寨卡病毒蛋白酶抑制剂及其使用方法与流程
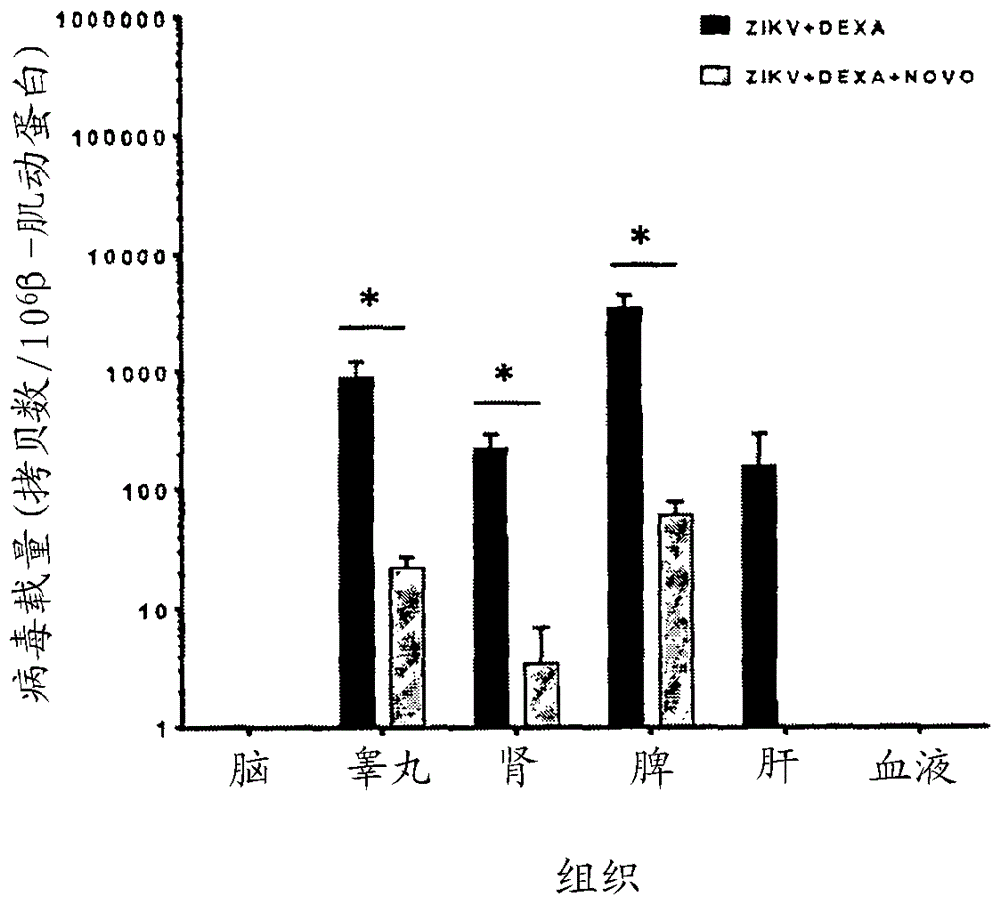
发明领域所公开的发明总体涉及病毒学领域,并且特别是寨卡病毒的领域。发明背景寨卡病毒(zikv)是新出现的人类致病性黄病毒,其主要通过感染的埃及伊蚊(aedesaegypti)或白纹伊蚊(aedesalbopictus)的叮咬而传播。尽管历史上zikv的传播主要限于南半球,但近来的气候变化已导致所述病毒向北进一步传播,且在美国大陆中持续传播的报告越来越多。所述病毒从孕妇传播到其胎儿的能力得到充分记录。因此,zikv已经在美洲引起了史无前例的先天性小头畸形和身体畸形的大规模流行(chan等人,j.infect.72,507-524(2016a))。急性zikv感染的症状包括发热、皮疹、头痛、关节痛、结膜炎和肌肉疼痛。尽管最初认为该疾病在受感染的成年人中完全是自限性的,但随着在美洲和其他地区的扩大流行,最近在成年患者中报告了越来越多的严重并发症(duffy等人,n.engl.j.med.360,2536-2543(2009);musso和gubler,clin.microbiol.rev.29,487-524(2016))。这些包括严重的神经系统并发症,例如格林-巴利综合征、脑膜脑炎和脊髓炎,血小板减少症和弥散性血管内凝血,伴有出血并发症、肝功能障碍、急性呼吸窘迫综合征、休克、多器官功能障碍综合征和死亡(arzuza-ortega等人,emerg.infect.dis.22,925-927(2016);azevedo等人,j.clin.virol.85,56-64(2016);cao-lormeau等人,lancet387,1531-1539(2016);carteaux等人,n.engl.j.med.374,1595-1596(2016);chraibi等人,j.clin.virol.83,61-62(2016);mecharles等人,lancet387,1481(2016);sarmiento-ospina等人,lancetinfect.dis.16,523-524(2016);soares等人,j.clin.virol.83,63-65(2016))。令人震惊的是,还描述了可能对雄性生育力具有长期影响的人类血精症病例和睾丸炎小鼠模型(chan等人,ebiomedicine14,112-122(2016c);foy等人,emerg.infect.dis.17,880-882(2011);govero等人,nature540,438-442(2016);ma等人,cell167,1511-24.e10(2016);musso等人,emerg.infect.dis.21,359-361(2015))。当前用于预防zikv感染的方法包括通过穿着长袖衬衫和裤子、停留在室内、使用纱窗和纱门以及使用杀虫剂和驱昆虫剂来预防蚊虫叮咬。通常还建议通过使用避孕套来防止病毒的性传播。由于目前尚无专门针对zikv感染的疫苗或药物,因此目前治疗的方法和建议主要由支持性护理组成。这些方法包括休息,饮用流体以防止脱水,以及服用非处方药例如对乙酰氨基酚以减轻疼痛和发热。怀孕患者中的zikv感染和严重zikv相关并发症的治疗选择仍然特别有限。为了鉴定立即可用的抗zikv治疗选择,已经通过使用基于细胞培养的抗病毒测定筛选药物文库而进行许多药物再利用计划(barrows等人,cellhostmicrobe20,259-270(2016);retallack等人,proc.natl.acad.sci.u.s.a.113,14408-14413(2016);xu等人,naturemed.22,1101-1107(2016))。但是,被发现具有体外抗zikv活性的这些临床批准药物的大多数是抗癌的或免疫调节性的药剂,其为免疫抑制性的或在妊娠期中是禁忌的(fda妊娠d类)。此外,此类筛选方法没有阐明这些药物的抗zikv机制,这对于进一步开发先导药物化合物的更安全且更有效的药物类似物是重要的。需要预防和治疗寨卡病毒感染的替代方法,尤其是考虑到它对发育中胎儿的影响。因此,本发明的一个目的是提供可以预防或治疗寨卡病毒感染而对患者的毒性作用较少的化合物。本发明的进一步目的是提供可以预防或治疗寨卡病毒感染而对患者的毒性作用较少的方法。本发明的进一步目的是提供用于鉴定或选择可以预防或治疗寨卡病毒感染而对患者的毒性作用较少的化合物的方法。本说明书中已包括的关于文件、法案、材料、装置、物品等的任何讨论均不应被视为承认任何或所有这些事项,由于其在本申请的每项权利要求的优先权日之前存在,而形成现有技术基础的一部分或是与本公开相关的领域中的公知常识。在本说明书全文中,词语“包含”("comprising"或者其变形例如"comprises"或"comprising"),应当被理解为暗示包括所述的要素、整数或步骤,或要素、整数或步骤的组,但不排除任何其它要素、整数或步骤,或要素、整数或步骤的组。发明简述公开了预防或治疗寨卡病毒(zikv)感染的方法和化合物。公开了预防和/或治疗zikv感染的材料和方法。在一些方式中,公开了用于预防和/或治疗受试者中的zikv感染的方法,所述方法包括将受试者诊断为患有zikv感染或处于发生zikv感染的风险中;和向受试者施用有效量的预防和/或治疗zikv感染的组合物。在一些方式中,组合物可以包含有效量的选自以下的化合物:新生霉素、洛匹那韦-利托那韦、利福平、醋酸去氨加压素和醋酸奥曲肽。还公开了鉴定可用于预防和/或治疗zikv感染的化合物的方法,所述方法包括对已知药物候选物的结构筛选和使用体外和体内zikv感染模型的评估。公开了预防或治疗寨卡病毒感染的方法,其中所述方法包括向患有寨卡病毒感染或处于发生寨卡病毒感染的风险中的受试者施用有效量的抑制寨卡病毒蛋白酶的组合物。作为另一实例,公开了用于预防和/或治疗受试者中的寨卡病毒感染的方法,其中所述方法包括将受试者诊断为患有寨卡病毒感染或处于发生寨卡病毒感染的风险中;和向受试者施用有效量的预防和/或治疗寨卡病毒感染的组合物。在一些方式中,组合物的量有效地抑制受试者中的寨卡病毒蛋白酶。在一些方式中,受试者是成年人。在一些方式中,受试者是怀孕的。在一些方式中,受试者是免疫受损的。通常认为免疫受损的受试者处于免疫系统对抗传染病和癌症的能力受损或完全不存在的状态。在一些方式中,组合物包括新生霉素、洛匹那韦-利托那韦、利福平、醋酸去氨加压素、醋酸奥曲肽或其组合。在一些方式中,组合物包括新生霉素、洛匹那韦-利托那韦或其组合。在一些方式中,该组合物包括新生霉素。在一些方式中,组合物包括有效量的新生霉素。在一些方式中,新生霉素的有效量小于300mg/kg/d。在一些方式中,所述方法还包括在施用前,将受试者诊断为患有寨卡病毒感染或处于发生寨卡病毒感染的风险中。在一些方式中,受试者处于发生寨卡病毒感染的风险中。在一些方式中,向处于寨卡病毒感染的风险中的受试者施用的组合物的量是有效地预防受试者中的寨卡病毒感染的量。在一些方式中,受试者患有寨卡病毒感染。在一些方式中,向患有寨卡病毒感染的受试者施用的组合物的量是有效地治疗受试者中的寨卡病毒感染的量。还公开了鉴定可用于预防或治疗寨卡病毒感染的化合物的方法。例如,公开了用于鉴定抑制寨卡病毒蛋白酶的化合物的方法,其中所述方法包括:(a)使蛋白酶底物、测试化合物和包含至少一种寨卡病毒蛋白酶的寨卡病毒分离物在第一测试室中接触;和(b)在第一测试室中测量裂解的蛋白酶底物的量。在一些方式中,如果在第一测试室中测量的裂解的蛋白酶底物的量小于在没有测试化合物的对照测试室中测量的裂解的蛋白酶底物的量,则可以将测试化合物鉴定为抑制寨卡病毒蛋白酶的化合物。在一些方式中,步骤(a)和(b)可以在一个或多个另外的测试室中进行,每个另外的测试室具有的测试化合物的量不同于第一测试室中的测试化合物的量。在一些方式中,如果在具有小于50μg/ml的测试化合物的测试室之一中测量的裂解的蛋白酶底物的量比在对照测试室中测量的裂解的蛋白酶底物的量少至少50%,则可以将测试化合物鉴定为抑制寨卡病毒蛋白酶的化合物。在一些方式中,第一测试室可以具有小于50μg/ml的测试化合物。在一些方式中,如果在第一测试室中测量的裂解的蛋白酶底物的量比在对照测试室中测量的裂解的蛋白酶底物的量少至少50%,则可以将测试化合物鉴定为抑制寨卡病毒蛋白酶的化合物。在一些方式中,测试化合物可以是被鉴定为如下化合物的化合物:使用分子动力学模拟,预测为与寨卡病毒蛋白酶形成稳定复合物的化合物。在一些方式中,分子动力学模拟可以使用蛋白质模型系统,所述蛋白质模型系统使用寨卡病毒蛋白酶的晶体结构和候选化合物的结构构建。还公开了鉴定抑制寨卡病毒感染的化合物的方法,其中所述方法包括:(a)使寨卡病毒易感细胞系、测试化合物和寨卡病毒分离物在第一测试室中接触,和(b)在第一测试室中测量寨卡病毒分离物对寨卡病毒易感细胞系的细胞病变作用水平。在一些方式中,如果在第一测试室中测量的细胞病变作用水平小于在没有测试化合物的对照测试室中测量的细胞病变作用水平,则可以将测试化合物鉴定为抑制寨卡病毒感染的化合物。在一些方式中,步骤(a)和(b)可以在一个或多个另外的测试室中进行,每个另外的测试室具有的测试化合物的量不同于第一测试室中的测试化合物的量。在一些方式中,如果在具有小于50μg/ml的测试化合物的测试室之一中测量的细胞病变作用水平比在对照测试室中测量的细胞病变作用水平少至少50%,则可以将测试化合物鉴定为抑制寨卡病毒感染的化合物。在一些方式中,第一测试室可以具有小于50μg/ml的测试化合物。在一些方式中,如果在第一测试室中测量的细胞病变作用水平比在对照测试室中测量的细胞病变作用水平少至少50%,则可以将测试化合物鉴定为抑制寨卡病毒感染的化合物。在一些方式中,测试化合物可以是被鉴定为如下化合物的化合物:使用分子动力学模拟,预测为与寨卡病毒非结构蛋白形成稳定复合物的化合物。在一些方式中,分子动力学模拟可以使用蛋白质模型系统,所述蛋白质模型系统使用寨卡病毒非结构蛋白的晶体结构和候选化合物的结构构建。还公开了鉴定可用于预防和/或治疗寨卡病毒感染的化合物的方法,其中所述方法包括提供蛋白酶底物,提供候选药物化合物,提供包含至少一种寨卡病毒蛋白酶的寨卡病毒分离物,使寨卡病毒分离物与不同量的至少一种候选药物化合物接触,使蛋白酶底物与寨卡病毒分离物和至少一种候选化合物接触,定量由至少一种寨卡病毒蛋白酶释放的肽的量,并且当可测量出zika蛋白酶活性降低50%的化合物浓度小于50μg/ml时,选择药物化合物作为可用于预防和/或治疗寨卡病毒感染。还公开了鉴定可用于预防和/或治疗寨卡病毒感染的化合物的方法,其中所述方法包括提供寨卡病毒非结构蛋白的晶体结构,提供候选药物化合物,构建蛋白质模型系统,进行分子动力学模拟,以预测寨卡病毒非结构蛋白-化合物复合物的稳定性,选择与寨卡病毒非结构蛋白形成稳定复合物的化合物,提供寨卡病毒易感细胞系,提供寨卡病毒分离物,在存在和不存在渐增浓度的至少一种所选化合物的情况下,使寨卡病毒易感细胞系与寨卡病毒分离物接触,确定寨卡病毒分离物对寨卡病毒易感细胞系的细胞病变作用,并且当可测量出细胞病变作用降低50%的化合物浓度小于50μg/ml时,选择化合物作为可用于预防和/或治疗寨卡病毒感染。所公开的方法和组合物的另外优点将在下面的描述中部分地阐述,并且部分地由所述描述理解,或者可以通过实践所公开的方法和组合物而获知。所公开的方法和组合物的优点将借助在所附权利要求中具体指出的要素及组合而被认识和实现。要理解的是,前文的概述和后续的详述都仅仅是示例性和解释性的,并不限制所要求保护的本发明。附图简述并入本说明书并构成本说明书一部分的附图示出了所公开的方法和组合物的几个实施方案,并且与说明书一起用于解释所公开的方法和组合物的原理。图1是显示用于鉴定可用于预防和治疗寨卡病毒感染的化合物的筛选和验证策略的图。图2a-2c是显示新生霉素对zikv-ns2b-ns3的蛋白酶活性的抑制的图。图2a是在不同浓度的新生霉素存在的情况下荧光强度相对于蛋白质浓度的图。图2b是归一化响应相对于抑肽酶浓度的对数的图。