在合适的装置中对活性成分、药学上可接受的载体和至少足以润湿混合物的液体的混合物进行模制来制备模制片剂。
用于生产片剂的药学上可接受的赋形剂包括惰性稀释剂、成粒剂和崩解剂、粘结剂和润滑剂。已知的分散剂包括马铃薯淀粉和羟乙酸淀粉钠。已知的表面活性剂包括月桂基硫酸钠。已知的稀释剂包括碳酸钙、碳酸钠、乳糖、微晶纤维素、磷酸钙、磷酸氢钙和磷酸钠。已知的成粒剂和崩解剂包括玉米淀粉和藻酸。已知的粘结剂包括明胶、阿拉伯树胶、预胶凝的玉米淀粉、聚乙烯吡咯烷酮和羟丙基甲基纤维素。已知的润滑剂包括硬脂酸镁、硬脂酸、硅石和滑石。
片剂可未经包衣或其可使用已知的方法包衣以实现在人体胃肠道中的延迟崩解,从而使活性成分持续释放和吸收。举例而言,例如甘油单硬脂酸酯或甘油二硬脂酸酯的材料可用于对片剂进行包衣。片剂还可包含甜味剂、调味剂、染色剂、防腐剂或这些的某种组合,以提供药学上优质的和可口的制剂。
可制备具有包衣或外壳,例如肠溶包衣和本领域中熟知的其它包衣的固体剂型,如片剂、糖衣丸剂、胶囊和颗粒。其还可包含遮光剂,并且还可具有使其以延迟方式释放活性化合物的组合物。可用的包埋组合物的实例为聚合物和蜡。活性化合物还可呈微囊化形式,适当时具有上述赋形剂中的一种或多种。
相似类型的固体组合物可在使用如乳糖或奶糖、以及高分子量聚乙二醇等作为赋形剂的软质或硬质明胶填充的胶囊中用作填料。可使用可生理降解的组合物如明胶来制备包含活性成分的硬质胶囊。此类硬质胶囊包含活性成分,并且还可包含附加成分,包括例如惰性固体稀释剂如碳酸钙、磷酸钙或高岭土。可使用可生理降解的组合物如明胶来制备包含活性成分的软质明胶胶囊。此类软质胶囊包含可与水或油介质如花生油、液体石蜡或橄榄油混合的活性成分。
可使用已知技术来制备口腔组合物,其专门在人类患者的小肠或大肠内释放口服给予的药剂。例如,用于递送至胃肠系统(包括结肠)的制剂包括肠溶衣系统,其基于例如仅在ph6及以上溶解的甲基丙烯酸酯共聚物如聚(甲基丙烯酸、甲基丙烯酸甲酯),从而所述聚合物只有在进入小肠时才开始溶解。所述聚合物制剂崩解的位置取决于小肠通过速率和所含聚合物的量。例如,相对较厚的聚合物包衣用于递送至结肠近端。还可使用能够提供位置特异性结肠递送的聚合物,其中所述聚合物依赖于大肠菌落来提供聚合物包衣的酶促降解从而释放药物。例如,偶氮聚合物、配糖物以及各种天然获得和修饰的多糖可用于此类制剂中。
用于口服给予的液体剂型包括药学上可接受的乳剂、溶液、悬浮液、糖浆和酏剂。除了活性化合物之外,液体剂型可包含本领域中常用的惰性稀释剂例如水或其它溶剂、等渗盐水、增溶剂和乳化剂,例如乙醇、异丙醇、碳酸乙酯、乙酸乙酯、苄醇、苯甲酸苄酯、丙二醇、1,3-丁二醇、二甲基甲酰胺、油类,具体为杏仁油、花生油、椰子油、棉籽油、落花生油、玉米胚油、橄榄油、蓖麻油、芝麻油、甘油、分馏的植物油、矿物油如液体石蜡、四氢糠醇、聚乙二醇、脱水山梨糖醇的脂肪酸酯或这些物质的混合物等。
除了这些惰性稀释剂之外,本发明的化合物还可包括佐剂如润湿剂、乳化剂和悬浮剂、缓和剂、防腐剂、缓冲剂、盐、甜味剂、调味剂、染色剂和芳香剂。除了活性化合物之外,悬浮液还可包含悬浮剂例如乙氧基化异硬脂醇、聚氧乙烯山梨糖醇酯或脱水山梨糖醇酯、微晶纤维素、氢化的食用脂、藻酸钠、聚乙烯吡咯烷酮、黄蓍胶、阿拉伯树胶、琼脂和纤维素衍生物如羧甲基纤维素钠、甲基纤维素、羟丙基甲基纤维素、偏氢氧化铝、膨润土或这些物质的混合物等。适于口服给予的本发明药物组合物的液体制剂可以液体形式或以旨在使用前用水或另一合适的载剂复溶的干燥产品的形式来制备、包装和出售。
