用于治疗慢性和神经性疼痛的D-对映体肽的制作方法
本发明涉及一种用作镇痛剂、用于疼痛治疗、用于治疗慢性和/或神经性疼痛和/或用于抑制n型神经元钙通道(ncc)的由肽组成或包含肽以及还由聚合物组成或包含聚合物的组合物,所述肽选自由rd2、d3、具有至少50%同一性的同系物和rd2或d3的衍生物组成的组或包含它们,所述聚合物包含具有至少50%同一性的rd2/d3同系物以及rd2和d3的衍生物或由它们组成。本发明还涉及一种与对照相比减少神经递质释放的方法以及一种采用上述组合物抑制n型ncc的方法。
背景技术:
德国疼痛研究学会的统计表明,在德国,有超过1200万的人患有长期的慢性疼痛和/或神经性疼痛。外周或中枢神经系统中的传入系统受到损害或发生疾病后就会出现神经性疼痛。尽管药物治疗使疼痛减少了50-80%,但是完全免于疼痛通常是不可能的。在所有的药物治疗方案中,大约20-40%的患者对治疗仅有不充分的反应(疼痛减少<30%,即所谓的无反应者),或者承受无法忍耐的副作用。
除了抗抑郁药(三环/四环抗抑郁药、选择性血清素/去甲肾上腺素摄取抑制剂)、长效阿片类药物、作用于神经元钠通道的抗惊厥药(例如,卡马西平)和局部治疗(利多卡因贴片、大剂量辣椒素贴片)以外,还可采用能对神经元钙通道产生作用的药物治疗,如加巴喷丁、普瑞巴林和齐考诺肽
齐考诺肽可用于患有极其严重的疼痛并且所有治疗方案都失败的患者。
然而,齐考诺肽的鞘内(i.t.)给药有引起严重的感染的风险,如可能致命的脑膜炎。由于生物体沿导管的渗入或输液系统的意外污染引起的脑膜炎是公知的鞘内药物给药的并发症,特别是使用外部系统引起的并发症。此外,在88%的患者中,齐考诺肽可引起严重的中枢神经系统副作用,如头晕、恶心、混乱、步态不稳、记忆障碍、视力模糊、头疼、虚弱、呕吐和嗜睡。尽管齐考诺肽存在高度复杂且危险的给药途径和严重的副作用,但是它还是可用于疼痛治疗,因为它是唯一的被批准的特异性n型钙通道抑制剂,该抑制剂已被证明是成功的,即使在有例如由肿瘤或aids引起的剧烈疼痛(甚至吗啡已不再提供缓解)的情况下。
技术实现要素:
本发明的目的是克服现有技术的不足。特别地,目的是提供具有较少副作用的组合物,以相应方式特异性且选择性地抑制n型ncc,并且对l型ncc的功能没有影响。待提供的组合物可以以简单的方式给药,优选口服给药。另外,为了防止长期疼痛的不敏感性和损害,对n型ncc的抑制是可逆的。高于皮摩尔范围的ic50值是期望的,以允许独立于受体生物体的特异性代谢的给药。当ic50值在皮摩尔范围内或以下时,活性化合物浓度即使有微小变化也会产生显著的影响。这很容易导致服药不足或过量,服药不足或过量可能分别取决于特异性代谢并会导致不期望的副作用或长期的损害。
另一目的是提供一种包含可确保血脑屏障的通路的活性化合物的组合物。
另一目的是提供组合物的稳定的肠内、静脉内、皮下、腹腔、鼻内和/或口服给药,这些给药方式在靶位、n型ncc表现出这种作用。
在实施方案中,该目的可通过以下实现:一种用作镇痛剂的组合物,其由肽组成或包含肽,还由聚合物组成或包含聚合物,所述肽选自由rd2、d3、具有至少50%同一性的同系物和rd2或d3的衍生物组成的组或包含它们,所述聚合物包含rd2、d3、具有至少50%同一性的同系物以及rd2和d3的衍生物或由它们组成。
本发明的另一主题是一种用于疼痛治疗的组合物,其由肽组成或包含肽,还由聚合物组成或包含聚合物,所述肽选自由rd2、d3、具有至少50%同一性的同系物和rd2或d3的衍生物组成的组或包含它们,所述聚合物包含rd2、d3、具有至少50%同一性的同系物以及rd2和d3的衍生物或由它们组成。
本发明的另一主题是一种用于治疗慢性和/或神经性疼痛的组合物,其由肽组成或包含肽,还由聚合物组成或包含聚合物,所述肽选自由rd2、d3、具有至少50%同一性的同系物和rd2或d3的衍生物组成的组或包含它们,所述聚合物包含rd2、d3、具有至少50%同一性的同系物以及rd2和d3的衍生物或由它们组成。