图2c是归一化响应相对于新生霉素浓度的对数的图。数据表示为平均值±平均值的标准误差(误差线)。ic50,半数最大抑制浓度(蛋白酶活性降低50%的药物浓度)。图3a-3g是归一化响应(zikv-ns2b-ns3蛋白酶活性的抑制%)相对于不同浓度的不同化合物的图。在蛋白酶抑制测定中,底物和zikvns2b-ns3蛋白酶的浓度分别固定为10μm和5nm,同时添加渐增浓度的(图3a)醋酸去氨加压素、(图3b)醋酸奥曲肽、(图3c)洛匹那韦-利托那韦、(图3d)利福平、(图3e)孟鲁司特、(图3f)西罗莫司和(图3g)他克莫司。将各小图中的数据针对未治疗组归一化为百分比。*表示p<0.05,且**表示p<0.01(通过student氏t检验与dmso对照组相比)。结果表示为平均值和平均值的标准误差(误差线)。实验一式三份进行,并重复两次以确认。图4a-4c是显示新生霉素的体外抗zikv活性的评估的图。图4a是vero细胞中对数病毒血液载量相对于新生霉素浓度的图。图4b是huh-7细胞中对数病毒血液载量相对于新生霉素浓度的图。图4c是噬斑减少(%)相对于新生霉素浓度的图。所有实验一式三份进行,并重复两次以确认。*表示p<0.05,且***表示p<0.0001(通过student氏t检验与dmso对照组相比)。数据表示为平均值±平均值的标准误差(误差线)。图5a和5b是显示洛匹那韦-利托那韦的体外抗zikv活性的图。图5a是vero细胞中对数归一化病毒载量相对于洛匹那韦-利托那韦浓度的图。图5b是huh-7细胞中对数归一化病毒载量相对于洛匹那韦-利托那韦浓度的图。所有实验均一式三份进行。*表示p<0.05,且***表示p<0.0001(通过student氏t检验与dmso对照组相比)。数据表示为平均值±平均值的标准误差(误差线)。图6a-6c是显示新生霉素的体内抗zikv活性的评估的图。图6a是体重变化相对于寨卡病毒接种后的天数的图。图6b是临床得分相对于寨卡病毒接种后的天数的图。图6c是存活率(%)相对于寨卡病毒接种后的天数的图。图7a和7b是体内研究中不同组小鼠的血液和主要器官中病毒载量的图。*表示p-值<0.05。数据表示为平均值±平均值的标准误差(误差线)。缩写:dexa,地塞米松;novo,新生霉素;zikv,寨卡病毒。图8是新生霉素的对数归一化病毒载量相对于药物添加时间的图。图9a以新生霉素和zikvns2b-ns3之间的蛋白质-配体相互作用的二维示意图,说明了与zikvns2b-ns3对接的新生霉素的分子模型。氢键和疏水接触面积分别由虚线和两个括号表示。标记了主要的相互作用残基。虚线表示氢与所涉及的受体原子之间的键。图9b是显示新生霉素的蛋白酶抑制的竞争模式的图。在测定中,zikvns2b-ns3蛋白酶的浓度固定为5nm,同时添加不同浓度的底物(100、50、25、12.5、6.25、3.125和0μm),以绘制酶动力学曲线。针对不同浓度的底物,分别测试了四种浓度的药物(1000、250、62.5和0µg/ml)。重复实验两次以确认。通过graphpadprism6在酶动力学模块中,针对抑制模式对数据进行分析。r2>0.95被认为是统计学显著的。发明详述通过参考以下对具体实施方案的详细描述及其中包括的实施例以及附图及其前后的描述,可以更容易地理解所公开的方法和组合物。i.化合物和组合物公开了用于预防和/或治疗寨卡病毒(zikv)感染的组合物。已经发现,用于发现抗zikv治疗的有用方法是通过重新使用意外地抑制zikv的关键酶(包括例如zikv蛋白酶、解旋酶和/或聚合酶)的临床批准的药物。基于这个发现,已经通过计算机芯片上虚拟筛选鉴定了对zikv蛋白酶zikv-ns2b-ns3具有高亲和力并且能够在体外和体内抑制zikv-ns2b-ns3活性的药物化合物。在一些方式中,所述组合物抑制寨卡病毒解旋酶。在一些方式中,所述组合物抑制寨卡病毒聚合酶。在特定方式中,所述组合物抑制寨卡病毒蛋白酶。a.新生霉素新生霉素,也称为阿巴霉素(albamycin)或卡卓霉素(cathomycin),是由放线菌雪白链霉菌(streptomycesniveus)产生的抗生素。它具有针对表皮葡萄球菌(staphylococcusepidermidis)的活性,但也可以用作在mrsa治疗中使用的抗葡萄球菌剂。新生霉素是细菌dna促旋酶的抑制剂,并且通过靶向参与能量转化的酶的gyrb亚基起作用。它充当gyrb催化的atp酶反应的竞争性抑制剂。gyra亚基参与dna切口和连接活性。b.洛匹那韦-利托那韦洛匹那韦-利托那韦是用于治疗和预防hiv/aids的固定剂量的组合药物。洛匹那韦和利托那韦均为hiv蛋白酶抑制剂。通常建议将所述药物与其他抗逆转录病毒药物一起使用。它可用于针刺伤或其他潜在暴露后的预防。它作为片剂或溶液口服。它通常用于妊娠期,并且它看起来是安全的。利托那韦通过减缓洛匹那韦的降解而起作用。c.利福平利福平(rifampicin),也称为甲哌利福霉素(rifampin),是由土壤细菌amycolatopsisrifamycinica产生的抗生素,并且用于治疗几种类型的细菌感染,例如结核病、麻风病和军团病。它几乎总是与其他抗生素一起使用,除了在已经暴露于那些细菌的人中给予以预防b型流感嗜血杆菌(haemophilusinfluenzae)和脑膜炎球菌病。利福平可以口服或静脉内给予,并且是妊娠期间活动性结核病的推荐治疗的一部分。这种药物通过抑制细菌的dna依赖性rna聚合酶起作用,因此阻止细菌产生rna。还显示出它针对牛痘病毒有效。d.醋酸去氨加压素去氨加压素是加压素的合成形式,加压素是减少尿液产生的激素。这种药物通常以醋酸去氨加压素的形式开出处方,并且通常用于治疗尿崩症、尿床、a型血友病、血管性血友病(vonwillebranddisease)和高血尿素水平。它可以在鼻中、通过注射至静脉内、通过口服或在舌下施用。在怀孕期间使用这种药物看起来是安全的。去氨加压素是一种抗利尿剂,并且因此通过限制排出在尿液中的水量起作用。它通过与v2受体结合而在肾收集管的水平发挥作用,所述v2受体信号传导用于水通道蛋白通道经由胞质囊泡易位至收集管的顶膜。这些水通道蛋白通道在远端肾单位中的存在,导致水从尿液中的重吸收增加,其被动地通过基底侧膜通道从肾单位重新分配到全身循环。去氨加压素也通过作用于v2受体而刺激从内皮细胞释放血管性血友病因子。如实施例中所示,醋酸去氨加压素还能够在体外和体内抑制zikv-ns2b-ns3活性。e.醋酸奥曲肽奥曲肽是在药理上模拟天然生长抑素的八肽,尽管它是比天然激素更有效的生长激素、胰高血糖素和胰岛素的抑制剂。奥曲肽用于治疗产生生长激素的肿瘤(肢端肥大症和巨人症),分泌甲状腺刺激激素的垂体瘤(促甲状腺激素瘤),与类癌综合症有关的腹泻和潮红发作,以及患有血管活性肠肽分泌性肿瘤的人的腹泻。奥曲肽也经常作为输注剂给予,用于肝硬化中食管静脉曲张引起的急性出血的管控。通过用铟-111标记,它也用于核医学成像(octreoscan),以非侵入性方式成像神经内分泌和其他表达生长抑素受体的肿瘤。奥曲肽还通过减少gi分泌和抑制gi动力,因此控制和减少其输出,来帮助瘘管的管控。如实施例中所示,醋酸去氨加压素还能够在体外和体内抑制zikv-ns2b-ns3活性。除了选择的分子以外,本文所述的药物组合物可包括但不限于载体、增稠剂、稀释剂、缓冲剂、防腐剂、表面活性剂等。药物组合物还可包含一种或多种活性成分,例如抗微生物剂、抗炎剂、麻醉剂等。公开了经验证的zikv-ns2b-ns3蛋白酶抑制剂,以在体外抑制zikv复制。在所述方法的一些方式中,zikv-ns2b-ns3蛋白酶抑制剂新生霉素和洛匹那韦-利托那韦可用于在体外和体内抑制zikv复制。已发现,在播散性zikv感染的动物模型中,使用新生霉素的治疗显著改善了小鼠的临床结局。在一些方式中,本文所述的组合物包含有效量的新生霉素、洛匹那韦-利托那韦、利福平、醋酸去氨加压素、醋酸奥曲肽,或其组合。在一些方式中,组合物包含有效量的新生霉素、洛匹那韦-利托那韦,或其组合。在一些方式中,组合物包含有效量的新生霉素。然而,如本领域技术人员通常所理解的,组合物可包含另外的化合物,并且因此不限于本文所述的化合物。在一些方式中,组合物包含表2中所列化合物中的至少一种化合物。如本文所用,术语“活性”是指生物学活性。如本文所用,术语“药理活性”是指肽或多肽的固有物理特性。这些特性包括但不限于半衰期、溶解度和稳定性以及其他药代动力学特性。ii.筛选和鉴定化合物的方法所公开的鉴定可用于预防或治疗寨卡病毒感染的化合物的方法可以以任何合适的方式进行。通常,鉴定方法的首要目标是鉴定具有一种或多种期望特征的化合物。这之后可以针对一种或多种其他期望特征的存在,筛选或鉴定那些化合物命中物。筛选和鉴定可以是针对特征的存在或特征的特定水平,或者超过阈值水平的特征水平。期望特征可以是与最终目标相关的任何特征。已经发现,用于鉴定可用于预防或治疗寨卡病毒感染的化合物的有用特征包括:(a)使用分子动力学模拟预测化合物与寨卡病毒非结构蛋白(例如寨卡病毒蛋白酶、解旋酶或聚合酶)形成稳定复合物,(b)使用分子动力学模拟预测化合物与寨卡病毒非结构蛋白(例如寨卡病毒蛋白酶、解旋酶或聚合酶)形成稳定性等于或高于稳定性阈值水平的复合物,(c)化合物抑制寨卡病毒非结构蛋白(例如寨卡病毒蛋白酶、解旋酶或聚合酶);(d)化合物抑制寨卡病毒非结构蛋白(例如寨卡病毒蛋白酶、解旋酶或聚合酶)达到或超过抑制的阈值水平,(e)化合物降低寨卡病毒分离物对寨卡病毒易感细胞系的细胞病变作用水平,(f)化合物降低寨卡病毒分离物对寨卡病毒易感细胞系的细胞病变作用水平至或超过细胞病变作用的阈值水平,(g)化合物对待使用该化合物治疗的受试者没有毒性或具有低毒性,(h)化合物对待使用该化合物治疗的受试者具有低于毒性阈值的毒性;(i)化合物对特定细胞或组织没有毒性或具有低毒性;和(j)化合物对特定细胞或组织具有低于毒性阈值的毒性。可以就例如化合物与结合配偶体相互作用的能态来评估稳定的复合物。也可以使用本领域技术人员已知的其他稳定性量度。有用的稳定性阈值水平可以是复合物的动力学行为,其可以通过例如进行分子动力学模拟来评估。例如,复合物可以稳定(a)至少,(b)持续或(c)大于10ns、15ns、20ns、25ns、30ns、35ns、40ns、45ns、50ns、55ns、60ns、65ns、70ns、75ns、80ns、85ns、90ns、95ns、100ns、110ns、120ns、130ns140ns150ns、160ns、170ns、180ns,190ns或200ns。在公开的研究中产生的轨迹揭示,所述复合物在50ns的事件期间是相当稳定的。可以就例如被抑制的蛋白质的活性降低(例如蛋白酶的蛋白酶活性降低)来评估抑制。为评估抑制而测量的活性可以是蛋白质的任何活性,但通常可以是蛋白质的主要活性。可以使用任何合适的测定技术来测量活性,例如通过使用对于所述活性的特殊的、模型的或指示性的底物。寨卡病毒对寨卡病毒易感细胞系的任何合适的细胞病变作用可用于评估细胞病变作用的降低。例如,所评估的细胞病变作用可以是细胞生长受限、细胞周期失调、诱导细胞肥大、诱导应激颗粒、er增殖增加,或凋亡。可以使用例如实施例中所述的测定法(也描述于chan等人(2017a))来评估此类细胞病变作用。也可以使用病毒产量减少或噬斑减少来评估细胞病变作用。病毒产量减少和噬斑减少可以使用例如实施例中所述的测定法(也描述于chan等人(2017a))来评估。有用的寨卡病毒分离物可以是例如波多黎各毒株prvabc59。有用的寨卡病毒易感细胞系包括例如vero细胞和huh-7细胞。化合物对细胞、组织和生物的毒性可使用已知用于此类目的的众多技术中的任一种进行评估。毒性可以通过例如化合物的杀灭作用,损伤或病理作用的发展,或对细胞、组织或生物的某些其他有害作用来评估。例如,可以通过确定化合物对合适的细胞或细胞系(例如vero细胞或huh-7细胞)的细胞毒性,来评估化合物的毒性。作为毒性的量度,例如可以测量化合物的50%有效细胞毒性浓度。可以使用例如实施例中所述的测定法(也描述于chan等人(2017a)),来评估细胞毒性。