一种用于计算基因的组织特异表达的鲁棒z-score打分方法与流程
本发明涉及生物学技术领域,更具体地,涉及一种用于计算基因的组织特异表达的鲁棒z-score打分方法。
背景技术:
很多人类疾病在病理上准确对应的组织或细胞还不清晰,这个问题长期以来妨碍了医学界对致病机理的更进一步理解。基因在组织中的特异表达谱系对解析组织特异性致病机理非常重要,为治疗和药品研发提供指导和有力依据。随着很多组织和细胞类型基因表达数据的持续积累和增加,迫切需要准确量化基因的组织特异性表达的强大有效的统计方法。
分析基因的组织特异表达图谱能够扩大在生命科学和人类疾病学领域的知识。国际大型的协作项目[1-3],为很多人类组织或细胞型产生转录组,如基因型组织表达项目(gtex,v7p)发布了52个组织或细胞型的11,688个转录组,并且初步揭示了大量基因在组织中的常规表达情况[4;5]。大量文献也表明基因的组织特异表达会涉及特定组织的致病机理[6]。antanaviciute等人甚至使用了组织特异基因表达图谱来寻找候选致病基因[7]。虽然这些进展令人鼓舞,但对于探究复杂特异表达模式还仅仅处于起步阶段,大多数与疾病相关基因的致病组织或细胞型仍然难以捉摸[8]。因此,现代生命科学和医学研究急需更好的方法对基于这些丰富基因表达资源进行深入探究基因的组织特异表达特性,进一步解析疾病的组织特异表达机理[9]。
理论上而言,组织特异表达是指基因在一个或少数的几个组织中相对于大多数组织而言较高表达或者较低表达的情况[10]。但是,通常难点就在相对差异量的衡量,因为在实际分析中这种少数与绝大多数(或者众数)的边界是模糊不确定的。比方说,如果误将某个特异表达的组织放进绝大多数组织的这个类就会缩小另外一个特异表达组织与绝大多数组织类的相对差异,从而就会得到一个偏低的特异表达量。目前,有几种度量基因在组织中特异表达的方法[11]。然而,很多早期的方法只能判断一个基因是否有特异表达,而不能度量具体在那些个别的组织中有多少的特异表达[12;13]。后来的方法,可以检测单个组织中具体的特异表达量。但不能对特异表达量进行统计显著性评估。这在实际应用中是很常见的需求。
大体而言,目前求算组织特异性的计算方法主要分为两大类:一类是计算基因是否具有组织特异性(如tau,gini,tsi,countsandhg),不区分该基因在不同组织中的特异性程度;另一类算法则分别计算基因在每一类组织中的特异性程度(z-score,spm,eeandpem)。每类方法在特定的应用场景都具有特定的优势,最近有一项研究分析和比较了以上常用9种组织特异性的算法,在各项测试中,tau在第一类方法中整体表现效果最好,而如果要确定基因在特定组织的特异性,pem也能得出可接受的结果。
但在实际应用中,往往需要知道基因在特定组织中的特异性值,故对第二类算法是现在方法学研究的重点。以下是几个常用第二类方法的简单介绍。用xi表示基因x在组织i中的表达量,n表示所用组织的数目。
z-score的算法如下:
μ表示基因表达的平均值,σ表示标准差。
spm(specificitymeasure):
ee(expressionenrichment):
pem(preferentialexpressionmeasure):
ee和pem中的si表示组织i中所有基因的表达量总和。
研究指出,用ee算法得出的基因组织特异性度分布与其他方法相比,呈现出过多基因分布于非特异表达的组织中,而只有少数基因存在于特异度较高的特异表达基因组织。而在进行5个随机组织的测试中,标准z-core表现出了最低的相关性,表明标准z-core对组织数目不稳定,不适用于数目较小的组织特异性测定;而在这四种算法中pem的相关性表现最好。ee整合了基因所在组织的整体表达量信息,去除了所在组织基因整体表达量对基因表达特异性的影响,而pem是在ee的基础之上用对数来进一步校正特异性指数;spm则是通过取平方来校正特异性指数。