本发明的另一主题是一种用于抑制n型神经元钙通道(ncc)的组合物,其由肽组成或包含肽,还由聚合物组成或包含聚合物,所述肽选自由rd2、d3、具有至少50%同一性的同系物和rd2或d3的衍生物组成的组或包含它们,所述聚合物包含rd2、d3、具有至少50%同一性的同系物以及rd2和d3的衍生物或由它们组成。
在本发明的实施方案中,所述肽主要由d-氨基酸构成。在本发明的含义内,术语“主要由d-氨基酸”是指根据本发明使用的肽包含至少50%、60%、优选75%、80%、81%、82%、83%、84%、特别优选85%、86%、87%、88%、89%、90%、91%、92%、93%、94%、95%、特别是96%、97%、98%、99%或100%的d-氨基酸,或者由其构成。
与诸如齐考诺肽的l-对映体肽相比,d-肽对蛋白酶的抵抗更强(体内),因此有口服给药的可能性,因为消除半衰期长。另外,它们通常不具有或仅具有最低免疫原性。
另一实施方案涉及用于每千克体重给药1μg至1g、优选每千克体重给药100μg至10mg、特别优选每千克体重给药0.5mg至5mg的上述组合物。
一个实施方案涉及用于肠内、静脉内、皮下、腹腔、鼻内或口服给药、优选口服给药的上述组合物。
有利地是,对于10nm至100μm、特别优选100nm至10μm、特别是100nm至1μm的n型ncc,上述组合物优选地具有1纳摩尔至1毫摩尔的ic50值。
在一变化方案中,根据本发明使用的组合物包含肽或由肽组成,所述肽选自由以下组成的组或包含以下:
ank1;(游离n-末端,优选酰胺化的c-末端):rkrirlvyhinr(seqidno:8);
ank2;(游离n-末端,优选酰胺化的c-末端):rkrirl06yhinr(seqidno:9);
ank3;(游离n-末端,优选酰胺化的c-末端):rkrirl06yhwnr(seqidno:10);
ank4;(游离n-末端,优选酰胺化的c-末端):rkrirlvyhwnr(seqidno:11);
ank5;(游离n-末端,优选酰胺化的c-末端):rkrvrlvyhkkr(seqidno:12);
ank6;(游离n-末端,优选酰胺化的c-末端):rkrirlvtkkkr(seqidno:13);
ank7;(游离n-末端,优选酰胺化的c-末端):rkrvrl02thikr(seqidno:14);
ank15;(游离n-末端,优选酰胺化的c-末端):rprvrl06yhwnr(seqidno:15);
ank16;(游离n-末端,优选酰胺化的c-末端):rkr07rlvtkrnr(seqidno:16);
ank17;(游离n-末端,优选酰胺化的c-末端):rkrirl06yhikr(seqidno:17);
ank18;(游离n-末端,优选酰胺化的c-末端):rpr07rlhtkkkr(seqidno:18);
其中,02:4-氟苯丙氨酸(d),06:苯基甘氨酸(d),07:d-高精氨酸。
在替代方案中,根据本发明和上述描述使用的肽的衍生物为环化的或酰胺化的肽。
在另一实施方案中,根据本发明使用的肽通过游离c末端与游离n末端的相互共价结合而连接,因而呈环状。该环合的优点还在于:游离c末端的羧基不再存在。所述肽有利地具有氨基酸序列,其中,例如通过缩合反应而将第一氨基酸与最后一个氨基酸共价结合,发生了线型分子的环化。当然,还存在环化的其它可能性,例如,通过将其他氨基酸彼此连接。仅作为示例,我们可以想到的是将第二氨基酸与最后一个氨基酸连接。还可以想到任何其他可能的连接。当肽的第一个与最后一个氨基酸彼此连接时,有利的是在肽链(氨基酸序列)中不存在开放端。该方法的另一个结果是:环化后产生无法再彼此区分的相同氨基酸序列的具有线性氨基酸序列的所有肽在此意义上都是相同的。