公开了鉴定可用于预防或治疗寨卡病毒感染的化合物的方法。例如,公开了用于鉴定抑制寨卡病毒蛋白酶的化合物的方法,其中所述方法包括:(a)使蛋白酶底物、测试化合物和包含至少一种寨卡病毒蛋白酶的寨卡病毒分离物在第一测试室中接触;和(b)在第一测试室中测量裂解的蛋白酶底物的量。在一些方式中,如果在第一测试室中测量的裂解的蛋白酶底物的量小于在没有测试化合物的对照测试室中测量的裂解的蛋白酶底物的量,则可以将测试化合物鉴定为抑制寨卡病毒蛋白酶的化合物。在一些方式中,步骤(a)和(b)可以在一个或多个另外的测试室中进行,每个另外的测试室具有的测试化合物的量不同于第一测试室中的测试化合物的量。在一些方式中,如果在具有小于50μg/ml的测试化合物的测试室之一中测量的裂解的蛋白酶底物的量比在对照测试室中测量的裂解的蛋白酶底物的量少至少50%,则可以将测试化合物鉴定为抑制寨卡病毒蛋白酶的化合物。在一些方式中,第一测试室可以具有小于50μg/ml的测试化合物。在一些方式中,如果在第一测试室中测量的裂解的蛋白酶底物的量比在对照测试室中测量的裂解的蛋白酶底物的量少至少50%,则可以将测试化合物鉴定为抑制寨卡病毒蛋白酶的化合物。还公开了用于系统性计算机芯片上、体外和体内筛选zikv感染的治疗以及开发新型抗zikv剂的方法。如实施例所示,在计算机芯片上进行由8227种药物化合物组成的大型化学文库的基于结构的虚拟筛选,以鉴定潜在的zikv-ns2b-ns3蛋白酶抑制剂。zikv-ns2b-ns3蛋白酶晶体结构被用作程序的输入(lei等人,science353,503-505(2016)),并且通过其对zikv-ns2b-ns3蛋白酶的预测结合亲和力,对前100种初步命中化合物进行排序(表2)。在一些方式中,测试化合物可以是被鉴定为如下化合物的化合物:使用分子动力学模拟,预测为与寨卡病毒蛋白酶形成稳定复合物的化合物。在一些方式中,分子动力学模拟可以使用蛋白质模型系统,所述蛋白质模型系统使用寨卡病毒蛋白酶的晶体结构和候选化合物的结构构建。分子动力学模拟以及用于此类模拟的平台是已知的,并且可以用于所公开的方法。通常,可以使用任何评估两种或更多种组分的结合相互作用的分子动力学模拟。例如,可以使用实施例中使用的分子动力学模拟leadfinder(biomoltech)。蛋白质模型系统是指在模型系统(例如分子动力学模拟)中存在并使用的蛋白质的结构信息。还公开了鉴定可用于预防和/或治疗寨卡病毒感染的化合物的方法,其中所述方法包括提供蛋白酶底物,提供候选药物化合物,提供包含至少一种寨卡病毒蛋白酶的寨卡病毒分离物,使寨卡病毒分离物与不同量的至少一种候选药物化合物接触,使蛋白酶底物与寨卡病毒分离物和至少一种候选化合物接触,定量由至少一种寨卡病毒蛋白酶释放的肽的量,并且当可测量出zika蛋白酶活性降低50%的化合物浓度小于50μg/ml时,选择药物化合物作为可用于预防和/或治疗寨卡病毒感染。示例性候选药物化合物包括但不一定限于表2中列出的药物化合物。在特定方式中,筛选方法的目的在于抑制寨卡病毒蛋白酶的化合物。此类方法可包括:(a)使蛋白酶底物、测试化合物和包含至少一种寨卡病毒蛋白酶的寨卡病毒分离物在第一测试室中接触;和(b)在第一测试室中测量裂解的蛋白酶底物的量,其中如果在第一测试室中测量的裂解的蛋白酶底物的量小于在没有测试化合物的对照测试室中测量的裂解的蛋白酶底物的量,则将测试化合物鉴定为抑制寨卡病毒蛋白酶的化合物。在一些方式中,步骤(a)和(b)在一个或多个另外的测试室中进行,每个另外的测试室具有的测试化合物的量不同于第一测试室中的测试化合物的量,其中如果在具有小于50μg/ml的测试化合物的测试室之一中测量的裂解的蛋白酶底物的量比在对照测试室中测量的裂解的蛋白酶底物的量少至少50%,则将测试化合物鉴定为抑制寨卡病毒蛋白酶的化合物。在一些方式中,第一测试室具有小于50μg/ml的测试化合物,其中如果在第一测试室中测量的裂解的蛋白酶底物的量比在对照测试室中测量的裂解的蛋白酶底物的量少至少50%,则将测试化合物鉴定为抑制寨卡病毒蛋白酶的化合物。在一些方式中,测试化合物是被鉴定为如下化合物的化合物:使用分子动力学模拟,预测为与寨卡病毒蛋白酶形成稳定复合物的化合物。在一些方式中,分子动力学模拟使用蛋白质模型系统,所述蛋白质模型系统使用寨卡病毒蛋白酶的晶体结构和候选化合物的结构构建。每当所述方法包括混合组合物或组分或试剂或使组合物或组分或试剂接触时,则进行所述方法产生许多不同的混合物。例如,如果所述方法包括3个混合步骤,如果所述步骤分开进行,则在这些步骤的每一个之后形成独特的混合物。另外,无论如何进行所述步骤,在所有步骤完成时都形成混合物。本公开设想了通过进行所公开的方法获得的这些混合物,以及包含任何公开的试剂、组合物或组分的混合物,例如本文所公开的。还公开了鉴定抑制寨卡病毒感染的化合物的方法,其中所述方法包括:(a)使寨卡病毒易感细胞系、测试化合物和寨卡病毒分离物在第一测试室中接触,和(b)在第一测试室中测量寨卡病毒分离物对寨卡病毒易感细胞系的细胞病变作用水平。在一些方式中,如果在第一测试室中测量的细胞病变作用水平小于在没有测试化合物的对照测试室中测量的细胞病变作用水平,则可以将测试化合物鉴定为抑制寨卡病毒感染的化合物。在一些方式中,步骤(a)和(b)可以在一个或多个另外的测试室中进行,每个另外的测试室具有的测试化合物的量不同于第一测试室中的测试化合物的量。在一些方式中,如果在具有小于50μg/ml的测试化合物的测试室之一中测量的细胞病变作用水平比在对照测试室中测量的细胞病变作用水平少至少50%,则可以将测试化合物鉴定为抑制寨卡病毒感染的化合物。在一些方式中,第一测试室可以具有小于50μg/ml的测试化合物。在一些方式中,如果在第一测试室中测量的细胞病变作用水平比在对照测试室中测量的细胞病变作用水平少至少50%,则可以将测试化合物鉴定为抑制寨卡病毒感染的化合物。在一些方式中,测试化合物可以是被鉴定为如下化合物的化合物:使用分子动力学模拟,预测为与寨卡病毒非结构蛋白形成稳定复合物的化合物。在一些方式中,分子动力学模拟可以使用蛋白质模型系统,所述蛋白质模型系统使用寨卡病毒非结构蛋白的晶体结构和候选化合物的结构构建。还公开了鉴定可用于预防和/或治疗寨卡病毒感染的化合物的方法,其中所述方法包括提供寨卡病毒非结构蛋白的晶体结构,提供候选药物化合物,构建蛋白质模型系统,进行分子动力学模拟,以预测寨卡病毒非结构蛋白-化合物复合物的稳定性,选择与寨卡病毒非结构蛋白形成稳定复合物的化合物,提供寨卡病毒易感细胞系,提供寨卡病毒分离物,在存在和不存在渐增浓度的至少一种所选化合物的情况下,使寨卡病毒易感细胞系与寨卡病毒分离物接触,确定寨卡病毒分离物对寨卡病毒易感细胞系的细胞病变作用,并且当可测量出细胞病变作用降低50%的化合物浓度小于50μg/ml时,选择化合物作为可用于预防和/或治疗寨卡病毒感染。在特定方式中,筛选方法的目的在于抑制寨卡病毒感染的化合物。此类方法可包括:(a)使寨卡病毒易感细胞系、测试化合物和寨卡病毒分离物在第一测试室中接触;和(b)在第一测试室中测量寨卡病毒分离物对寨卡病毒易感细胞系的细胞病变作用水平,其中如果在第一测试室中测量的细胞病变作用水平小于在没有测试化合物的对照测试室中测量的细胞病变作用水平,则将测试化合物鉴定为抑制寨卡病毒感染的化合物。在一些方式中,步骤(a)和(b)在一个或多个另外的测试室中进行,每个另外的测试室具有的测试化合物的量不同于第一测试室中的测试化合物的量,其中如果在具有小于50μg/ml的测试化合物的测试室之一中测量的细胞病变作用水平比在对照测试室中测量的细胞病变作用水平少至少50%,则将测试化合物鉴定为抑制寨卡病毒感染的化合物。在一些方式中,第一测试室具有小于50μg/ml的测试化合物,其中如果在第一测试室中测量的细胞病变作用水平比在对照测试室中测量的细胞病变作用水平少至少50%,则将测试化合物鉴定为抑制寨卡病毒感染的化合物。在一些方式中,测试化合物是被鉴定为如下化合物的化合物:使用分子动力学模拟,预测为与寨卡病毒非结构蛋白形成稳定复合物的化合物。在一些方式中,分子动力学模拟使用蛋白质模型系统,所述蛋白质模型系统使用寨卡病毒非结构蛋白的晶体结构和候选化合物的结构构建。如本领域技术人员已知的,设想了筛选化合物的另外的方法,且因此不限于本文所述的方法。测试室是指管、孔、容器、基板上的斑点等,其中可以保持或放置不同测定或样品的组分。分离的测试室是指不同的测试室,其中可以放置或进行混合物、反应或测定,而不干扰其他测试室中的其他混合物、反应或测定,或与之组合。包含或包括组分的测试室的实例包括孔板、管、容器、表面、载片等。如本文所用,术语“使…接触”是指将组分(例如化合物或分子)添加、混合、组合等至另一组分的任何方式、模式或技术。使…接触还包括采取将导致一组分与另一组分接触的动作。术语“命中物”是指在测定中显示期望特性的测试化合物。术语“测试化合物”是指待通过一种或多种筛选方法作为推定的调节剂测试的化学物质。测试化合物可以是任何化学物质,例如无机化学物质、有机化学物质、蛋白质、肽、碳水化合物、脂质,或其组合。通常,使用各种预定浓度的测试化合物进行筛选,例如0.01微摩尔、1微摩尔和10微摩尔。测试化合物对照可包括在不存在测试化合物的情况下测量信号,或与已知调节靶标的化合物的比较。如本文所用,术语“候选化合物”是指化学物质,其结构待通过一种或多种筛选方法,使用分子动力学模拟测试为与靶标结构形成稳定的复合物。如本文所用,术语“调节”是指与适当的对照相比,化合物以某种可测量的方式改变活性的能力。由于测定中存在化合物,因此与不存在这些化合物的对照相比,活性可以增加或降低。与不存在化合物的情况下的活性水平相比,活性的增加优选为至少25%,更优选至少50%,最优选至少100%。类似地,与不存在化合物的情况下的活性水平相比,活性的降低优选为至少25%,更优选至少50%,最优选至少100%。增加已知活性的化合物是“激动剂”。减少或阻止已知活性的化合物是“拮抗剂”。术语“抑制”是指降低或减少活性或表达。这可以是活性或表达的完全抑制,或部分抑制。抑制可以与对照或标准水平进行比较。抑制可以是1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99,或100%。iii.使用组合物的方法还公开了预防和/或治疗寨卡病毒感染的方法。在一些方式中,所述方法包括向患有寨卡病毒感染或处于发生寨卡病毒感染的风险中的受试者施用有效量的组合物,所述组合物包含有效量的结合寨卡病毒非结构蛋白的化合物。在一些方式中,所述方法包括施用表2的化合物中的至少一种化合物。如本文所用,术语“预防”是指在疾病或病况的临床症状发作之前施用化合物,以防止与疾病或病况相关的异常的身体表现。在寨卡病毒感染的上下文中,术语“预防”是指在寨卡病毒感染的临床症状发作之前施用化合物,以防止与寨卡病毒感染相关的异常的身体表现。如本文所用,术语“治疗”("treatment"和"treating")是指旨在治愈、改善、稳定或预防疾病、病理状况或病症的受试者的医学管控。该术语包括积极治疗,即特异性针对疾病、病理状况或病症的改善的治疗,并且还包括病因治疗,即针对消除相关疾病、病理状况或病症的病因的治疗。此外,该术语包括姑息治疗,即设计用于缓解症状而不是治愈疾病、病理状况或病症的治疗;预防性治疗,即旨在最小化或部分或完全抑制相关疾病、病理状况或病症的发展的治疗;和支持性治疗,即用于补充另一种针对相关疾病、病理状况或病症的改善的特定疗法的治疗。可以理解的是,治疗虽然旨在治愈、改善、稳定或预防疾病、病理状况或病症,但实际上并不需要导致治愈、改善、稳定或预防。可以如本文所述和本领域已知适合于所涉及的疾病、病理状况或病症的那样测量或评估治疗的效果。此类测量和评估可以定性和/或定量的方式进行。因此,例如,疾病、病理状况或病症的特性或特征和/或疾病、病理状况或病症的症状,可以减少到任何效果或任何量。在患有寨卡病毒感染的受试者的上下文中,术语“治疗”("treatment"和"treating")是指旨在治愈、改善或稳定寨卡病毒感染的受试者的医学管控。