然而,从生物学上的解释来说标准z-core无疑是最明确的。当基因在某组织中相对于大多数组织有特异高的表达时,标准z-core方法可以得到一个正的特异表达值,而当基因在某组织中相对于大多数组织有特异低的表达时,标准z-core方法可以得到一个负的特异表达值。而且标准z-core方法理论上逼近正态分布,可以用正态分布近似计算特异表达的统计显著性程度。然而,标准z-core方法的理论缺陷也很明显。它用到了所有样本计算的平均值和方差,其中包含有具有特异表达的组织。当样本量较少的时候,特异表达的组织样本平均值和方差的估算影响特别大,这也就是为什么有研究指出标准z-core不适用于数目较小的组织特异性测定的根本原因。其实另外的三个方法(spm,ee和pem)也用了所有的样本计算,也会有类似的问题,只是因为它们求算的是商而不是差,影响程度会相对较小。而且,几乎没有有效区分少数与众数的方法,当某个基因在一个以上组织中都有特异表达的时候,现有方法检出特异表达的效能也常常比较低下[14]。这些问题最终的结局就是导致特异表达敏感度的降低。此外,三个方法也不利于计算统计显著性程度。
技术实现要素:
本发明为解决现有技术提供的打分方法存在的对基因的组织特异表达敏感度较低和无法直接评估统计显著性的缺陷,提供了一种用于计算基因的组织特异表达的鲁棒z-score打分方法。
为实现以上发明目的,采用的技术方案是:
一种用于计算基因的组织特异表达的鲁棒z-score打分方法,包括以下步骤:
s1.取某个基因或转录本在n个组织的表达值按从小到达的顺序进行排序;
s2.为每个表达值构建线性拟合模型:
y(i)=β0+β1*i+e(i)
其中β0和β1是回归系数,其初始值利用最小二乘法生成,i∈[1…n]表示表达值排序的序号,y(i)表示排序后的第i个表达值,e(i)表示第i个表达值的残差;
s3.计算每个表达值的权值:
其中,w(i)是权值,k是加权阈值;
s4.根据计算得到的权值利用加权最小二乘法对β0、β1、e(i)进行更新;并令
s5.重复执行步骤s2~s4,直至β0和β1恒定,将β0和β1代入式(1)中,得到表达值y(i)的估计值
s6.对w(i)进行标准化:
利用权值
s7.计算鲁棒z-score:
s8.利用鲁棒z-score计算组织特异表达值的统计显著性pi值:
其中,φ(x)是标准正态分布的分布函数;
s9.设定一个阈值,若pi小于设定的阈值,则拒绝零假设,认为该基因或细胞型在序号为i的组织上有特异表达;如果pi大于或等于设定的阈值,则接受零假设,认为该基因或细胞型在编号为i的组织上没有特异表达。
优选地,所述阈值为0.05。
与现有技术相比,本发明的有益效果是:
本发明提供的方法对基因的组织特异表达有更高的敏感度,且能够直接评估统计显著性的有点,解决了现有技术的缺陷。
附图说明
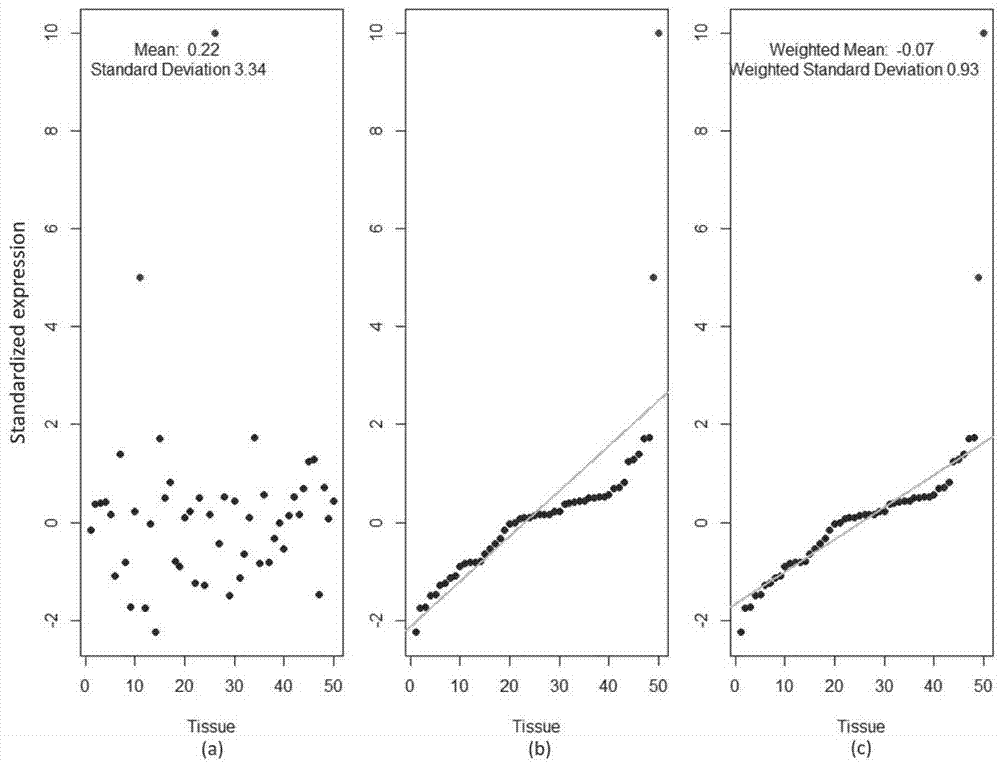
图1为基因或者转录本在50个组织的标准表达值的示意图。
图2为基因或者转录本在上千个组织中的表达值的示意图。
图3为方法的流程示意图。
具体实施方式
附图仅用于示例性说明,不能理解为对本专利的限制;
以下结合附图和实施例对本发明做进一步的阐述。