实例是:公知的肽d3的线性氨基酸序列为rprtrlhthrnr(seqidno:2)。由n末端氨基基团与c末端羧基之间的酰胺键连接的相应环化的肽“cd3”不能再与序列prtrlhthrnrr、rtrlhthrnrrp、trlhthrnrrpr、rlhthrnrrprt、lhthrnrrprtr、hthrnrrprtrl、thrnrrprtrlh、hrnrrprtrlht、rnrrprtrlhth、nrrprtrlhthr或rrprtrlhthrn的环化的肽进行区分。此外,cd3还可进一步衍生自这些序列中的每一个。
rd2:ptlhthnrrrrr(seqidno:1)的情况与序列tlhthrrrrrp、lhthnrrrrrpt、hthnrrrrrptl、thnrrrrrptlh、hnrrrrrptlht、nrrrrrptlhth、rrrrrptlhthn、rrrrptlhthnr、rrrptlhthnrr、rrptlhthnrrr、rptlhthnrrrr的环化的肽的情况相似,命名为“crd2”。
环化的肽的生产属于现有技术,并且例如,可根据de102005049537a1中所述的方法来进行。
有利的是,通过肽的第一个与最后一个氨基酸的环化的结果是:肽链中不再存在任何“开放端”,所述“开放端”通常是例如通过氨肽酶和羧肽酶对细胞、动物或人类中的肽降解活性进行攻击的点。
在本发明的另一实施方案中,根据本发明使用的肽在游离c末端被酰胺化,从而具有酸酰胺基团以代替羧基基团。因而,酸酰胺基团(-conh2基团)替代羧基基团(-cooh基团)而排列在c末端处。肽有利地在游离c末端被酰胺化。这使得另一目的可以特别有利地实现,因为提供了一种不具有过剩负电荷的肽,其能以更好的亲和性与靶向分子结合并能以简单的方式获得。
在本发明的替代实施方案中,肽具有以下基团:coh、cocl、cobr、conh-烷基和conh-烷基胺基(净正电荷)代替羧基基团,其中,本发明不限于此。
另外,在一个实施方案中,根据本发明使用的每个肽在n末端和/或c末端由一个或两个氨基酸相对于原始序列而被加长或缩短。在一个替代实施方案中,一个末端具有一个或两个附加的氨基酸,而另一个末端具有一个或两个较少的氨基酸。肽优选地在c末端通过选自氨基酸rlht、优选地为r的一个氨基酸相对于原始序列而被加长。
优选的衍生物为环化的(环)肽,酰胺化的肽和/或在c末端通过一个氨基酸、优选地为精氨酸r、特别是d3rrprtrlhthrnrr(seqidno:5),以及环化的d3r而被加长的肽序列。另外优选的衍生物为环化的rd2及其各个线性酰胺化的形式和/或在c末端通过一个氨基酸、优选地为精氨酸r而被加长的肽序列rd2rptlhthnrrrrr(seqidno:19)。
因此,本发明的另一主题也是肽rd2rptlhthnrrrrrr(seqidno:19),基于在c末端通过一个氨基酸、优选r而延伸的rd2的13聚体。酰胺化的和/或环化形式的crd2r(类似于crd2)也是本发明的主题。本发明的另一主题是肽rd2r,因此也是酰胺化和/环化的形式,优选crd2r,可用于医药中或用作药用产品,优选用于疼痛的治疗或治疗疼痛,特别是慢性和神经性疼痛。根据本发明使用的另外的肽为:rd2rd2、ptlhthnrrrrrptlhthnrrrrr(seqidno:3)、rd2d3ptlhthnrrrrrrprtrlhthrnr(seqidno:4)、环-rd2rd2、ptlhthnrrrrrptlhthnrrrrr、环-d3r、rprtrlhthrnrr、d3p、rprtrlhthrnrp(seqidno:6)和环-d3p以及d3a、rprtrlhthrnra(seqidno:7)和环-d3a。