已知的分散剂或润湿剂包括天然存在的磷脂如卵磷脂,环氧烷与脂肪酸、与长链脂族醇、与衍生自脂肪酸和己糖醇的偏酯、或与衍生自脂肪酸和己糖醇酐的偏酯的缩合产物(例如分别为聚氧乙烯硬脂酸酯、十七基乙烯氧基鲸蜡醇、聚氧乙烯山梨糖醇单油酸酯和聚氧乙烯脱水山梨糖醇单油酸酯)。已知的乳化剂包括卵磷脂和阿拉伯树胶。已知的防腐剂包括甲基、乙基或正丙基-对-羟基苯甲酸、抗坏血酸和山梨酸。已知的甜味剂包括例如,甘油、丙二醇、山梨醇、蔗糖和糖精。已知的用于油性悬浮液的增稠剂包括例如蜂蜡、硬石蜡和鲸蜡醇。
活性成分在水性或油性溶剂中的液体溶液可以基本上与液体悬浮液相同的方式进行制备,主要的不同之处在于活性成分是溶解而非悬浮在溶剂中。本发明药物组合物的液体溶液可包含关于液体悬浮液所述的各组分,应理解悬浮剂未必有助于活性成分在溶剂中的溶解。水性溶剂包括例如水和等渗盐水。油性溶剂包括例如杏仁油、油性酯、乙醇、植物油如花生油、橄榄油、芝麻油或椰子油、分馏的植物油和矿物油如液体石蜡。
本发明药物组合物可以适于含服给予的制剂来制备、包装或出售。此类制剂例如可用常规方法制成片剂或锭剂的形式,并且可例如包含有效量的活性成分,含口腔可溶解或降解组合物和任选地一种或多种本文所述附加成分中的剩余部分。或者,适于含服给予的制剂可包括含活性成分的粉末或气雾化或雾化的溶液或悬浮液。此类粉末状、气雾化或雾化制剂在分散时优选具有约0.1-约200nm的平均粒径或液滴大小,并且还可包含本文所述附加成分的一种或多种。
为了在非人动物中施用,本发明的化合物可以糊剂或粒料的形式制备。糊剂制剂可通过将化合物分散在药学上可接受的油如花生油、芝蔴油、玉米油等中来制备。包含治疗有效量的化合物的粒料可通过将化合物与稀释剂如碳蜡、卡洛巴蜡等混合来制备,并且可添加润滑剂如硬脂酸镁或硬脂酸钙以改善造粒工艺。当然,认识到可将不止一种粒料给予动物以达到所需剂量水平。
本发明化合物是否直接给予细胞、含细胞的组织、接触细胞的体液或化合物从中扩散或运送至细胞的位置并不关键。以一定的量和途径将化合物给予患者从而所述化合物的量通过该途径足以使细胞中的脂质直接或间接抵达细胞就足够了。最小量随化合物的特征而变化。
可使用的具体剂量和剂量范围取决于多种因素,包括患者的需求、所治疗病症的严重程度以及所给予化合物的药理活性。根据本公开,对具体患者的剂量范围和最佳剂量的确定完全在本领域技术人员的普通技术范围之内。应理解普通医师、牙医或兽医将易于确定和开出有效量的化合物处方,以在患者中移动脂质贮存、导致体重减轻或抑制食欲。在前述中,医师或兽医可例如首先开出较低剂量处方,随后增加剂量直至获得适当响应。然而还应理解,对于任何特定个人的具体剂量水平将取决于多种因素,包括所用具体化合物的活性、人的年龄、体重、一般健康状况、性别和饮食、给药时间、给药途径、分泌速率、任何药物组合以及所治疗失调的严重程度。
本发明组合物还可包括一种或多种药学上合适的赋形剂,如溶剂和共溶剂、增溶剂、润湿剂、悬浮剂、增稠剂、乳化剂、螯合剂、缓冲液、ph调节剂、抗氧化剂、还原剂、抗微生物防腐剂、增量剂、保护剂、张力调节剂和特殊添加剂。
ii.三种激动剂对脂肪肉瘤细胞系的激动效率
在一个实施方式中,5nm到20nm的6-ecdca对93t-449高分化脂肪肉瘤细胞系抑制率为:10%到60%(见表1)。
在一个实施方式中:10nm到25nm的way-362450对93t-449高分化脂肪肉瘤细胞系抑制率为:7%到25%(见表2)。
在一个实施方式中,5nm到20nm的gw4064对93t-449高分化脂肪肉瘤细胞系抑制率为:13%到47%(见表3)。