假设一个基因在n个不同的组织y1,...,yn有表达值,而且绝大部分的表达值(或称为众多数)近似服从正态分布,然而表达值在一个或者少数几个组织中特别低或高。那么目标是求少数的这一个(或几个)组织相对绝大多数组织特异多或少的表达量。如图1中a)所示,例如有一个基因在50个组织有表达,这50个表达值就是图中的50个点,其中两个组织具有高的特异表达,其余的48个表达没有特异表达。图中所示的是非常理想的情况,而实际情况却远没有这么明显区分。通常情况下,少数表达值(有特异表达组织)与大多数表达值(无特异表达组织)的界限是非常的模糊的,如图2中a)所示,因此很难追踪到。尤其是特异表达值越多,难度越大。因此,要实现对特异表达组织或细胞的精准定位,就要解决实际情况下少数值与众数之间没有清晰明确的界限的问题,而这一界限是没有客观的标准定义的。如果界限太松,相对值就会偏小,反之则会偏大。
目前广泛用于评估组织特异表达的一种方法就是技术背景中所提到的标准z-score算法。标准z-score算法通过计算单个组织中的表达值与所有表达值的平均值来近似推断相对特异值。但标准z-score算法没有考虑特异表达值与绝大多数非特异表达值之间的本质差异,而是以全部取值(特异值和非特异值一起)的平均值替代了众多数非特异值的平均值。因为这两者是不能等同的,所以导致了计算偏差。特别是在小样本和高特异性的情形下,因为特异值对平均值的影响较大时,偏差会更大,所以该方法通常不够有效。本发明针对这一瓶颈问题,提出了有效的解决方法,不是简单地规避分界问题,而是采用了更为科学的计算解决了分界问题,还可以准确量化表达值。
为了克服以上技术问题,本发明提供了一种用于计算基因的组织特异表达的鲁棒z-score打分方法,该方法能够准确量化基因在单个组织中相对于大多数值的特异性表达值。这是通过为每个表达值产生权值,且与鲁棒线性回归相结合实现的。
如图3所示,本发明提供的打分方法包括有以下步骤:
第一步、假定有n个不同的组织样本,用常规方法估算某个基因或转录本在每个样本中的表达量均值yi和标准差si。对该基因在n个组织的表达均值yi按从小到达的顺序进行排序。受众多数正态分布的值能在排序后近似形成一条直线的启发,先对看似在杂乱无章的表达值数据先排序,例如对如图2a)中一个基因或者转录本在上千个组织中的表达值排序。排序后,每个表达值都会有一个序号,序号从小到大,即表达值的秩。如图2b)所示,表达值都近似形成一条直线。
第二步、鲁棒线性回顾及求权值
(1)为每个表达值构建线性拟合模型:
y(i)=β0+β1*i+e(i)
其中β0和β1是回归系数,其初始值利用最小二乘法生成,i∈[1…n]表示表达值排序的序号,y(i)表示排序后的第i个表达值,e(i)表示第i个表达值的残差;
本发明采用了鲁棒线性回归模型中广泛应用的huber方法的m估计法,m估计对正态和非正态分布都有效,而且比其他方法能更灵活强势地应对极端值相对变化。这非常适应处理表达值相对变化的情况,这也是目前需要解决的关键问题之一;
计算每个表达值的权值:
其中,w(i)是权值,k是加权阈值;
(2)根据计算得到的权值利用加权最小二乘法对β0、β1、e(i)进行更新;并令
(3)重复执行步骤s2~s4,直至β0和β1恒定,将β0和β1代入式(1)中,得到表达值y(i)的估计值
第三步、求取观测值加权后的均值和方差
对w(i)进行标准化:
利用权值
第四步、求鲁棒z-score
鲁棒z-score量化了每一个组织中的表达值与大部分表达值均值间的偏差,且鲁棒z-score用于计算表达特异性显著水平的p值。
第五步、鲁棒z-score计算表达特异性显著水平的p值
用鲁棒z-score量化后,偏差的统计显著性近似标准正态分布,
显然,本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围之内。
参考文献
[1]melem,ferreirapg,reverterf,etal.humangenomics.thehumantranscriptomeacrosstissuesandindividuals[j].science,2015,348(6235):660-5.