在本发明含义内的“同源序列”或“同系物”表示氨基酸序列与至少50%、55%、60%、65%、70%、71%、72%、73%、74%、75%、76%、77%、78%、79%、80%、81%、82%、83%、84%、85%、86%、87%、88%、89%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%或100%的单体的上述氨基酸序列之一具有同一性。除了术语“同一性”以外,术语“同系物”或“同源性”也以相同的含义在本发明中使用。两个氨基酸序列或多肽序列之间的同一性通过以下方式计算:采用基于smith,t.f.和waterman,m.s(adv.appl.math.2:482-489(1981))的算法的程序bestfit进行比对;为氨基酸设置以下参数:空位创造罚分:8和空位延伸罚分:2;以及为核酸设置以下参数:空位创造罚分:50和空位延伸罚分:3。优选地,两个核酸序列或多肽序列之间的同一性由在各个序列全长范围内的核酸序列/多肽序列的同一性定义,计算方法如下:采用基于needleman,s.b.和wunsch,c.d.(j.mol.biol.48:443-453)的算法的程序gap进行比对;为氨基酸设置以下参数:空位创造罚分:8和空位延伸罚分:2;以及为核酸设置以下参数:空位创造罚分:50和空位延伸罚分:3。
在本发明的含义内,当两个氨基酸序列具有相同的氨基酸序列时,它们是相同的。
在替代实施方案中,同源性或同一性由相应肽的各个全长来确定。在另一替代实施方案中,仅通过部分区域进行比对,从而确定同一性或同源性。
在该替代实施方案中,根据本发明使用的多肽具有至少20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、71%、72%、73%、74%、75%、76%、77%、78%、79%、80%、81%、82%、83%、84%、85%、86%、87%、88%、89%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%或100%的同源性。
在本发明的另一实施方案中,所述组合物包含聚合物或由聚合物组成,或包含上述另外的肽或由上述另外的肽组成,所述聚合物包含rd2、d3、具有至少50%同一性的同系物以及rd2和d3的衍生物或由它们组成。根据本发明使用的聚合物包含上述单体肽中的2个、3个、4个、5个、6个、7个、8个、9个、10个或更多个。
优选使用选自由rd2d3、d3rd2、d3d和rd2rd2或同系物或衍生物组成的组或包含它们的二聚体。
线性或环化形式的上述序列中的2个、3个、4个、5个、6个、7个、8个、9个、10个或更多个的任意所需组合可以形成所述聚合物。
例如,通过化学合成或肽合成来生产聚合物。
在本发明的一个变化方案中,所述肽以线性方式彼此连接,特别是如上所述。在另一个变化方案中,所述肽以支链方式彼此连接,形成根据本发明的聚合物。根据本发明,支链聚合物可以是树状聚合物,其中,单体通过共价或非共价结合而彼此连接。或者,所述肽也可以与平台分子(诸如,例如peg或糖)连接,从而形成支链聚合物。
单体肽在有或无额外的连接体的情况下以线性方式进行头对头、尾对尾或头对尾连接,并且在替代实施方案中,单体肽在有或无额外的连接体的情况下(例如,一个或多个氨基酸)通过剩余两端的全部共价连接而被环化。
在本发明的实施方案中,根据本发明使用的肽与另外的氨基酸、连接体、间隔体和/或官能团共价连接。
在本发明的替代实施方案中,这些另外的氨基酸、连接体、间隔体和/或官能团不会改变所述肽的性质。在另外的替代实施方案中,所述另外的氨基酸、连接体、间隔体和/或官能团可使所述肽的性质发生变化。在该实施方案中,根据本发明的聚合物相对于n型ncc的选择性和/或亲和性得到增强。
连接体被理解为是通过共价键与肽结合的一个或多个分子,其中,这些连接体也可以通过共价键彼此连接。
官能团被理解为是与肽共价结合的分子。在一个变化方案中,所述官能团包括生物素基团。这允许与链霉亲和素有强的共价结合。
