治疗帕金森病中运动障碍的D3多巴胺受体激动剂的制作方法
发明背景
从功能和结构观点,g蛋白偶联受体(gpcr)家族是最重要的蛋白质类型之一。人基因组包含近950个编码gpcr的基因,其中近450个基因已经被暗示作为治疗靶标。结合至gpcr的配体诱导多种受体构象,并且不同的配体可稳定不同的受体构象(kenakin&miller,2010,pharmacol.rev.62(2):265-304)。功能选择性的概念基于下述假设:不同的受体构象募集不同的信号传导蛋白,其导致一种信号传导途径相对于另一种优先活化(mailman,2007,trendspharmacol,sci.28(8):390-396)。除了选择信号传导途径,激动剂诱导的受体构象也可潜在影响受体信号传导性能。
在gpcr中,多巴胺受体亚族已经吸引了生物学家和药理学家的注意。在中枢神经系统中,广泛表达多巴胺受体并且其参与移动、认知、情感和神经内分泌的控制。在外周系统中,多巴胺受体更主要存在于肾脏、脉管系统和垂体,其中它们主要影响钠稳态、血管紧张度和激素分泌。尽管有许多优先活化一种而不是其他信号传导级联的功能选择性配体的例子,但是改变受体信号传导性能的功能选择性配体是稀少的,并且对于多巴胺受体还未描述。
神经递质多巴胺经多巴胺受体的5个主要亚型控制哺乳动物的大量生理和行为功能。基于药理学和功能,它们宽泛地分成“d1类”和“d2类”多巴胺受体。d1类由d1和d5受体组成,而d2类由d2、d3和d4受体组成。d3受体主要与百日咳毒素敏感型gα-蛋白(gi/go)结合(ahlgren-beckendorf&levant,2004,j.recept.signaltransduct.res.24(3):117-130)。当转染入不同的细胞系时,d3受体结合至腺苷酸环化酶v同工型(robinson&caron,1997,mol.pharmacol.52:508-514)并且启动信号传导事件,包括有丝分裂原活化蛋白(map)激酶的磷酸化(cussac等,1999,mol.pharmacol.56(5):1025-103)。d2和d3多巴胺受体也调节钾和钙通道功能(seabrook等,1994,br.j.pharmacol.111:391-393;werner等,996,mol.pharmacol.49:656-661)。转染的d3受体稳固结合至天然表达的g蛋白偶联内向整流钾通道(girk)和电压门控p/q型钙通道,并且抑制自发性动作电位的激发和att-20神经内分泌细胞系中的分泌活性(kuzhikandathil&oxford,1999,j.neurosci.19(5):1698-1707;kuzhikandathil&oxford,2000,j.gen.physiol.115:697-706;kuzhikandathil等,1998,mol.cellneurosci.12:390-402)。d3受体在att-20细胞中进一步结合至天然表达的腺苷酸环化酶v(kuzhikandathil&bartoszyk,2006,neuropharm.51:873-884)、map激酶(westrich&kuzhikandathil,2007,biochim.biophys.acta-mcr1773:1747-1758)和离子通道(kuzhikandathil&oxford,1999,j.neurosci.19(5):1698-1707;kuzhikandathil&oxford,2000,j.gen.physiol.115:697-706;kuzhikandathil等,1998,moi.cellneurosci.12:390-402;kuzhikandathil等,2004,mol.cellneurosci.26:144-155)。
d3多巴胺受体的表达在许多病理条件下以及在长期的治疗之后被改变。在帕金森病中,左旋多巴诱导的运动障碍与壳(putamen)和苍白球内段、正常表达d2受体的区域中d3r表达的特异性上调有关(bezard等,2003,nat.med.9(6):762-767;guigoni等,2005,parkinsonismrelateddisorders11suppl1,s25-29)。在啮齿动物模型中,与左旋多巴治疗相关的行为敏感由上调的d3受体介导(guillin等,2001,nature411(6833):86-89)。在神经分裂症中,d3受体表达水平在基底核中增加两倍。也已经报道抗紧张剂治疗改变了d3受体的表达。长期可卡因使用者的纹状体和黑质以及伏核中,d3受体的密度增加。压力和抑郁诱导的d3受体表达下调在长期的抗抑郁剂治疗后逆转。