[2]ogasawarao,otsujim,watanabek,etal.bodymap-xs:anatomicalbreakdownof17millionanimalestsforcross-speciescomparisonofgeneexpression[j].nucleicacidsres,2006,34(databaseissue):d628-31.
[3]liux,yux,zackdj,etal.tiger:adatabasefortissue-specificgeneexpressionandregulation[j].bmcbioinformatics,2008,9:271.
[4]lix,kimy,tsangek,etal.theimpactofrarevariationongeneexpressionacrosstissues[j].nature,2017,550(7675):239-243.
[5]tanmh,liq,shanmugamr,etal.dynamiclandscapeandregulationofrnaeditinginmammals[j].nature,2017,550(7675):249-254.
[6]lagek,hansennt,karlbergeo,etal.alarge-scaleanalysisoftissue-specificpathologyandgeneexpressionofhumandiseasegenesandcomplexes[j].procnatlacadsciusa,2008,105(52):20870-5.
[7]antanaviciutea,dalyc,crinnionla,etal.genetier:prioritizationofcandidatediseasegenesusingtissue-specificgeneexpressionprofiles[j].bioinformatics,2015,31(16):2728-35.
[8]calderond,bhaskara,knowlesda,etal.inferringrelevantcelltypesforcomplextraitsbyusingsingle-cellgeneexpression[j].amjhumgenet,2017,101(5):686-699.
[9]rapaportf,khaninr,liangy,etal.comprehensiveevaluationofdifferentialgeneexpressionanalysismethodsforrna-seqdata[j].genomebiol,2013,14(9):r95.
[10]liangs,liy,bex,etal.detectingandprofilingtissue-selectivegenes[j].physiolgenomics,2006,26(2):158-62.
[11]kryuchkova-mostaccin,robinson-rechavim.abenchmarkofgeneexpressiontissue-specificitymetrics[j].briefbioinform,2017,18(2):205-214.
[12]kadotak,nishimuras,bonoh,etal.detectionofgeneswithtissue-specificexpressionpatternsusingakaike'sinformationcriterionprocedure.physiolgenomics.2003:251-9.
[13]kadotak,yej,nakaiy,etal.roku:anovelmethodforidentificationoftissue-specificgenes[j].bmcbioinformatics,2006,7:294.
[14]xiaosj,zhangc,zouq,etal.tisged:adatabasefortissue-specificgenes[j].bioinformatics,2010,26(9):1273-5.
- 还没有人留言评论。精彩留言会获得点赞!
- 一种经腺病毒基因修饰的肿瘤特异性细胞毒性t淋巴细胞的培养方法
- CRISPR-Cas9特异性敲除猪SLA-1基因的方法及用于特异性靶向SLA-1基因的sgRNA的制作方法
- 一种特异性靶向QKI基因的sgRNA及其应用
- 一种泰国库蠓的特异基因及其分子鉴定方法
- 一种美丽库蠓特异基因及其分子鉴定方法
- 一种特异于禽类原始生殖细胞的基因转移系统及方法
- CRISPR-Cas9特异性敲除猪SALL1基因的方法及用于特异性靶向SALL1基因的sgRNA的制作方法
- CRISPR-Cas9特异性敲除猪CMAH基因的方法及用于特异性靶向CMAH基因的sgRNA的制作方法
- CRISPR-Cas9特异性敲除猪SLA-2基因的方法及用于特异性靶向SLA-2基因的sgRNA的制作方法
- 一种安宁铗蠓的特异基因及其分子鉴定方法