在处于发生寨卡病毒感染的风险中的受试者的上下文中,术语“治疗”("treatment"和"treating")是指旨在预防寨卡病毒感染的受试者的医学管控。a.受试者在一些方式中,所述方法包括向患有寨卡病毒感染或处于发生寨卡病毒感染的风险中的受试者施用。如果受试者已被诊断患有寨卡病毒感染和/或患有与寨卡病毒感染有关的任何症状,例如包括但不限于发热、皮疹、头痛、关节痛、结膜炎、肌肉痛、严重的神经系统并发症(例如格林-巴利综合征,脑膜脑炎和脊髓炎)、血小板减少症和弥散性血管内凝血连同出血性并发症、肝功能障碍、急性呼吸窘迫综合征、休克和多器官功能障碍综合征,则通常认为受试者患有寨卡病毒感染。如果受试者已旅行至寨卡病毒高发的区域,如果受试者已遭受蚊虫叮咬,如果受试者与感染寨卡病毒的人发生未保护的性行为,如果受试者已由受污染的血液输血,或者如果受试者在实验室或医疗机构中暴露于寨卡病毒,则通常认为受试者处于发生感染寨卡病毒的风险中。如本文所用,“受试者”包括但不限于动物、植物、细菌、病毒、寄生虫和任何其他生物或实体。受试者可以是脊椎动物,更具体地是哺乳动物(例如人、马、猪、兔、狗、绵羊、山羊、非人灵长动物、牛、猫、豚鼠或啮齿动物)、鱼、鸟或爬行动物或两栖动物。受试者可以是无脊椎动物,更具体地是节肢动物(例如,昆虫和甲壳类动物)。所述术语不表示特定的年龄或性别。因此,旨在涵盖成年和新生的受试者,以及胎儿,无论是男性还是女性。患者是指患有疾病或病症的受试者。术语“患者”包括人和兽医受试者。在一方面,本文所述的化合物可以施用于需要改善或缓解公认的医疗状况的受试者,包括人类或动物,所述动物包括但不限于小鼠、狗、猫、马、牛或绵羊等。如本文所用,术语“监测”是指本领域中可以测量活性的任何方法。如本文所用,术语“需要治疗”是指护理人员(例如,在人类的情况下的医师、护士、护士从业者或个人;在动物(包括非人类哺乳动物)的情况下的兽医)做出的受试者需要或将受益于治疗的判断。该判断是基于各种因素做出的,所述各种因素在护理人员专业范围内,但包括以下知识:受试者因所公开化合物可治疗的病况而生病或将要生病。所公开的方法包括基于测量、检测、比较、分析、测定、筛选等,对受试者,疾病,病况,状态等的确定、鉴定、指示、关联、诊断、预后等(可以统称为“鉴定”)。例如,受试者可以被确定为或鉴定为处于寨卡病毒感染的风险中,受试者可以被诊断、确定或鉴定为患有寨卡病毒感染,化合物可以被确定为或鉴定为抑制寨卡病毒蛋白酶的化合物,化合物可以被确定为或鉴定为抑制寨卡病毒感染的化合物等。所公开的方法可以包括例如鉴定处于被寨卡病毒感染的风险中的受试者,或已被诊断为患有寨卡病毒感染的受试者。此类受试者包括健康的,具有免疫能力的或免疫受损的成年人、老年人、儿童和婴儿。在一些方式中,受试者是男性。在一些方式中,受试者是女性。在特定方式中,受试者是孕妇。出于多种原因,此类鉴定是有用的。例如,并且特别地,此类鉴定允许基于所做出的特定鉴定且相关于所做出的特定鉴定而采取特定行动。例如,在特定受试者中诊断特定疾病或病况(并且在其他受试者中没有诊断该疾病或病况)具有鉴定将受益于基于诊断的治疗、行动、行为等的受试者的非常有用的作用。例如,对于所鉴定的受试者中的特定疾病或病况的治疗,显著不同于没有做出这种鉴定(或不考虑鉴定)的所有受试者的治疗。需要或可受益于治疗的受试者将接受治疗,且不需要或不会受益于治疗的受试者将不接受治疗。因此,本文还公开了包括在所公开的鉴定之后并基于所公开的鉴定采取特定行动的方法。例如,公开了包括创建鉴定记录(例如,以物理形式,例如纸、电子或其他形式)的方法。因此,例如,基于所公开的方法创建鉴定记录在物理上且有形地不同于仅进行测量、检测、比较、分析、测定、筛选等。这种记录具有特别的巨大价值和重要性,因为它允许将所述鉴定以有形的形式固定,所述有形的形式可以例如传达给其他人(例如,可以基于所述鉴定对受试者进行治疗、监测、随访、建议等的那些);保留以供后续使用或审查;用作数据以评估受试者的集合、治疗功效,基于不同测量、检测、比较、分析、测定、筛选等的鉴定准确性等等。例如,此类鉴定记录的使用可以例如由与做出所述鉴定记录的个体或实体相同的个体或实体、与做出所述鉴定记录的个体或实体不同的个体或实体,或与做出所述鉴定记录的个体或实体相同的个体或实体和不同的个体或实体的组合做出。所公开的创建记录的方法可以与本文公开的任一种或多种其他方法组合,且特别是与所公开的鉴定方法的任一个或多个步骤组合。作为另一实例,公开了包括基于一个或多个其他鉴定做出一个或多个进一步鉴定的方法。例如,可以基于其他鉴定,来鉴定特定的治疗、监测、随访、建议等。例如,将受试者鉴定为患有具有高水平特定组分或特征的疾病或病况,可以进一步鉴定为可以或应当使用基于或针对高水平组分或特征的疗法治疗的受试者。可以创建此类进一步鉴定的记录(例如,如上所述),并且可以任何合适的方式使用。此类进一步鉴定可以例如直接基于其他鉴定,此类其他鉴定的记录,或组合。此类进一步鉴定可以由例如与做出其他鉴定的个体或实体相同的个体或实体、与做出其他鉴定的个体或实体不同的个体或实体,或与做出其他鉴定的个体或实体相同的个体或实体和不同的个体或实体的组合做出。所公开的做出进一步鉴定的方法可以与本文公开的任一种或多种其他方法组合,且特别是与所公开的鉴定方法的任一个或多个步骤组合。作为另一实例,公开了包括将在任何公开的方法中鉴定的受试者治疗、监测、随访、建议等的方法。还公开了包括将其中已经从任何所公开的方法做出鉴定记录的受试者治疗、监测、随访、建议等的方法。例如,可以基于鉴定和/或基于鉴定记录,来使用特定的治疗、监测、随访、建议等。例如,可以使用基于或针对高水平组分或特征的疗法,来治疗被鉴定为患有具有高水平特定组分或特征的疾病或病况的受试者(和/或已经做出这样的鉴定记录的受试者)。此类治疗、监测、随访、建议等可以例如直接基于鉴定,此类鉴定记录,或组合。例如,此类治疗、监测、随访、建议等可以由与做出鉴定和/或鉴定记录的个体或实体相同的个体或实体、与做出鉴定和/或鉴定记录的个体或实体不同的个体或实体,或与做出鉴定和/或鉴定记录的个体或实体相同的个体或实体和不同的个体或实体的组合进行。所公开的治疗、监测、随访、建议等的方法可以与本文公开的任一种或多种其他方法组合,且特别是与所公开的鉴定方法的任一个或多个步骤组合。b.施用的形式和模式如本文所用,术语“提供”是指将化合物或分子添加至本领域已知物质的任何方式。提供的实例可以包括使用移液器、移液管、注射器、针头、管、枪等。这可以是手动或自动的。它可以包括以任何方式转染或以任何其他提供核酸至皿、细胞、组织、无细胞系统的方式,并且可以是体外或体内的。取决于是否期望局部或全身治疗以及待治疗的区域,可以以多种方式向受试者施用本文所述的化合物和药物组合物。因此,例如,本文所述的化合物或药物组合物可以作为眼科溶液和/或软膏剂施用至眼睛表面。此外,化合物或药物组合物可以经阴道地、经直肠地、鼻内、口服,通过吸入,或胃肠外,例如通过皮内、皮下、肌内、腹膜内、直肠内、动脉内、淋巴管内、静脉内、鞘内和气管内途径,施用至受试者。肠胃外施用,如果使用,通常以注射为特征。可注射剂可以以常规形式制备,作为液体溶液或悬浮液,适用于在注射前溶解或悬浮在液体中的固体形式,或作为乳剂。肠胃外施用的最近修订方法包括使用缓慢释放或持续释放系统,使得维持恒定剂量。参见例如美国专利号3,610,795,其通过引用并入本文。用于肠胃外施用的制剂包括无菌水溶液或非水溶液,悬浮液和乳剂,其还可以含有缓冲剂、稀释剂和其他合适的添加剂。非水溶剂的实例是丙二醇、聚乙二醇、植物油例如橄榄油和可注射有机酯例如油酸乙酯。水性载体包括水、醇/水溶液、乳剂或悬浮液,包括盐水和缓冲介质。肠胃外媒介物包括氯化钠溶液、林格氏右旋糖、右旋糖和氯化钠、乳酸林格氏液或固定油。静脉内媒介物包括流体和营养补充剂、电解质补充剂(诸如基于林格氏右旋糖的那些)等。还可以存在防腐剂和其它添加剂,例如,如抗微生物剂、抗氧化剂、螯合剂和惰性气体等。用于局部施用的制剂可以包括软膏剂、洗剂、乳膏剂、凝胶剂、滴剂、栓剂、喷雾剂、液体和粉剂。常规药学载体,水性、粉末或油性基质,增稠剂等可以是必需的或期望的。用于口服施用的组合物可以包括粉末或颗粒剂、在水或非水介质中的悬浮剂或溶液、胶囊、药囊(sachet)或片剂。增稠剂、调味剂、稀释剂、乳化剂、分散助剂或粘合剂可以是期望的。c.剂量在一些方式中,所述方法包括向患有寨卡病毒感染或处于发生寨卡病毒感染的风险中的受试者施用有效量的组合物,所述组合物包含有效量的抑制寨卡病毒非结构蛋白活性的化合物。使寨卡病毒非结构蛋白活性抑制至少50%的化合物的量可以是从低的约0.1µg/ml、约0.2µg/ml或约0.5µg/ml到高的约100µg/ml、约200µg/ml或约500µg/ml的量。例如,抑制寨卡病毒非结构蛋白活性的化合物的量可以是约0.1µg/ml至约100µg/ml,约0.2µg/ml至约200µg/ml,约0.5µg/ml至约500µg/ml,约0.1µg/ml至约10µg/ml,约0.1µg/ml至约20µg/ml,约0.1µg/ml至约50µg/ml,约0.5µg/ml至约20µg/ml,约0.5µg/ml至约50,约0.5µg/ml至约100µg/ml,约0.5至约200µg/ml,约0.5µg/ml至约500µg/ml,约15µg/ml至约25µg/ml,约15µg/ml至约50,约18µg/ml至约30µg/ml,约18µg/ml至约50µg/ml。在一些方式中,剂量可以是在受试者的一种或多种体液中产生如下浓度的量:所述浓度从低的约0.1μg/ml,约0.2μg/ml或约0.5μg/ml,至高的约100μg/ml,约200μg/ml或约500μg/ml。例如,剂量可以在受试者的一种或多种体液中产生如下浓度的量:约0.1µg/ml至约100µg/ml,约0.2µg/ml至约200µg/ml,约0.5µg/ml至约500µg/ml,约0.1µg/ml至约10µg/ml,约0.1µg/ml至约20µg/ml,约0.1µg/ml至约50µg/ml,约0.5µg/ml至约20µg/ml,约0.5µg/ml至约50,约0.5µg/ml至约100µg/ml,约0.5至约200µg/ml,约0.5µg/ml至约500µg/ml,约15µg/ml至约25µg/ml,约15µg/ml至约50,约18µg/ml至约30µg/ml,约18µg/ml至约50µg/ml。在一些方式中,剂量可以是在受试者的一种或多种体液中产生0.1μg/ml至500µg/ml范围的浓度的量。