多巴胺受体是用于治疗各种神经学和精神病学病症的靶标,比如帕金森病、神经分裂症、药瘾、抑郁、双相性精神障碍、注意力缺陷多动障碍、图雷特综合征、亨廷顿病和偏头痛。
帕金森氏疾病(也称为帕金森病)是中枢神经系统的退行性疾病,其通常损伤患者的运动技能、语言和其他功能(jankovic,2008,j.neurol.neurosurg.psychiatr.79(4):368-76)。帕金森氏疾病的特征为肌肉硬化、颤抖、身体运动缓慢(运动迟缓)和在极端情况下身体运动丧失(运动不能症)。帕金森氏疾病的主要症状是基底核对运动皮质刺激降低的结果,通常是由于不足的多巴胺形成和作用导致,多巴胺是在大脑的多巴胺能神经元(尤其黑质)中产生。次要症状可包括高水平的认知功能障碍和轻微的语言问题。帕金森氏疾病是慢性的并且渐进的。目前,帕金森氏疾病不能治愈,但是药物可提供症状的缓解。
最广泛使用的治疗形式是l-多巴(左旋多巴)。左旋多巴通过l-芳族氨基酸脱羧酶(也称为多巴-脱羧酶)在多巴胺能神经元中转化成多巴胺。但是,仅仅1-5%的左旋多巴进入多巴胺能神经元。剩余的左旋多巴通常在其他地方代谢成多巴胺,造成大量的副作用(“symptomaticpharmacologicaltherapyinparkinson′sdisease”,parkinson′sdisease,london:royalcollegeofphysicians,2006,pp,59-100))。由于反馈抑制,施用左旋多巴导致左旋多巴内生形成的减少,并且因此最终无法达到预期目标。左旋多巴也可与卡比多巴((2s)-3-(3,4-二羟苯基)-2-肼基-2-甲基丙酸)共同施用,后者防止左旋多巴在身体的其他地方代谢。已经发现多巴胺激动剂溴隐亭(bromocriptine)、培高利特(pergolide)、普拉克索(pramipexole)、罗匹尼罗(ropinirole)、吡贝地尔(piribedil)、卡麦角林(cabergoline)、阿扑吗啡(apomorphine)和利舒脲(lisuride)适度有效。
左旋多巴诱导的运动障碍(lid)是长期使用左旋多巴的特别严重的副作用。在5-10年的左旋多巴治疗之后,这些运动波动(motorfluctuation)出现在一半以上的帕金森氏疾病患者中,受影响的患者百分数随着时间增加,并且lid被认为是潜在地不可逆的。运动障碍最常出现在峰值左旋多巴血浆浓度的时间并且因此称为峰值-剂量运动障碍。随着患者的进展,他们可表现出双相运动障碍,其在药物浓度升高或下降时发生。通过使用其他治疗比如溴隐亭(parlodeltm)缓和运动障碍的试图表现出无效。为了避免运动障碍,具有青年发作形式疾病的患者通常对开始左旋多巴治疗犹豫不决,直到非常必要,因为担心稍后遭受严重的运动障碍。目前,没有药物治疗方式治疗遭受帕金森氏疾病患者的lid。
有趣地,在与多巴胺能系统相关的大部分疾病比如帕金森氏疾病中存在多巴胺受体表达的改变。多巴胺受体表达的改变也在这些神经学和精神学病症的长期治疗之后观察到。在d3多巴胺受体的情况下,在帕金森病、神经分裂症、抑郁和药瘾中已经报道了表达的改变。在长期药物治疗之后,研究已经报道帕金森病中lid中和神经分裂症中抗紧张剂诱导的迟发性运动障碍中d3受体的上调。
在帕金森氏疾病的治疗中使用左旋多巴作为治疗剂的能力由于将最终发展左旋多巴诱导的运动障碍(lid)的可能性而严重受阻。在本领域需要新型治疗剂,其在遭受帕金森氏疾病的患者中治疗、缓解或预防左旋多巴诱导的运动障碍。本发明实现了该需要。
技术实现要素:
本发明包括药物组合物,其包括至少一种药学上可接受的载体和至少一种选自下述的化合物:
式(i)的化合物:
其中式(i)中:
r1、r2和r3独立地选自h、氰基、羟基、氨基、乙酰氨基、卤基、烷氧基、硝基、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基、取代的杂环基、芳基、取代的芳基、芳基-(c1-3)烷基、取代的芳基-(c1-3)烷基、羧基、烷基羧基、甲酰基、烷基-羰基、芳基-羰基和杂芳基-羰基;
r4和r5独立地选自h、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基、取代的杂环基、芳基、取代的芳基、芳基-(c1-3)烷基和取代的芳基-(c1-3)烷基;和,