在本发明的实施方案中,肽包括所谓的间隔体。间隔体被理解为是通过共价键与肽结合并具有特定的物理和/或化学性质的分子,通过所述间隔体,肽的性质被改变。在一个实施方案中,使用亲水或疏水间隔体,优选亲水间隔体。亲水间隔体选自包括聚乙二醇、糖、丙三醇、聚l-赖氨酸或β-丙胺酸的分子。
在替代的实施方案中,肽与另外的物质连接。
在一个变化方案中,这些物质为根据2012年9月颁发的德国药物法规§2或§4(19)定义的药物或活性化合物。在一个替代实施方案中,活性化合物是被用作药物活性物质的治疗活性物质。
在另一个变化方案中,所述物质是用于控释活性化合物的载体(也称为载药系统),在一个方面,所述载体的目的一方面是防止活性化合物从体内快速排出。大多数传统的活性化合物都很小,因此只会短时间留在血流中,因为它们的小尺寸意味着它们的尺寸低于肾阈值,因此可与血液分离并排出。另一方面,这些载体系统可特异性地将活性化合物运送至特定器官或细胞的附近并以受控的方式在那里释放它们。
在另外的变化方案中,所述物质是增强肽的作用的化合物。在替代实施方案中,此类化合物为氨基吡唑和/或氨基吡唑衍生物。在本发明的含义内,氨基吡唑衍生物为3-氨基吡唑-5-羧酸或3-硝基吡唑-5-羧酸,以及其中杂环ch基团被-cr-或-n-或-o-或-s-取代的所有衍生物,还可以为由它们衍生的所有肽二聚体、三聚体或四聚体。在另外的替代实施方案中,这些是改善肽的溶解性和/或其通过血脑屏障的通路的化合物。
所述物质与肽连接,其中,所述连接是共价键或不是共价键。本文中,所述连接通过氢键、亲水、疏水或静电相互作用或键合和/或空间固定化而发生。
本发明的另一主题是一种用于与对照相比减少神经递质释放的方法,其特征在于,将一种用作镇痛剂的组合物与n型ncc接触,所述组合物由肽组成或包含肽,还由聚合物组成或包含聚合物,所述肽选自由rd2、d3、具有至少50%同一性的同系物和rd2或d3的衍生物组成的组或包含它们,所述聚合物包含rd2、d3、具有至少50%同一性的同系物以及rd2和d3的衍生物或由它们组成。
具体地,与对照相比,通过根据本发明的方法来减少通过ncc的钙流入。
本发明的另一主题是一种用于与对照相比抑制n型ncc的方法,其特征在于,使n型ncc与由肽组成或包含肽以及还可由聚合物组成或包含聚合物的组合物接触,所述肽选自由rd2、d3、具有至少50%同一性的同系物和rd2或d3的衍生物组成的组或包含它们,所述聚合物包含rd2、d3、具有至少50%同一性的同系物以及rd2和/或d3的衍生物。
在本发明的含义内,短语“与…接触”意指存在允许根据本发明使用的肽与n型ncc之间的相互作用的条件,特别是允许物理和/或化学相互作用(诸如,例如形成配位键或氢键)的条件。
选择对照是试验方法的常规部分。使用与待研究的生物体相同的另外的生物体作为对照。将待研究的生物体用根据本发明的组合物进行治疗或在最广泛的意义上与之接触,而对照不经受此过程。根据本发明,术语生物体还包括部分生物体、组织、组织样本、单个细胞或细胞培养物、细胞的部分如优选n型或l型的含膜ncc,或包含人造膜(如优选n型和/或l型的双脂质膜)的神经元钙通道。对照和待研究的生物体都选自上述所列。在另一替代实施方案中,根据临床研究的规范定义对照。
在本发明的含义内,抑制作用是指与对照相比通过根据本发明使用的组合物降低了优选n型神经元钙通道的活性。根据本发明,对照不与组合物接触。作为替代实施方案,对照还可与公知的抑制剂接触。
基于使用公知的抑制剂与根据本发明使用的组合物的对比,本领域普通技术人员还设法得出了关于根据本发明的组合物的功效的结论。
在本发明的一个实施方案中,在与ic50值相当或相等的浓度下确定抑制作用。
根据本发明使用的组合物将n型ncc抑制至少10%、15%、20%、25%、优选30%、特别优选40%、50%、60%、70%、80%、特别是90%或100%。