例如,所述浓度可以:(a)在如下范围内,(b)大约如下范围内,(c)从,或(d)大约从0.1µg/ml、0.15µg/ml、0.2µg/ml、0.25µg/ml、0.3µg/ml、0.35µg/ml、0.4µg/ml、0.45µg/ml、0.5µg/ml、0.55µg/ml、0.6µg/ml、0.65µg/ml、0.7µg/ml、0.75µg/ml、0.8µg/ml、0.85µg/ml、0.9µg/ml、0.95µg/ml、1µg/ml、2µg/ml、3µg/ml、4µg/ml、5µg/ml、6µg/ml、7µg/ml、8µg/ml、9µg/ml、10µg/ml、12µg/ml、14µg/ml、15µg/ml、16µg/ml、18µg/ml、20µg/ml、25µg/ml、30µg/ml、35µg/ml、40µg/ml、45µg/ml、50µg/ml、55µg/ml、60µg/ml、65µg/ml、70µg/ml、75µg/ml、80µg/ml、85µg/ml、90µg/ml、95µg/ml、100µg/ml、110µg/ml、120µg/ml、125µg/ml、130µg/ml、140µg/ml、150µg/ml、160µg/ml、170µg/ml、175µg/ml、180µg/ml、190µg/ml、200µg/ml、220µg/ml、240µg/ml、250µg/ml、260µg/ml、280µg/ml、300µg/ml、320µg/ml、340µg/ml、350µg/ml、360µg/ml、380µg/ml、400µg/ml、420µg/ml、440µg/ml、450µg/ml、460µg/ml、480µg/ml、或500µg/ml中任一者,(a)至,(b)至大约,(c)至,或(d)至大约0.1µg/ml、0.15µg/ml、0.2µg/ml、0.25µg/ml、0.3µg/ml、0.35µg/ml、0.4µg/ml、0.45µg/ml、0.5µg/ml、0.55µg/ml、0.6µg/ml、0.65µg/ml、0.7µg/ml、0.75µg/ml、0.8µg/ml、0.85µg/ml、0.9µg/ml、0.95µg/ml、1µg/ml、2µg/ml、3µg/ml、4µg/ml、5µg/ml、6µg/ml、7µg/ml、8µg/ml、9µg/ml、10µg/ml、12µg/ml、14µg/ml、15µg/ml、16µg/ml、18µg/ml、20µg/ml、25µg/ml、30µg/ml、35µg/ml、40µg/ml、45µg/ml、50µg/ml、55µg/ml、60µg/ml、65µg/ml、70µg/ml、75µg/ml、80µg/ml、85µg/ml、90µg/ml、95µg/ml、100µg/ml、110µg/ml、120µg/ml、125µg/ml、130µg/ml、140µg/ml、150µg/ml、160µg/ml、170µg/ml、175µg/ml、180µg/ml、190µg/ml、200µg/ml、220µg/ml、240µg/ml、250µg/ml、260µg/ml、280µg/ml、300µg/ml、320µg/ml、340µg/ml、350µg/ml、360µg/ml、380µg/ml、400µg/ml、420µg/ml、440µg/ml、450µg/ml、460µg/ml、480µg/ml,或500µg/ml中任一者,所有范围均包括端点。在一些方式中,化合物的剂量是在受试者的一种或多种体液中产生如下浓度的量,所述浓度:(a)可以是,或(b)可以是约500µg/ml、480µg/ml、460µg/ml、450µg/ml、440µg/ml、420µg/ml、400µg/ml、380µg/ml、360µg/ml、350µg/ml、340µg/ml、320µg/ml、300µg/ml、280µg/ml、260µg/ml、250µg/ml、240µg/ml、220µg/ml、200µg/ml、190µg/ml、180µg/ml、175µg/ml、170µg/ml、160µg/ml、150µg/ml、140µg/ml、130µg/ml、125µg/ml、120µg/ml、110µg/ml、100µg/ml、95µg/ml、90µg/ml、85µg/ml、80µg/ml、75µg/ml、70µg/ml、65µg/ml、60µg/ml、55µg/ml、50µg/ml、45µg/ml、40µg/ml、35µg/ml、30µg/ml、25µg/ml、20µg/ml、18µg/ml、16µg/ml、15µg/ml、14µg/ml、12µg/ml、10µg/ml、9µg/ml、8µg/ml、7µg/ml、6µg/ml、5µg/ml、4µg/ml、3µg/ml、2µg/ml、1µg/ml、0.95µg/ml、0.9µg/ml、0.85µg/ml、0.8µg/ml、0.75µg/ml、0.7µg/ml、0.65µg/ml、0.6µg/ml、0.55µg/ml、0.5µg/ml、0.45µg/ml、0.4µg/ml、0.35µg/ml、0.3µg/ml、0.25µg/ml、0.2µg/ml、0.15µg/ml或0.1µg/ml。在一些方式中,浓度可以在0.1µg/ml至10µg/ml的范围。在一些方式中,浓度可以在0.1µg/ml至5µg/ml的范围。在一些方式中,浓度可以在0.1µg/ml至1µg/ml的范围。术语“高”、“较高”、“增加”、“提升”或“升高”是指例如与对照相比,高于基础水平的增加。术语“低”、“较低”、“减少”或“降低”是指例如与对照相比,低于基础水平的减少。如本文提供的,术语“有效量”的化合物是指无毒但足以提供期望结果的量的化合物。如下文将指出的,取决于受试者的物种、年龄和一般状况,所治疗疾病的严重程度,所使用的特定化合物,其施用模式等,所需的确切量将因受试者而异。因此,不可能指定确切的“有效量”。但是,本领域普通技术人员仅使用常规实验即可确定适当的有效量。在用于预防寨卡病毒感染的化合物和组合物的上下文中,如本文提供的,术语“有效量”的化合物是指无毒但足以预防寨卡病毒感染的量的化合物。在用于治疗患有寨卡病毒感染的受试者的化合物和组合物的上下文中,如本文提供的,术语“有效量”的化合物是指无毒但足以治疗寨卡病毒感染的量的化合物。本文描述的化合物的剂量或量足够大以在发生递送的方法中产生期望的效果。剂量不应太大而引起不良的副作用,例如不希望的交叉反应,过敏反应等。通常,剂量将随受试者的年龄、状况、性别和疾病的程度而变化,并且可以由本领域技术人员确定。剂量可由个体医师基于所涉及受试者的临床状况来调整。可以改变剂量、剂量时间表和施用途径。可以通过评估病史、体征、症状和客观实验室检查的特定方面来确定根据本文所述方法施用特定剂量的化合物或组合物的功效,其中已知所述特定方面可用于评估需要治疗寨卡病毒感染或其他疾病和/或病况的受试者的状态。这些体征、症状和客观实验室检查将取决于所治疗或预防的特定疾病或病况而不同,如治疗此类患者的任何临床医生或在本领域进行实验的研究人员所知晓的。例如,如果基于与适当对照组的比较和/或疾病在一般人群或特定个体中的正常进展的知识:(1)显示受试者的身体状况得到改善(例如,肿瘤已部分或全部消退),(2)显示疾病或病况的进展稳定、减慢或逆转,或(3)减少或消除对用于治疗疾病或病况的其他药物的需求,则特定的治疗方案将被视为有效的。“药学上可接受的”是指不是生物学上或其他方面不期望的材料,即,所述材料可以与选择的化合物一起施用于受试者,而不引起任何不希望的生物学作用或以有害的方式与包含它的药物组合物的任何其他组分相互作用。应当理解,除非另有说明,否则所公开的方法和组合物不限于特定的合成方法,特定的分析技术或特定的试剂,并且因此可以变化。还应理解,本文所用的术语仅用于描述具体实施方案的目的,而并非旨在用于进行限制。实施例材料与方法在计算机芯片上对化学文库和分子对接的基于结构的虚拟筛选通过使用ambertools(amber2017,universityofcalifornia,sanfrancisco)(case等人,2017),将存放在drugbankv5.0.1中的所有化合物设置用于对接模拟。使用zikvns2b-ns3蛋白酶的晶体结构(蛋白质数据库(pdb)代码5lc0)构建蛋白质模型系统(lei等人,2016)。通过去除盐并中和其质子化状态,通过mmff94力场计算部分电荷,添加氢原子并使能量最小化(默认参数)(halgren,1995),来计算分子参数。使用zikvns2b-ns3蛋白酶的晶体结构(蛋白质数据库(pdb)代码5lc0)构建蛋白质模型系统(lei等人,2016)。在早期阶段,使用蛋白质制备向导(proteinpreparationwizard)模块分配键序(bondorder),添加氢,并包含帽末端,如maestro(schrödingerrelease2016-4:maestro,schrödinger,llc,newyork,usa)中所实施的(sastry等人,2013)。随后,使用propka3.1定义所有侧链的质子化状态。最后,在amber99力场方案内分配所有原子上的部分电荷,如ambertools(amber2017,universityofcalifornia,sanfrancisco)中实施的(case等人,2017)。使用leadfinder软件v1.1.16进行对接模拟(stroganov等人,2008)。所有对接参数均设置为默认值用于计算。保留对于每种化合物产生的排序最高的对接评分,用于进一步分析。分子动力学模拟进行分子动力学模拟以预测新生霉素-zikvns2b-ns3复合物的稳定性。使用与opls-2005力场组合的desmond(desmondmoleculardynamicssystem,d.e.shawresearch,和maestro-desmondinteroperabilitytools,schrödinger,llc,newyork,usa)进行计算(shivakumar等人,2010)。病毒株和滴定从最近南美流行病患者获得的zikv的临床分离物(波多黎各毒株prvabc59),由美国疾病控制与预防中心的brandyrussell和barbarajohnson惠赠。对病毒进行培养和滴定,如我们此前所述并稍作修改(chan等人,2016b;zhou等人,2014)。在补充1%胎牛血清(fbs,gibcotm,lifetechnologiescorporation,massachusetts,usa)和100单位/ml青霉素加100μg/ml链霉素的最低必需培养基(mem)中,通过在vero细胞(atcc)中的三次额外传代扩增病毒以制备病毒工作储备液(4×10650%组织培养感染剂量(tcid50)/ml)。对于病毒滴定,将zikv的等分试样应用于96孔板中的汇合的vero细胞上,用于tcid50测定,如此前所述并稍作修改(chan等人,2016b;zhou等人,2014)。简言之,将zikv的10倍系列稀释液一式四份接种在vero细胞单层中,并在补充青霉素/链霉素的mem和1%fbs中培养。观察板的cpe6天。用reed和münch终点法计算病毒滴度。一个tcid50被解释为在50%的接种孔中引起细胞病变作用的病毒量。细胞系和药物化合物vero和huh-7细胞系分别从美国典型培养物保藏中心(americantypeculturecollection)和日本冈山大学的jcrb细胞库获得,如前所述(chan等人,2013a;chan等人,2016b)。使用抑肽酶(sigma-aldrich,missouri,usa)、醋酸去氨加压素(ferringpharmaceuticals,saintprex,switzerland)、洛匹那韦-利托那韦(abbottlaboratories,illinois,usa)、孟鲁司特(merck&co.,inc.,newjersey,usa)、新生霉素钠(sigma-aldrichchemiegmbh,steinheim,germany)、醋酸奥曲肽(novartis,basel,switzerland)、利福平(gruppolepetitsrl,milan,italy)、西罗莫司(pfizer,newyorkcity,usa)和他克莫司(astellaspharma,tokyo,japan),用于体外和/或体内研究。细胞活力测定和cpe抑制测定通过噻唑蓝溴化四唑(mtt)测定确定所选药物在vero和huh-7细胞中的50%有效的细胞毒性浓度(cc50),如我们此前所述并稍作修改(chan等人,2017a;yuan等人,2016)。简言之,将vero或huh-7细胞(4×104个细胞/孔)与不同浓度的单个药物一起孵育24小时,然后添加10μl/孔的5mg/mlmtt底物。如上所述,将单层孵育4小时(避光)。最后,添加100μl的10%十二烷基硫酸钠(sds)和0.01mhcl,并在37℃与5%co2下进一步孵育过夜。在od570读取活性,且参考波长在od640。为了确认新生霉素的抗病毒活性,还进行了基于mtt的cpe抑制测定,如此前所述并稍作修改(chan等人,2017a)。简言之,将药物化合物用无血清mem稀释,并添加至96孔培养板中汇合的vero细胞(4×104细胞/孔),一式三份,在37℃进行2小时。孵育后,除去含药物的培养基,并向每个孔中添加zikv(感染复数,moi=0.0002)以及新鲜的药物-化合物培养基。在37℃吸附1小时后,除去病毒-化合物混合物,并且将细胞用mem洗涤两次,以除去未结合的病毒。