在一个实施方式中:10nm到100nm的int-767对93t-449高分化脂肪肉瘤细胞系抑制率为:20%到57%(见表4)。
在一个实施方式中:10nm到100nm的px-104对93t-449高分化脂肪肉瘤细胞系抑制率为:22%到77%(见表5)。
在一个实施方式中:10μm到100μm的cdca对93t-449高分化脂肪肉瘤细胞系抑制率为:20%到74%(见表6)。
在一个实施方式中:10μm到100μm的udca对93t-449高分化脂肪肉瘤细胞系抑制率为:4%到44%(见表7)。
iii.fxr激动剂对脂肪肉瘤细胞系的迁移有抑制效果
在另一个实施方式中,6-ecdca5nm对脂肪肉瘤细胞系的迁移侵袭能力具有抑制作用:5nm6-ecdca在transwell细胞迁移实验中,抑制了94t-778细胞系的穿膜侵袭能力,见图1。
在另一个实施方式中,gw40645nm对脂肪肉瘤细胞系的侵袭能力具有抑制作用:5nmgw4064a在transwell细胞迁移实验中,抑制了94t-778细胞系的穿膜侵袭能力,见图2。
在另一个实施方式中,way-36245010nm对脂肪肉瘤细胞系的侵袭能力具有抑制作用:10nmway-362450在transwell细胞迁移实验中,抑制了94t-778细胞系的穿膜侵袭能力,见图3。
在另一个实施方式中,int-76710nm对脂肪肉瘤细胞系的侵袭能力具有抑制作用:10nmint-767在transwell细胞迁移实验中,抑制了94t-778细胞系的穿膜侵袭能力,见图4。
在另一个实施方式中,px-10410nm对脂肪肉瘤细胞系的侵袭能力具有抑制作用:10nmpx-104在transwell细胞迁移实验中,抑制了94t-778细胞系的穿膜侵袭能力,见图5。
在另一个实施方式中,cdca10μm对脂肪肉瘤细胞系的侵袭能力具有抑制作用:10μmcdca在transwell细胞迁移实验中,抑制了94t-778细胞系的穿膜侵袭能力,见图6。
在另一个实施方式中,udca10μm对脂肪肉瘤细胞系的侵袭能力具有抑制作用:10μmudca在transwell细胞迁移实验中,抑制了94t-778细胞系的穿膜侵袭能力,见图7。
iv.fxr激动剂促进多种脂肪肉瘤细胞系分化及脂肪堆积
在另一个实施方式中,6-ecdca促进多种脂肪肉瘤细胞系脂肪堆积:6-ecdca在5nm的工作浓度下,多种脂肪肉瘤细胞系的油红实验可见差异,6-ecdca激动的细胞组油红染色率明显高于对照组(见图8)。
v.口服fxr激动剂后可在多种脂肪肉瘤pdx小鼠上抑制肿瘤增速
在一个实施方式中,口服给予6-ecdca后具有抑制肿瘤增长速度的效果:在6-ecdca口服(5mg/kg/day)给药4周小鼠中,口服6-ecdca有效抑制肿瘤增长趋势,并使截尾时肿瘤平均体积明显小于对照组(见图9和图10)。
在一个实施方式中,口服给予gw4064后具有抑制肿瘤增长速度的效果:在gw4064口服(10mg/kg/day)给药4周小鼠中,口服gw4064有效抑制肿瘤增长趋势,并使截尾时肿瘤平均体积明显小于对照组(见图9和图10)。
在一个实施方式中,口服给予way-362450后具有抑制肿瘤增长速度的效果:在way-362450口服(5mg/kg/day)给药4周小鼠中,口服way-362450有效抑制肿瘤增长趋势,并使截尾时肿瘤平均体积明显小于对照组(见图9和图10)。