在一个实施方案中,根据本发明,通过所谓的膜片钳技术来确定抑制作用。在该技术中,在不同电压下测量穿过钙通道的离子电流。因此,可基于离子电流的减小来确定神经元钙通道的抑制作用。在替代实施方案中,离子电流减少了至少10%、15%、20%、25%、优选30%、35%、特别优选40%、42%、44%、46%、48%、50%、52%、54%、56%、58%、60%、70%、80%、特别是90%或100%。优选地,在等于ic50值的肽浓度下确定该减少。在一个实施方案中,在150nm的浓度下通过将相应的通道与根据本发明使用的组合物接触来进行膜片钳测量。
根据本发明的方法的特征在于与对照相比,l型ncc的功能未被修改。换言之,与对照相比,通过l型ncc的钙流入未被改变。根据本发明使用的肽特异性和/或选择性地作用于n型ncc。
本发明的另一主题还是将根据本发明使用的肽作为芋螺毒素的替代物的用途。
以这种方式,可在体外(离体)进行与ncc或包含ncc的膜或包含ncc的相应来源的接触。
本发明还涉及慢性和/或神经性疼痛的治疗,即用于疼痛缓解的治疗,其中,以每千克体重1μg至1g的浓度,通过肠内、静脉内、皮下、腹膜内、鼻内或口服给药、优选口服给药,将本发明使用的肽施用于待治疗的个体。
本发明因此还涉及一种利用本领域普通技术人员公知的传统方法将根据本发明使用的肽并入药物制剂中的方法。因此,本发明的主题也是上述肽在药物制剂中的用途。因此,包含根据本发明的上述肽的药物制剂也是本发明的主题。
此外,本发明的另一主题是根据权利要求1所述的组合物用于制备镇痛剂、用于疼痛治疗的药物和/或用于治疗慢性和神经性疼痛的药物的用途。
本发明的另一主题是根据权利要求1所述的组合物用于抑制n型神经元钙通道ncc的用途。
在根据本发明的方法中,还可使用上文描述的另外的肽、衍生物和同系物。
以任意上述的肽进行进一步的实施例。
具体实施方式
实施例:
1.rd2(c末端酰胺化)
a.蛋白构建体:
电压依赖性钙通道的两个不同的α1成孔单元(cavα1)的编码区与荧光蛋白在框架内如下融合(c末端融合):兔cav1.2(uniprotkb:p15381)的单元与yfp(cav1.2-yfp)融合,而人单元cav2.2(uniprotkb:q00975-1)与gfp(cav2.2-gfp)融合。β-亚基cavβ2e(uniprotkb:q8vgc3-2)与mrfp(cavβ2e-mrfp)连接,以及cavβ4(uniprotkb:o00305.2)与mcherry(cavβ4-mcherry)连接。
b.细胞转染:
由于cavα1亚基的正常功能和表面表达需要与cavβ亚基关联,所以用cav1.2-yfp和cavβ2e-mrfp或cav2.2-gfp和cavβ4-mcherry瞬时共转染tsa201细胞。采用lipofectamine2000(tm)(invitrogen)进行转染,利用荧光信号识别被成功转染的细胞。转染后24-48小时进行电生理学放电。
c.电生理学:
使用装配有patchmaster软件(hekaelektronik)的epc-10放大器,采用全细胞膜片钳技术测量离子电流。使用钡作为载体。在sutterp-100拔出器(harvardapparatus)上拔出电阻值为0.9-2mω的硼硅酸盐玻璃移液管,其尖部用narishigemf-830显微拉制仪进行表面热抛光。所使用的外部测量溶液为:140mmtea-mesos、10mmbacl2和10mmhepes(ph7.3);所使用的内测量溶液为:135mmcs-meso3、10mmegta、5mmcscl2、1mmmgcl2、4mmmgatp、0.4mmna2gtp和10mmhepes(ph7.3)。采用软件fitmaster(heka)、origin(originlab)和excel(微软)的组合进行数据分析。所有的数据均显示为平均值±sem。采用p/4协议(漏减法)校正离子电流。
为了研究rd2的药理学作用,分离细胞并转移至含有或不含有测试物质的灌注流中。在恒定灌注下进行观察以确保测试物质的浓度恒定。将rd2溶解于dmso中,终浓度为1mm,并在使用前不久将其溶于外部测量溶液中至150nm。采用公知的cav1.2和cav2.2钙通道阻滞剂尼莫地平(10μm)和ω-芋螺毒素(1nm)进行对照实验。