随后,向细胞添加含抗病毒化合物的培养基,在5%co2加湿环境中于37℃下进一步孵育6天。通过倒置光学显微镜检查cpe(chan等人,2016b)。此后,如上所述,依次添加mtt底物和sds-hcl。使用excel插件ed50v10中的sigmaplot(spss)计算半数最大抑制浓度(ic50)和cc50。病毒载量减少测定进行病毒载量减少测定和噬斑减少测定以评估抗病毒活性。简言之,使用不同浓度的药物或二甲基亚砜(dmso)对照处理感染zikv的vero细胞(moi=0.05),在感染后48小时收集细胞培养上清液(vero和huh-7),然后进行总核酸提取和定量逆转录-pcr(qrt-pcr),如此前所述并稍作修改(chan等人,2013;chan等人,2016b;chan等人,2017a)。使用excel插件ed50v10中的sigmaplot(spss)计算ic50。病毒载量减少测定实验一式三份进行,并重复两次以确认。噬斑减少测定进行噬斑减少测定,如此前所述并稍作修改(chan等,2017a)。简言之,在进行测定的前一天,将vero细胞以2×105细胞/孔接种在24孔组织培养板中。孵育24小时后,将20-40噬斑形成单位(pfu)的zikv添加到具有有或没有添加药物化合物的细胞单层,并将平板在37℃5%co2中进一步孵育1h,然后通过抽吸培养基并用mem洗涤一次,除去未结合的病毒颗粒。然后,将单层用含有mem中1%低熔点琼脂糖(cambrexcorporation,newjersey,usa)和适当浓度的药物化合物的培养基覆盖,倒置并如上所述再孵育96小时。然后,将孔用10%甲醛(bdh,merck,darmstadt,germany)固定过夜。除去琼脂糖塞后,将单层用0.7%的结晶紫(bdh,merck)染色并计数噬斑。对于每种药物化合物浓度,确定相对于对照(即,不添加化合物)孔的噬斑抑制百分比。使用excel插件ed50v10中的sigmaplot(spss)计算ic50。噬斑减少测定实验一式三份进行,并重复两次以确认。药物添加时间测定对新生霉素进行药物添加时间测定,以确定药物干扰了病毒周期的哪个阶段(chan等人,2017a;kato等人,2016)。简言之,将vero细胞接种在24孔板中(2×105细胞/孔)。用zikv(moi=2)接种细胞,且然后孵育1小时以使病毒内化。然后,去除病毒接种物,并用pbs洗涤细胞两次。在感染后0、3、6、9、12和14小时,将100µg/ml新生霉素加入感染的细胞中,然后在37℃5%co2中孵育直至感染后18小时。对于“-1至0hpi”的时间点,在感染前1小时添加100µg/ml新生霉素,并在0hpi去除,然后进行zikv接种和细胞孵育直至18hpi。对于“0-1hpi”的时间点,在0hpi将100μg/ml的新生霉素与zikv接种一起添加,然后在1hpi去除药物并将细胞孵育直至18hpi。在18hpi,收集每个时间点实验的细胞培养上清液,以使用qrt-pcr测量病毒载量。包括二甲基亚砜(0.5%,0µg/ml新生霉素)作为阴性对照。实验一式三份进行,并重复两次以确认。蛋白质产生和纯化产生重组zikvns2b-ns3蛋白酶,如此前所述并稍作修改(lei等人,2016)。简言之,将zikv-ns2b(残基49-95)和zikv-ns3(残基1-170)基因的编码区融合并克隆到pet-28b(+)表达载体中(novagen,merck&co.,newjersey,usa)。将质粒转化到大肠杆菌菌株bl21(de3)中,并在2×yt培养基中过表达。当培养物的od600达到0.8时,通过在20℃加入0.5mmiptg过夜诱导zikvns2b-ns3蛋白酶基因的过表达。通过his-标签亲和色谱从可溶性裂解物纯化zikvns2b-ns3蛋白酶,并在10mmtris-hcl,20%甘油,1mm3-[((3-胆酰胺基丙基)二甲基铵]-1-丙磺酸盐(chaps),ph8.5中透析过夜。通过vivaspin20离心浓缩器(gehealthcare,littlechalfont,uk)浓缩蛋白酶,并在-80℃下储存。使用抗his-标签抗体通过western印迹检测纯化的蛋白质。使用牛血清白蛋白作为标准品,通过bradford蛋白质测定试剂盒(bio-radlaboratories,california,usa)测定蛋白质的浓度。基于荧光的蛋白酶抑制测定产生重组zikvns2b-ns3蛋白酶,如此前所述并稍作修改(lei等人,2016)。为了检测zikvns2b-ns3蛋白酶的活性,在96孔黑色微孔板(greinerbio-one,kremsmünster,germany)中进行基于荧光的酶促测定。使用7-氨基-4-甲基香豆素底物bz-nle-lys-lys-arg-amc(lei等人,2016)。使用victor™x3多标记酶标仪(perkinelmer,massachusetts,usa),在360nm激发下,在460nm测量来自释放的amc的荧光信号。为了检测每种药物是否抑制zikvns2b-ns3蛋白酶活性,在测定中固定5nmzikvns2b-ns3蛋白酶和10µm底物。zikvns2b-ns3蛋白酶与不同浓度的化合物在37℃孵育10分钟。在50μl反应缓冲液中制备蛋白酶和单个药物的混合物,并分散到每个孔中,然后在50μl缓冲液中添加20μm底物以启动裂解。10分钟后,测量荧光强度。反应和稀释缓冲液由10mmtris-hcl,20%甘油,1mmchaps,5%dmso,ph8.5组成。包括dmso(5%)作为阴性对照。小鼠模型和新生霉素治疗的体内评估在我们最近使用地塞米松-免疫抑制小鼠和播散的zikv感染建立的zikv感染的动物模型中,评估新生霉素的体内治疗效果(chan等人,2016c)。获得了香港大学教学与研究中活体动物使用委员会的批准。简言之,将6-8周龄的雄性balb/c小鼠随机分组,并给予病毒接种、地塞米松和新生霉素治疗的不同方案(表1)。使用磷酸盐缓冲盐水(pbs)将病毒储备液稀释至期望浓度,并且对接种物进行反滴定以验证给予的剂量。在病毒接种当天,通过腹膜内途径向氯胺酮(100mg/kg)和甲苯噻嗪(10mg/kg)麻醉下的小鼠接种200μlpbs中等同于6×106tcid50(3.24×106噬斑形成单位)的剂量的病毒。阴性对照组中的小鼠被注射相同体积的pbs。每天三次监测小鼠的疾病的临床体征,并在每次观察时分配数值评分,如前所述(chan等人,2016c)。在5dpi,每组处死三只小鼠用于病毒学、组织学和免疫组织化学分析。在14dpi处死剩余小鼠,或当体重减轻≥20%或体重减轻≥10%且临床体征≥1时,实施安乐死。在尸检时收集主要器官的样品,用于病毒学,组织学和免疫组织化学分析,如我们此前所述(chan等人,2016c.chan等人,2017b)。还收集了血液样品用于rna提取和qrt-pcr分析。表1:在本研究中接受病毒接种、地塞米松和新生霉素治疗的不同方案的小鼠组a。a在所有组中均使用6-8周龄的balb/c雄性小鼠,因为在雄性小鼠中观察到比雌性小鼠更严重的寨卡病毒疾病,如我们此前在这种已建立的动物模型中报道的(chraibis.等人,jclinvirol83:61-62;2016)。b组1的小鼠由于体重减轻≥10%且临床症状≥1,在12dpi被安乐死。缩写:dpi,接种后的天数;ip,腹膜内;m,雄性;sc,皮下。统计分析所有统计分析均使用graphpadprism软件(graphpadsoftware,inc)进行。通过对数秩检验分析kaplan-meier生存曲线。使用student氏t-检验确定单个治疗组之间体重减轻和病毒滴度中的显著差异,如此前报道的(chan等人,2016c)。p-值<0.05被认为是统计学显著的。实施例1:在计算机芯片上对化学文库和分子对接的基于结构的虚拟筛选。通过使用分子操作环境v2008(molecularoperatingenviromentv2008,moe,chemicalcomputinggroupinc.,montreal,canada),将存放在drugbankv5.0.1中的所有化合物设置用于对接模拟(wishart等人,2006)。使用zikv-ns2b-ns3蛋白酶的晶体结构(蛋白质数据库(pdb)代码5lc0)构建蛋白质模型系统(lei等人,2016)。通过去除盐并中和其质子化状态,通过mmff94力场计算部分电荷,添加氢原子并使能量最小化(默认参数)(halgren,1995),来计算分子参数。使用zikv-ns2b-ns3蛋白酶的晶体结构(蛋白质数据库(pdb)代码5lc0)构建蛋白质模型系统(lei等人,2016)。在早期阶段,使用蛋白质制备向导模块分配键序,添加氢,并包含帽末端,如maestro(schrödingerrelease2016-4:maestro,schrödinger,llc,newyork,usa)中所实施的(sastry等人,2013)。随后,使用propka3.1定义所有侧链的质子化状态。最后,在amber99力场方案内分配所有原子的部分电荷,如moev2008中实施的。使用leadfinder软件v1.1.16进行对接模拟(stroganov等人,2008)。所有对接参数均设置为默认值用于计算。保留对于每种化合物产生的排序最高的对接评分,用于进一步分析。为了鉴定zikv-ns2b-ns3蛋白酶的潜在抑制剂,使用leadfinder软件筛选来自drugbank数据库的总共8227个条目,其中使用zikv-ns2b-ns3蛋白酶的晶体结构作为程序的输入(lei等人,2016)(图1)。前100种初步命中化合物按照其与zikv-ns2b-ns3蛋白酶的预测结合亲和力排序(表2)。对具有8277种药物化合物的化学文库进行计算机芯片上的虚拟筛选。在具有与zikv-ns2b-ns3蛋白酶的最高预测结合亲和力的前100种初步命中化合物中,选择了8种属于不同药物类别的临床批准药物,以验证抗zikv-ns2b-ns3蛋白酶的活性。未选择其他药物化合物,因为它们大多数不是临床批准的药物,不具有众所周知的药代动力学和药效学特性,和/或与这8种药物属于相同的药物类别(表2)。其中,5种(62.5%)在基于荧光的酶促测定中被证明具有抗zikv-ns2b-ns3蛋白酶活性。然后,在这5种药物中,选择对于在妊娠期和/或患有严重zikv相关并发症的患者中具有高临床使用潜力的3种进行进一步评估。然后,通过病毒载量减少,细胞病变作用抑制和/或噬斑减少测定评估这3种药物(新生霉素、洛匹那韦-利托那韦和利福平)的抗zikv活性。新生霉素和洛匹那韦-利托那韦在病毒载量减少测定中展示出抗zikv活性。基于其比洛匹那韦-利托那韦更高的选择性指数,随后选择新生霉素在小鼠模型中进行进一步评估。表2:通过leadfinder软件,以zikv-ns2b-ns3蛋白酶为靶标鉴定的前100种初步drugbank命中物。