在一个实施方式中,口服给予int-767后具有抑制肿瘤增长速度的效果:在int-767口服(15mg/kg/day)给药4周小鼠中,口服int-767有效抑制肿瘤增长趋势,并使截尾时肿瘤平均体积明显小于对照组(见图9和图10)。
在一个实施方式中,口服给予px-104后具有抑制肿瘤增长速度的效果:在px-104口服(10mg/kg/day)给药4周小鼠中,口服px-104有效抑制肿瘤增长趋势,并使截尾时肿瘤平均体积明显小于对照组(见图9和图10)。
在一个实施方式中,口服给予cdca后具有抑制肿瘤增长速度的效果:在cdca口服(40mg/kg/day)给药4周小鼠中,口服cdca有效抑制肿瘤增长趋势,并使截尾时肿瘤平均体积明显小于对照组(见图9和图10)。
在一个实施方式中,口服给予udca后具有抑制肿瘤增长速度的效果:在udca口服(50mg/kg/day)给药4周小鼠中,口服udca有效抑制肿瘤增长趋势,并使截尾时肿瘤平均体积明显小于对照组(见图9和图10)。
vi.口服fxr激动剂后可在脂肪肉瘤pdx小鼠模型上促进脂肪肉瘤细胞系脂肪性分化及脂肪堆积
在一个实施方式中,口服给予6-ecdca后具有促进脂肪肉瘤细胞系脂肪性分化及脂肪堆积的效果:在6-ecdca口服(5mg/kg/day)给药4周小鼠中,口服6-ecdca的实验裸小鼠,其肿瘤发生了较对照组更明显的脂肪源性的分化(见图11)。
实施例
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于以任何方式限制本发明的范围。通过上述说明和以下实施例,在本文示出和描述的那些之外的本发明的各种修改对于本领域的技术人员显而易见,并且落在所附权利要求的范围之内。
除非另行定义,所有专业与科学用语与本领域熟练人员所熟悉的意义相同。此外,任何与所记载内容相似或均等的方法及材料皆可应用于本发明中。文中所述的较佳实施方法与材料仅作示范之用。
实施例1.fxr激动剂的体外实验可抑制脂肪肉瘤细胞生长和迁移
本实施例的目的是证明fxr激动剂6-ecdca、way-362450、gw4064、int-767、px-104、cdca、udca可以在体外抑制脂肪肉瘤细胞的生长增殖和迁移。
在本实施例中,fxr激动剂均采购自selleck或sigma公司,粉剂,纯度大于等于97%;脂肪肉瘤细胞系包括,高分化型细胞株93t—449、94t—778,以及未分化多形性细胞株sw872。所有细胞株均采购自美国模式培养物集存库(americantypeculturecollection,atcc)。93t449细胞株以及94t-778细胞株采用以,高糖rpmi培养基+10%胎牛血清+双抗+支原体预防剂配成的完全培养基培养。sw872细胞株采用以l-15培养基+10%太牛血清+双抗+支原体预防剂配成的完全培养基在air-free的孵箱中培养。结果发现6-ecdca、way-362450、gw4064、int-767、px-104、cdca、udca可以分别在体外抑制脂肪肉瘤细胞的生长增殖(表1,表2,表3,表4,表5,表6,表7)和迁移(图1,图2,图3,图4,图5,图6,图7)。
实验方法:
1、将贴壁生长的脂肪肉瘤细胞配制成细胞悬液,接种于6孔板内。
2、待6孔板内细胞生长到约50%~60%时,分组分别滴加fxr激动剂,以及阴性对照组(只加dmso),滴加后充分混匀培养基,每组三个副孔,培养12h后,pbs清洗后换液再培养24h。
3、消化细胞,用细胞计数板计数3次以上,或者用台盼蓝染色后电子细胞计数仪计数2次。
4、计算浓度后,取9×104个细胞接种于放有transwell小室的24孔板,每组3个副孔,行transwell细胞迁移实验。