d.结果:
rd2对cav2.2介导的离子电流的影响
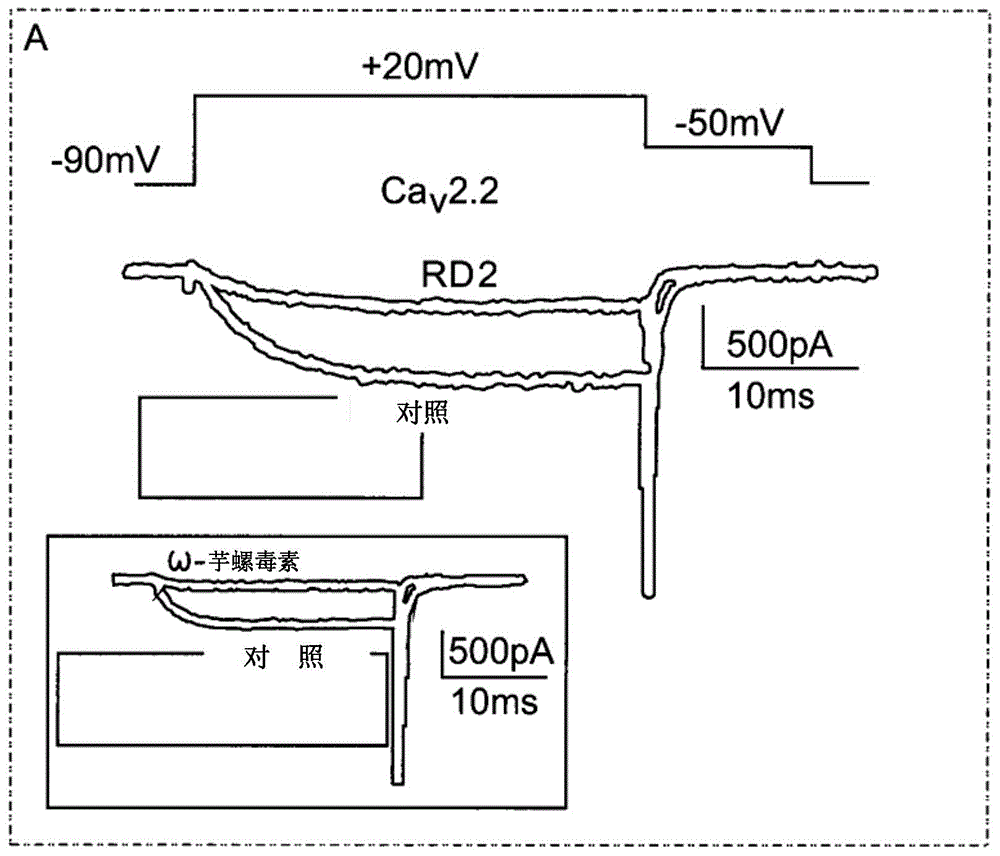
cav2.2位于突触前神经末梢并介导中枢突触的神经传递。cav2.2介导的离子电流被表达cav2.2/cavβ4的tsa201细胞吸收。使用包含rd2(150nm)的外部测量溶液但不单独使用外部测量溶液的细胞灌注导致离子电流显著减小(图1a、b;其中,a:在与包含150mm的rd2的组合物接触前和接触后,基于-90mv至+20mv的保持电位,在40ms脉冲期间产生的由cav2.2通道介导的电流强度的代表性图;b:在-90mv至+20mv的连续脉冲期间,在代表性细胞中,每5秒进行抑制的过程)。电流-电压图(i-v)表明,在所有测试电压下,离子电流减少了至多55%(图1c:在rd2(n=7)存在和不存在的情况下表达cav2.2的细胞的电流强度与电压的平均比)。由于离子电流的阻断并不伴随通道的电压依赖性激活的变化,所以假设了rd2的孔阻滞机制(图1d:被激活的通道的比例对图c中相同细胞的电压图(激活曲线);作为对照;因此通道的功能或机制无变化,因为曲线彼此重叠,没有移位)。在施用1nm的cav2.2阻滞剂ω-芋螺毒素后离子电流被中断,这证实了它们是通过cav2.2通道介导的。(图1a插入内容)。
rd2对cav1.2介导的离子电流的影响
cav1.2是主要在心脏中表达的l型钙通道,负责心肌细胞的电激活与肌丝收缩的耦合。离子电流被cav1.2和cavβ2e共表达细胞吸收,并且在暴露于150nm的rd2后保持基本上未变化(图2a:在与包含150mm的rd2的组合物接触前和接触后,基于-90mv至+10mv的保持电位,在40ms脉冲期间产生的由cav1.2通道介导的电流强度的代表性图。图2b:用150nm的rd2灌注时电流强度无变化)。相反地,用10μm尼莫地平(一种公知的cav1.2通道阻滞剂)进行灌注导致离子电流被几乎完全阻断。因此,cav1.2通道的激活的电压依赖性不受rd2的存在的影响(图2c和图2d)。
e.结论
在150nm的浓度下对rd2测试其阻断由cav2.2和cav1.2通道介导的离子电流的能力。在表达cav2.2和cav1.2通道的tsa201细胞中使用全细胞膜片钳技术进行该测试。rd2阻断cav2.2通道,而在测试浓度下cav1.2通道对rd2不敏感(图3)。
2.d3(c末端酰胺化)
a.蛋白构建体
电压依赖性钙通道cav2.2(uniprotkb:q00975-1)的人α1成孔单元(cavα1)的编码区与gfp融合,形成cav2.2-gfp。β-亚基cavβ4(uniprotkb:o00305.2)与mcherry融合,形成cavβ4-mcherry。
b.细胞转染:
由于cavα1亚基的正常功能和表面表达需要与cavβ亚基关联,所以用cav2.2-gfp和cavβ4-mcherry瞬时共转染tsa201细胞。采用lipofectamine
2000(tm)(invitrogen)进行转染,利用荧光信号识别被成功转染的细胞。转染后24-48小时进行电生理学放电。
c.电生理学:
使用装配有patchmaster软件(hekaelektronik)的epc-10放大器,采用全细胞膜片钳技术测量离子电流。使用钡作为载体。在sutterp-100拔出器(harvardapparatus)上拔出电阻值为0.9-2mω的硼硅酸盐玻璃移液管,其尖部用narishigemf-830显微拉制仪进行表面热抛光。所使用的外部测量溶液为:140mmtea-meso3、10mmbacl2和10mmhepes(ph7.3);所使用的内测量溶液为:135mmcs-meso3、10mmegta、5mmcscl2、1mmmgcl2、4mmmgatp、0.4mmna2gtp和10mmhepes(ph7.3)。采用软件fitmaster(heka)、origin(originlab)和excel(微软)的组合进行数据分析。所有的数据均显示为平均值±sem。采用p/4协议(漏减法)校正离子电流。