drugbankid药物化合物名称db00035去氨加压素db00093加压素db00104奥曲肽db00183五肽胃泌素db00309长春地辛db00314卷曲霉素db00416碘化二甲箭毒db00471孟鲁司特db00503利托那韦db00520卡泊芬净db00565苯磺酸顺阿曲库胺db00570长春碱db00646制霉菌素db00732苯磺酸阿曲库铵db00864他克莫司db00877西罗莫司db00947氟维司群db00954地红霉素db01045利福平db01051新生霉素db01135多库氯铵db01199筒箭毒碱db01201利福喷丁db01220利福昔明db01226米库氯铵db01229紫杉醇db01232沙奎那韦db01248多西他赛db01249碘克沙醇db01282卡贝缩宫素db01338哌库铵db01599普罗布考db01663λ-双(2,2'-联吡啶)-(5-甲基-2-2'-联吡啶)-c9-金刚烷钌(ii)db01721茚地那韦药物的类似物db01747粪生素db01934n-甲基-n-(10-甲基十一烷酰基)-d-丝氨酰-l-丙氨酰-n~1~-[(7s,10s,13s)-13-羧基-3,18-二羟基-10-甲基-8,11-二氧代-9,12-二氮杂三环[13.3.1.1~2,6~]二十碳-1(19),2(20),3,5,15,17-己烯-7-基]-n~1~-甲基甘氨酰胺db019844-[2-(3-苄氧基羰基氨基-4-环己基-1-羟基-2-氧代-丁基氨基)-5-胍基-戊酰基氨基]-4-(1-羧基-2-环己基-乙基氨甲酰)-丁酸db02003δ-双(2,2'-联吡啶)咪唑钌(ii)db02092亚油酸胆固醇酯db02136头孢菌素类似物db021699,10-脱表硫代-9,10-二脱氢棘皮海绵素db022263,8-二氨基-6-苯基-5-[6-[1-[2-[(1,2,3,4-四氢-9-吖啶基)氨基]乙基]-1h-1,2,3-三唑-4-基]己基]-菲啶鎓db022593-(3,5-二溴-4-羟基-苯甲酰)-2-乙基-苯基呋喃-6-磺酸(4-氨磺酰基-苯基)-酰胺db02378mmi-175db02460氢咕啉酸db02477[环己基乙基]-[[[[4-[2-甲基-1-咪唑基-丁基]苯基]乙酰基]-丝氨酰]-赖氨酰]-胺db02555sp4160db025815-[2,3-二氯-4-(5-{1-[2-(2-胍基-4-甲基-戊酰基氨基)-乙酰基]-哌啶-4-基}-1-甲基-1h-吡唑-3-基)-苯氧基甲基]-呋喃-2-甲酸db02638特利加压素db02702xv638db02704(2r,3r,4r,5r)-3,4-二羟基-n,n'-双[(1s,2r)-2-羟基-2,3-二氢-1h-茚-1-基]-2,5-双(2-苯基乙基)己二胺db03021ulapualideadb03063n-(1-苄基-3-{[3-(1,3-二氧代-1,3-二氢-异吲哚-2-基)-丙酰基]-[2-(六氢-苯并[1,3]二氧杂环戊烯-5-基)-乙基]-氨基}-2-羟基-丙基)-4-苄氧基-3,5-二甲氧基-苯甲酰胺db03208β-1,2,3,4,6-五-o-没食子酰基-d-吡喃葡萄糖db032322-[(2e,6e,10e,14e,18e,22e,26e)-3,7,11,15,19,23,27,31-八甲基三十二烷-2,6,10,14,18,22,26,30-八烯基]苯酚db032764-[(10s,14s,18s)-18-(2-氨基-2-氧乙基)-14-(1-萘甲基)-8,17,20-三氧代-7,16,19-三氮杂螺[5.14]二十碳-11-烯-10-基]苯基膦酸db033113-(3,5-二溴-4-羟基-苯甲酰)-2-乙基-苯并呋喃-6-磺酸[4-(噻唑-2-基氨磺酰基)-苯基]-酰胺db03395依那吉仑db03492λ-双(2,2'-联吡啶)咪唑锇(ii)db03616kabiramidecdb03621l-709,587db036482-{n'-[2-(5-氨基-1-苯基氨甲酰-戊基氨甲酰)-己基]-肼基甲基}-己酸(5-氨基-1-苯基氨甲酰-戊基)-酰胺db03850茉莉酰胺adb03871λ-双(2,2'-联吡啶)咪唑钌(ii)db03875δ-双(2,2'-联吡啶)-(5-甲基-2-2'-联吡啶)-c9-金刚烷钌(ii)db03932lfa703db03933c-1027芳香化发色团db040422-[4-(羟基-甲氧基-甲基)-苄基]-7-(4-羟基甲基-苄基)-1,1-二氧代-3,6-双-苯氧甲基-1λ6-[1,2,7]噻二氮杂环庚烷-4,5-二醇db04124奥迪霉素db04220利福霉素cgp4832db04269cyclotheonamideadb04348牛磺胆酸db04408ncs-发色团db04547bea409db04550δ-双(2,2'-联吡啶)咪唑锇(ii)db046062-[2-乙磺酰氨基-3-(5-丙氧基-1h-吲哚-3-基)-丙酰氨基]-戊二酸5-酰胺1-(4-氨基甲酰氨基-苄酰胺)db04629海兔双内酯adb04697反式-4-(胍基甲基)-环己烷-l-基-d-3-环己基丙氨酰-l-氮杂环丁烷-2-基-d-酪氨酰-l-高精氨酰胺db04711胆影酸db04738motuporindb04741粘噻唑db04748肟基芳基磺酰胺db04774reidispongiolideadb04775reidispongiolidecdb04785链霉素db04869olcegepantdb04877老刺木胺db04894伐普肽(vapreotide)db04972canfosfamidedb05128氨基康定db05296莫特沙芬镥db05297dha-紫杉醇db05399agi-1067db05403cep-1347db05428莫特沙芬钆db05434abt-510db05465mln-518db05868biln2061db06287替西罗莫司db06290西美普韦(simeprevir)db06419塞红霉素db06439泰洛沙泊db06636艾沙康唑鎓db06663帕瑞肽db06695达比加群酯db06749人参皂甙rb1db06750人参皂甙rg1db06772卡巴他赛db06827紫霉素db07165n-(5-氯-苯并[b]噻吩-3-基甲基)-2-[6-氯-氧代-3-(2-吡啶-2-基-乙基氨基)-2h-吡嗪-1-基]-乙酰胺db071695r-(3,4-二氯苯基甲基)-3-(2-噻吩磺酰氨基)-4-氧代-2-硫代噻唑烷db08823多杀菌素db08834牛磺熊去氧胆酸db08871艾日布林db08993恩维霉素db09065可比司他db09102达卡他韦db09134碘佛醇db09204阿罗洛尔db09270辅酶q10db09296奥比他韦db09297帕瑞他韦db09313碘克沙酸db11575格佐普韦(grazoprevir)db11581维纳妥拉实施例2:属于不同药物类别的初步命中化合物的zikv-ns2b-ns3蛋白酶抑制的验证。为了验证初步命中化合物是否抑制zikv-ns2b-ns3蛋白酶活性,我们选择了8种属于不同药物类别的临床批准的药物(即醋酸去氨加压素、洛匹那韦-利托那韦、孟鲁司特、新生霉素、醋酸奥曲肽、利福平、西罗莫司、他克莫司)进行进一步评估。未选择其他药物化合物,因为它们大多数不是临床批准的药物,不具有众所周知的药代动力学和药效学特性,和/或与这8种药物属于相同的药物类别。我们进行了基于荧光的蛋白酶抑制测定,所述测定在存在或不存在药物的情况下记录荧光信号。为了检测纯化的zikv-ns2b-ns3蛋白的酶促活性,我们首先使用四种浓度的蛋白酶底物(0.1μm,1μm,10μm和100μm)进行比较,并观察到向底物添加蛋白质后荧光强度的剂量依赖性增加。图2a显示了蛋白质剂量依赖性荧光强度,表明所述蛋白质对荧光底物(bz-nle-lys-lys-arg-amc)的裂解。出于优化测定目的,测试了四种浓度的底物(100µm,10µm,1µm和0.1µm)。包括与100µm底物一起孵育的pet32a(+)-空白蛋白,作为模拟纯化酶对照。结果显示为与基线水平相比添加zikv-ns2n-ns3蛋白后荧光强度的变化倍数。肽底物的裂解释放出荧光信号,其在添加50nm蛋白时达到平台期。与基线对照相比,在输入10μm和100μm底物的情况下,检测到高至25倍的荧光强度增加。这些结果表明,纯化的zikv-ns2b-ns3蛋白保持蛋白酶活性。为了检测药物对zikv-ns2b-ns3蛋白酶活性的抑制作用,我们使用抑肽酶作为阳性对照,测量了在存在一系列的不同浓度的药物的情况下的荧光信号(图2b)(phoo等人,2016)。在固定浓度的10μm底物和5nmzikv-ns2b-ns3蛋白酶下,我们观察到5/8种药物(62.5%)的随浓度升高的蛋白酶活性的剂量依赖性降低,包括新生霉素(图2c)、醋酸去氨加压素、醋酸奥曲肽、洛匹那韦-利托那韦和利福平(图3a-3g和表3)。在图2b和2c中,展示了渐增浓度的抑肽酶(阳性对照)和新生霉素阻断了蛋白酶的裂解活性,如通过荧光强度值的降低而测量的。底物和zikv-ns2b-ns3蛋白酶的浓度分别固定为10μm和5nm。我们的结果证实了这些药物抑制zikv-ns2b-ns3蛋白酶活性,并且计算机芯片上虚拟筛选可用于预测潜在的zikv-ns2b-ns3蛋白酶抑制剂。表3:每种药物针对zikvns2b-ns3蛋白酶活性的ic50的汇总。“-”表示没有抑制。ic50,半数最大抑制浓度(蛋白酶活性降低50%时的药物浓度)。实施例3:所选zikv-ns2b-ns3蛋白酶抑制剂的体外抗zikv活性的验证。在经验证的zikv-ns2b-ns3蛋白酶抑制剂中,我们进一步选择了三种具有良好副作用特征的临床批准药物,其可潜在用于妊娠患者和/或患有严重zikv-相关并发症的患者中(新生霉素,洛匹那韦-利托那韦和利福平),进行体外抗zikv活性测试。在可以稳健支持zikv复制的两种不同细胞系(vero和huh-7细胞)中评估药物的细胞毒性和抗病毒效力(chan等人,2016b)。新生霉素、洛匹那韦-利托那韦和利福平在vero细胞中的50%有效细胞毒性浓度(cc50)分别为850.50μg/ml、30.00μg/ml和400.00μg/ml,而在huh-7细胞中分别为1103.18μg/ml、32.12µg/ml和400.00µg/ml。在病毒载量减少测定中,在新生霉素处理(半数最大抑制浓度(ic50)在vero细胞中为26.12±0.33μg/ml,且在huh-7细胞中为38.14±4.53μg/ml)和洛匹那韦-利托那韦处理(ic50在vero细胞中为4.78±0.41μg/ml,且在huh-7细胞中为3.31±0.36μg/ml)的培养上清液中观察到病毒滴度的剂量依赖性降低(图4a,4b,5a和5b),但在利福平处理的培养上清液中则没有观察到(数据未显示)。在图4a中,在zikv接种(0.05moi)后48小时,通过定量逆转录聚合酶链式反应(qrt-pcr)定量使用新生霉素的vero细胞中的zikv病毒载量减少。在图4b中,在zikv接种(0.05moi)后48小时,通过qrt-pcr定量使用新生霉素的huh-7细胞中的zikv病毒载量减少。平均病毒滴度在新生霉素≥25.00μg/ml和洛匹那韦-利托那韦≥50.00μg/ml时显著降低(p<0.05)。鉴于新生霉素(28.92-32.56)比洛匹那韦-利托那韦(6.28-9.70)更高的选择性指数(cc50/ic50),我们进一步验证了新生霉素在细胞病变作用(cpe)抑制和噬斑减少测定中的抗zikv活性。在cpe抑制测定中,新生霉素保护vero细胞免于发展zikv诱导的cpe(ic50=53.26±2.48µg/ml)。在噬斑减少测定中,新生霉素在浓度≥50.00µg/ml时实现100%的噬斑减少(ic50=15.21±0.14µg/ml)(图4c)。通过噬斑减少测定确定的新生霉素的半数最大抑制浓度(ic50)显示在图4c中。实施例4:使用新生霉素的治疗改善了具有播散性zikv感染的地塞米松免疫抑制的小鼠的临床结局。从计算机芯片上和体外发现出发,我们进一步评估了新生霉素在我们最近建立的具有播散性zikv感染的地塞米松免疫抑制小鼠的动物模型中的体内治疗作用(chan等人,2016c)。从感染后(dpi)1-13天,我们使用皮下新生霉素100mg/kgq12h治疗地塞米松免疫抑制的小鼠,因为它们很好地耐受该剂量,而没有发生临床症状,并且具有<5%的体重损失(图6a)。图6a显示,在zikv接种(0.05moi)后48小时,通过定量逆转录聚合酶链式反应(qrt-pcr)定量使用新生霉素的vero细胞中的zikv病毒载量减少。图6b显示,在zikv接种(0.05moi)后48小时,通过qrt-pcr定量使用新生霉素的huh-7细胞中的zikv病毒载量减少。图6c显示,通过噬斑减少测定确定的新生霉素的半数最大抑制浓度(ic50)。所有实验一式三份进行,并重复两次以确认。*表示p<0.05,而***表示p<0.0001(通过student氏t检验与dmso对照组相比)。数据表示为平均值±平均值的标准误差(误差线)。