5、取3×104个细胞接种于96孔板,每孔2000个细胞,每组三个副孔,行五日cck-8细胞增殖实验。
6、取各个组1×105个细胞,按照试剂盒说明,加入trizol提取rna,进行逆转录pcr以及qpcr,检测激动剂的激动效率。rt-qpcr的试剂盒来自takara公司。
表1:6-ecdca浓度依赖抑制脂肪肉瘤细胞生长
表2:way-362450浓度依赖抑制脂肪肉瘤细胞生长
表3:gw4064浓度依赖抑制脂肪肉瘤细胞生长
表4:int-767浓度依赖抑制脂肪肉瘤细胞生长
表5:px-104浓度依赖抑制脂肪肉瘤细胞生长
表6:cdca浓度依赖抑制脂肪肉瘤细胞生长
表7:udca浓度依赖抑制脂肪肉瘤细胞生长
实施例2.6-ecdca(奥贝胆酸)体外实验诱导脂肪肉瘤细胞分化
本实施例的目的是证明fxr激动剂可以诱导脂肪肉瘤细胞分化。在本实例中,细胞培养步骤同实例2,油红o染色试剂盒来自上海博古生物有限公司。细胞处理同实施例2,待6孔板内细胞生长到约50%~60%时,分组分别滴加激动剂6-ecdca以及阴性对照组(只加dmso),滴加后充分混匀培养基,每组三个副孔,培养12h后,pbs清洗后换液再培养48h。根据试剂盒说明书进行油红o染色。检测原理为:脂肪肉瘤细胞内含有的脂滴会被油红应用染色液染为橘红色,非脂肪组织不会染色,之后用苏木精复染。不同细胞中,油红染色率高的细胞株为脂肪堆积明显的细胞,即细胞脂肪源性表达增加,分化级别增加。实验结果如图8所示,深灰色区域为油红染色区。
实施例3.体内实验口服fxr激动剂可以抑制脂肪肉瘤增长速度
本实施例的目的是证明fxr激动剂6-ecdca、way-362450、gw4064、int-767、px-104、cdca、udca在体外实验中可以抑制脂肪肉瘤增速。在本实施例中,发明人建立了去分化型和粘液型脂肪肉瘤人源肿瘤移植的裸小鼠模型(pdx),小鼠种属为balb/cnude♀6-8wbk,瘤体于右侧腹部皮下种植。建模成功后挑选肿瘤体积仿佛的小鼠,分别灌胃给予fxr激动剂6-ecdca、way-362450、gw4064、int-767、px-104、cdca、udca,并设溶剂组为阴性对照,每日定时给药,具体给药方案如表格所示。
分组及给药剂量如表格所示:
本实例具体实验步骤如下:
1、在连续培养28天后,按照动物伦理委员会要求,在所有动物瘤体超过3000mm3,对实验动物进行统一处理,co2箱内处死。
2、处死实验小鼠后立即完整剥离皮下肿瘤,称重,测量体积,切块后行冰冻切片+油红o染色。
3、留取实验动物照片,并统计数据。实验结果如图10和11所示,可见奥贝胆酸体内实验中仍然体现了脂肪肉瘤的抑制效果。
本实例中,6-ecdca、way-362450、gw4064、int-767、px-104、cdca、udca处理组在去分化脂肪肉瘤(图9),粘液型脂肪肉瘤(图10)两种分型的脂肪肉瘤pdx模型中,都显示了显著的肿瘤抑制效果,尤其在粘液型中抑制效果更为突出。通过在新鲜肿瘤组织的速冻切片油红染色,发明人发现,经6-ecdca(奥贝胆酸)处理的粘液性脂肪肉瘤组织,油红o染色率比对照组更高,意味着经6-ecdca口服处理的实验裸小鼠,其肿瘤发生了较对照组更明显的分化(图11),且肿瘤生长速度显著低于对照组。
本发明所涉及的多个方面已做如上阐述。然而,应理解的是,在不偏离本发明之精神与范围的前提下,对上述描述的任何修饰都是允许的。同样,类似的情况也包括在权利要求中。
- 还没有人留言评论。精彩留言会获得点赞!