为了研究d3的药理学作用,分离细胞并转移至含有或不含有测试物质的灌注流中。在恒定灌注的条件下进行观察以确保测试物质的浓度恒定。将d3溶解于dmso中,终浓度为1mm,并在使用前不久将其溶于外部测量溶液中至150nm。采用公知的cav2.2钙通道阻滞剂ω-芋螺毒素(1nm)进行对照实验。
d.结果:
d3对cav2.2介导的离子电流的影响
cav2.2介导的离子电流被表达cav2.2/cavβ4的tsa201细胞吸收。使用包含d3(150nm)的外部测量溶液但不单独使用外部测量溶液的细胞灌注导致离子电流显著减小(图4a、b;其中,a:在与包含150mm的d3的组合物接触前和接触后,基于-90mv至+20mv的保持电位,在40ms脉冲期间产生的由cav2.2通道介导的电流强度的代表性图;4b:用外部ringer’s溶液再灌注后,由d3诱导的抑制是可逆的。在-90mv至+20mv的连续脉冲期间,每5秒记录电流强度一次)。电流-电压图(i-v)表明,在所有测试电压下,离子电流减少了至多54%(图4c)。由于离子电流的阻断并不伴随通道的电压依赖性激活的变化,所以假设了d3的孔阻滞机制(图4d)。在施用1nm的cav2.2阻滞剂ω-芋螺毒素后离子电流被中断,这证实了它们是通过cav2.2通道介导的。(图4a插入内容)。
e.结论
d3阻断cav2.2通道(图5)。
3.cd3(环化的d3)和crd2(环化的rd2)
与实施例1和2类似地进行实验。
cd3r和crd2r对cav2.2和cav1.2通道的影响的概述(图6)。两个条形图汇总了从电流强度-电压示意图获得的平均离子电流(对于cav2.2为+20mv,对于cav1.2为+10mv,**p≤0.01)。
cd3r对cav2.2和cav1.2通道的影响(图7)。
在暴露于150nm的cd3r(7a,左图)之前和之后,在-90mv至+20mv的40ms脉冲期间,触发了由cav2.2通道介导的代表性离子电流放电。右图7a示出了在-90mv至+20mv的连续脉冲期间在代表性细胞每5秒进行阻断的过程。7b:在存在和不存在cd3r(n=8)的情况下表达cav2.2的细胞和被激活的通道的部分的平均电流强度与电压比对电压(激活曲线)。7c:在暴露于150nm的cd3r(7c,左图)之前和之后,在-90mv至+20mv的40ms脉冲期间,触发由cav2.1通道介导的典型离子电流放电。右图7c示出了在-90mv至+20mv的连续脉冲期间在代表性细胞中每5秒阻断的过程。7d:在存在和不存在cd3r(n=8)的情况下平均电流强度与电压比,以及表达cav2.1的细胞的激活曲线(n=7)。
crd2r对cav2.2和cav1.2通道的影响(图8)。
8a:在暴露于150nm的crd2r(8a,左图)之前和之后,在-90mv至+20mv的40ms脉冲期间,触发由cav2.2通道介导的代表性离子电流放电。右图8a示出了在-90mv至+20mv的连续脉冲期间在代表性细胞中每5秒阻断的过程。8b:在存在和不存在crd2r(n=7)的情况下表达cav2.2的细胞和被激活的通道的部分的平均电流强度与电压比对电压(激活曲线)。8c:在暴露于150nm的crd2r(左图)之前和之后,在-90mv至+20mv的40ms脉冲期间,触发由cav2.1通道介导的代表性离子电流放电。右图8c示出了在-90mv至+20mv的连续脉冲期间在代表性细胞中每5秒阻断的过程。8d:在存在和不存在crd2r(n=8)的情况下平均电流强度与电压比,以及表达cav2.1的细胞的激活曲线(n=6)。
- 还没有人留言评论。精彩留言会获得点赞!