在治疗6-8天后,较高剂量(150mg/kgq12h或200mg/kgq12h)的新生霉素在小鼠中导致嗜睡,进食不良和体重损失>10%的发生,并且因此没有在我们的体内治疗研究中使用。与我们之前的发现一致,地塞米松停药(10dpi)后不久,具有地塞米松免疫抑制和zikv接种的对照小鼠发生≥10%体重损失和高临床评分,这就需要在12dpi时对所有小鼠的安乐死(图6a-6c)。还评估了新生霉素对zikv感染的治疗效果。与未治疗的对照组小鼠(其发生播散性zikv感染且在5dpi收集的器官组织(大脑,睾丸和肾脏)中具有大量zikvns1抗原表达,并且在10dpi地塞米松停药后不久,发生临床恶化和多器官炎症,需要在12dpi安乐死)相比,用新生霉素治疗的小鼠在5dpi收集的其器官组织中具有极少的zikvns1抗原表达,而在12dpi地塞米松停药后几乎没有炎性浸润。在感染的早期(5dpi)和晚期(14dpi)期间,血液和主要器官组织(包括睾丸和肾脏,这对zikv的传播至关重要)中的平均病毒载量均显著减少。尽管新生霉素通过非发炎的脑膜渗透进入中枢神经系统可能受限,但病毒血症的早期控制和药物通过可能发炎的脑膜的渗透可能有助于新生霉素治疗的小鼠的改善的临床结局。对于免疫组织化学分析,每个器官都完全包埋在一个石蜡块中,且每块检查在每个器官最大直径的一个全面石蜡切片。在具有zikv接种且没有新生霉素治疗的地塞米松免疫抑制的小鼠的脑、睾丸和肾脏中,可见明显的炎性浸润和变形的组织结构。具有zikv接种和新生霉素治疗的小鼠和具有模拟感染和新生霉素治疗的对照小鼠,没有或具有轻度炎性浸润,并保留组织结构。这些小鼠的主要器官组织的免疫组织化学(5dpi)和苏木精和曙红(h&e)(12dpi)染色分别显示了丰富的zikv-ns1抗原表达和明显的炎性浸润物。相比之下,所有具有zikv接种的新生霉素治疗、地塞米松免疫抑制的小鼠均存活,且在地塞米松停药后发生≤8%的体重损失和极少的临床症状,并在13-14dpi时逐渐恢复(存活率,新生霉素治疗的小鼠100%对比未经治疗的对照小鼠0%,p<0.05)。与临床参数相印证,通过免疫组织化学染色在器官(5dpi)中很少检测到zikv-ns1抗原表达,并且在这些经新生霉素治疗的小鼠的h&e染色组织(14dpi)中看到极少的炎性浸润物。在具有地塞米松免疫抑制和新生霉素治疗的模拟感染对照小鼠的器官组织中的免疫组织化学染色中没有zikv-ns1抗原表达,且在h&e染色中没有炎性浸润物,表明这些变化并非由于药物诱导的作用。图7a和7b显示,在zikv接种后5天(来自两个独立实验,每组n=3)(图7a)和zikv接种后14天(来自两个独立实验,每组n=5)(图7b),具有新生霉素治疗的小鼠与未治疗的小鼠相比,具有减少的zikv血液和组织病毒载量。通过qrt-pcr测定小鼠的血液和组织中的zikvrna拷贝数,并通过β-肌动蛋白归一化。在5dpi和14dpi时,新生霉素治疗的小鼠的血液和大多数主要器官组织的平均病毒载量显著低于未治疗的对照小鼠的那些(具有≥2-log的减少)(图7a和7b)。实施例5:新生霉素抑制zikv复制周期的进入后事件。为了研究被新生霉素中断的zikv复制周期的阶段,我们通过如下进行药物添加时间测定:在病毒复制周期期间的不同时间点将感染zikv的细胞暴露于新生霉素,然后在接种后18小时(hpi)测量病毒滴度。以感染复数(moi)为2的zikv感染vero细胞,在病毒复制阶段(0、3、6、9或12hpi)期间添加新生霉素时,在12小时内病毒载量显著降低(p<0.05)(图8)。当在病毒吸附阶段(0-1hpi)或在释放阶段(14hpi)添加新生霉素时,没有观察到显著的抑制活性。当用新生霉素预处理vero细胞(-1至0hpi)时,zikv附着也没有受影响。由于先前已确定单个zikv生命周期的持续时间为12-14h左右,其中细胞内病毒rna产生的启动在~10-12hpi发生,且在12hpi后组装并释放后代病毒粒子(zmurko等人,2016年;chan等人,2017a),我们的药物添加时间结果表明,新生霉素干扰在zikv复制周期期间,在病毒内化后和出芽前的阶段内的进入后事件。实施例6:预测新生霉素与zikv-ns2b-ns3蛋白酶的关键酶促位点相互作用。为了更好地表征新生霉素与zikv-ns2b-ns3蛋白酶之间的结构相互作用,进行了分子对接以预测新生霉素在zikv-ns2b-ns3蛋白内的结合位点。如图9a中所示,通过残基met51*(*表示zikv-ns2b残基)、ser81*和lys54,在配体和蛋白质之间形成三个氢键,并且在化合物与残基his51和val155之间形成大面积的疏水稳定作用。有趣的是,在另一份报告中(lei等人,2016),这些预测的相互作用残基中的两个(ser81*和his51)也被鉴定为硼酸盐抑制剂cn-716与zikv-ns2b-ns3蛋白酶蛋白之间的接触残基。这个结果表明,如蛋白酶抑制剂cn-716,新生霉素可能也损害zikv-ns2b-ns3催化效率。为了探索蛋白酶抑制的模式,在存在或不存在药物的情况下进行酶动力学实验。如图9b中所示,新生霉素表现为竞争性zikvns2b-ns3蛋白酶抑制剂(全局r2=0.99)。分子动力学模拟提供了关于新生霉素-zikv-ns2b-ns3复合物的稳定性的更多见解,并显示与药物-蛋白质结合相关的几何波动可被快速稳定。总体而言,这些发现表明,新生霉素与其在zikv-ns2b-ns3的结合口袋的结合是高度稳定的,并且药物潜在地通过降低其催化效率而抑制zikv-ns2b-ns3蛋白酶活性。实施例7:在体内动物模型中成功预防和/或治疗zika病毒感染。最近溴隐亭作为抗zikv抑制剂的鉴定,证实zikv-ns2b-ns3蛋白酶作为zika治疗剂开发的可用药的靶标(chan等人,2017a)。在本申请中,采用计算机芯片上的基于结构的方法来快速筛选大型化学文库,并且鉴定出对zikv-ns2b-ns3蛋白酶具有抑制活性的许多其他临床批准的药物。在体外抗病毒测定和免疫缺陷小鼠模型中进一步验证了新生霉素的抗zikv活性。新生霉素,也称为阿巴霉素(albamycin)或卡卓霉素(cathomycin),是氨基香豆素抗生素,其通过靶向细菌dna促旋酶的gyrb亚基,以竞争性地抑制gyrb催化的腺苷三磷酸酶反应,从而发挥其抗细菌作用(主要针对葡萄球菌属)(kirby等人,1956)。此前已经描述了新生霉素对各种dna病毒的复制的抑制活性,所述dna病毒包括疱疹病毒(单纯疱疹病毒、巨细胞病毒、爱泼斯坦-巴尔病毒、卡波济肉瘤相关的疱疹病毒和牛痘病毒),猿猴病毒40和鸭乙型肝炎病毒(civitico等人,1990;droge等人,1985;furlini等人,1983;gonzalez-molleda等人,2012;pessina等人,1992;sekiguchi等人,1997;wu等人,2014)。提出的新生霉素针对这些dna病毒的抗病毒机制是通过阻断宿主拓扑异构酶来抑制病毒复制和组装(sekiguchi等人,1997;wu等人,2014)。我们的发现描述新生霉素抑制属于黄病毒科的rna病毒的复制的新型机制。分子对接和分子动力学模拟表明,新生霉素与zikv-ns2b-ns3之间的结合是高度稳定的。预测与新生霉素相互作用的his51残基是zikv-n2sb-ns3蛋白酶活性的高度保守的催化残基(phoo等人,2016)。这些结果表明,新生霉素可以针对不同的zikv亚型/毒株提供交叉保护。基于功能性荧光的蛋白酶抑制测定证实,新生霉素抑制zikv-ns2b-ns3蛋白酶活性。zikv-ns2b-ns3蛋白酶是一种有吸引力的抗病毒靶标,因为它在病毒复制期间加工病毒多蛋白以产生结构性和非结构性病毒蛋白中起关键作用(zhu等人,2016)。还在我们最近建立的小鼠模型中,评估了新生霉素对zikv感染的治疗功效(chan等人,2016c)。与未治疗的对照小鼠(其发生播散性zikv感染且在5dpi收集的器官组织中具有大量zikvns1抗原表达,并且在10dpi地塞米松停药后不久,发生临床恶化和多器官炎症,需要在12dpi安乐死)相比,用新生霉素治疗的小鼠在5dpi收集的其器官组织中具有极少的zikv-ns1抗原表达,而在12dpi地塞米松停药后几乎没有炎性浸润物。在感染的早期(5dpi)和晚期(14dpi)期间,血液和主要器官组织(包括睾丸和肾脏,这对zikv的传播至关重要)中的平均病毒载量均显著减少。尽管新生霉素通过非发炎的脑膜渗透进入中枢神经系统可能受限,但病毒血症的早期控制和药物通过可能发炎的脑膜的渗透可能有助于新生霉素治疗的小鼠的改善的临床结局。新生霉素于2011年在美国停售,因为随着许多其他较新的抗葡萄球菌抗生素可供使用,它不再被认为是有效的抗细菌剂。然而,新生霉素的治疗在临床上通常是良好耐受的,并且如果潜在的益处大于副作用,则可以考虑在孕妇中使用(fda妊娠c类)(kirby等人,1956)。最近还报道了其他几种非免疫抑制性且属于fda妊娠b或c类的临床批准药物,具有体外抗zikv作用。这些包括阿奇霉素、氯喹、达托霉素、伊维菌素、甲氟喹、氯硝柳胺和索非布韦(barrows等人,2016;bullard-feibelman等人,2016;delvecchio等人,2016;retallack等人,2016;xu等人,2016)。但是,除索非布韦外,这些都没有被证明在动物模型中有效(bullard-feibelman等人,2016)。重要的是,这些药物中的大多数具有常规的口服治疗剂量无法达到的ic50值。口服新生霉素的常规治疗剂量可获得的cmax:ic50比率(1.17-4.11)明显高于口服甲氟喹(0.58)、口服伊维菌素(0.30)、口服阿奇霉素(0.26)和口服氯喹(0.12)的那些(barrows等人,2016;drusano等人,1986;retallack等人,2016;xu等人,2016)。口服和肠胃外的氯硝柳胺是不溶的(zhang等人,2015)。达托霉素仅可用于静脉内施用。索非布韦价格昂贵(≥1000美元/片)。而且,尚未阐明这些药物中的大多数的抗zikv活性的机制。这些药物的局限性使我们的发现对于将新生霉素进一步开发成临床上有用的抗zikv治疗选择尤为重要。然而,在特殊情况下使用时,这些药物仍可用于治疗不同组的zikv感染患者(例如:500mg静脉内剂量的阿奇霉素针对zikv的cmax:ic50比率可高达2.31)。应考虑进一步评估它们(无论是单独还是组合)在动物模型和临床试验中的体内作用。在初步命中化合物中成功地鉴定出多种zikv-ns2b-ns3蛋白酶抑制剂,并证实新生霉素作为有效的抗zikv药物,已证明了我们的组合的计算机芯片上、体外和体内平台在即刻可用的临床批准药物中发现zikv酶抑制剂的能力。应考虑将相同的方法用于筛选其他大型化学数据库的潜在zikv解旋酶或聚合酶抑制剂,以扩大zikv感染的治疗选择。应理解所公开的方法和组合物并不限于所描述的具体方法、方案和试剂,因为这些可以变化。还应理解,本文所用的术语仅用于描述具体实施方案的目的,并无意限制仅受随附权利要求限制的本发明的范围。除非另有说明,本文所用的所有技术和科学术语都具有所公开的方法和组合物所属领域技术人员通常所理解的相同含义。虽然任何类似于或等同于本文所述那些的方法和材料也可以用于本方法和组合物的实践或测试,但是特别有用的方法、装置和材料是如所述的。本文所引用的出版物以及它们所引用的材料明确地通过引用并入本文。本文任何内容都不应解释为承认本发明无权凭借在先发明而先于此类公开内容。并未承认任何参考文献构成现有技术。对于参考文献的讨论描述了其作者所声称的内容,而申请人保留对所引用文献的准确性和针对性进行质疑的权利。应当清楚地理解,虽然本文提及许多出版物,但此类参考文献并不构成承认任何这些文献形成本领域一般常识的一部分。本领域技术人员仅仅使用常规实验,将认识到或者能够确定,本文所述的方法和组合物的具体实施方案的许多等同方案。此类等同方案旨在由以下权利要求书涵盖。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1