n是2、3、4或5;
式(iia)或(iib)的化合物:
其中式(iia)或(iib)中:
r1和r2独立地选自h、氰基、羟基、氨基、乙酰氨基、卤基、烷氧基、硝基、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基、取代的杂环基、芳基、取代的芳基、芳基-(c1-3)烷基、取代的芳基-(c1-3)烷基、羧基、烷基羧基、甲酰基、烷基-羰基、芳基-羰基和杂芳基-羰基;
r3和r4独立地选自h、c1-6烷基、芳基、杂芳基和取代的c1-6烷基;
r5选自h、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基、取代的杂环基、芳基、取代的芳基、芳基-(c1-3)烷基和取代的芳基-(c1-3)烷基;和,
m是1、2、3或4;
2,7-二氨基-5-(4-氟苯基)-4-氧-3,4,5,6-四氢吡啶并[2,3-d]嘧啶-6-腈;
(z)-2-(1h-苯并[d]咪唑-2-基)-n′-羟基-3-(4-甲氧苯基)丙脒(propanimidamide);其混合物,或其药学上可接受的盐。
在一种实施方式中,式(i)中r1、r2和r3独立地选自h、氰基、卤基、烷氧基、硝基、c1-6烷基和羧基。在另一实施方式中,式(i)中r4和r5独立地选自h、c1-6烷基和取代的c1-6烷基。在仍另一实施方式中,式(i)中n是2。在仍另一实施方式中,式(iia)或(iib)中m是1或2。
在一种实施方式中,所述至少一种化合物选自:2-氨基-4-(2-氯苯基)丁-1-醇;2-(3-氨基己基)苯酚;4-(2-氯苯基)-2-甲氨基-丁烷(也称为4-(2-氯苯基)-n-甲基丁-2-胺);4-(2-氯苯基)-丁-2-胺;4-(2-氟苯基)丁-2-胺;4-(2-溴苯基)丁-2-胺;4-(2-碘苯基)丁-2-胺;4-(2-甲氧苯基)丁-2-胺;2-(3-氨基丁基)苯酚;3-(3,4-二乙氧苯基)丙-1-胺;4-(4-氯苯基)丁-2-胺;4-(4-甲氧苯基)丁-2-胺;2-(5-氯-1-甲基-1h-吲哚-3-基)乙胺;1-(5-氟-1-甲基-1h-吲哚-3-基)丙-2-胺;1-(5-甲氧基-1-甲基-1h-吲哚-3-基)丙-2-胺;2,7-二氨基-5-(4-氟苯基)-4-氧-3,4,5,6-四氢吡啶并[2,3-d]嘧啶-6-腈;(z)-2-(1h-苯并[d]咪唑-2-基)-n′-羟基-3-(4-甲氧苯基)丙脒;其混合物,或其药学上可接受的盐。
本发明也包括在遭受帕金森病的患者中治疗、缓解或预防左旋多巴诱导的运动障碍的方法。该方法包括向患者施用包括治疗有效量的选自下述的至少一种化合物的药物组合物:
式(i)的化合物:
其中式(i)中:
r1、r2和r3独立地选自h、氰基、羟基、氨基、乙酰氨基、卤基、烷氧基、硝基、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基、取代的杂环基、芳基、取代的芳基、芳基-(c1-3)烷基、取代的芳基-(c1-3)烷基、羧基、烷基羧基、甲酰基、烷基-羰基、芳基-羰基和杂芳基-羰基;
r4和r5独立地选自h、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基、取代的杂环基、芳基、取代的芳基、芳基-(c1-3)烷基和取代的芳基-(c1-3)烷基;和,
n是2、3、4或5;
式(iia)或(iib)的化合物:
其中式(iia)或(iib)中:
r1和r2独立地选自h、氰基、羟基、氨基、乙酰氨基、卤基、烷氧基、硝基、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基、取代的杂环基、芳基、取代的芳基、芳基-(c1-3)烷基、取代的芳基-(c1-3)烷基、羧基、烷基羧基、甲酰基、烷基-羰基、芳基-羰基和杂芳基-羰基;
r3和r4独立地选自h、c1-6烷基、芳基、杂芳基和取代的c1-6烷基;
r5选自h、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基、取代的杂环基、芳基、取代的芳基、芳基-(c1-3)烷基和取代的芳基-(c1-3)烷基;和,
m是1、2、3或4;
2,7-二氨基-5-(4-氟苯基)-4-氧-3,4,5,6-四氢吡啶并[2,3-d]嘧啶-6-腈;
(z)-2-(1h-苯并[d]咪唑-2~基)-n′-羟基-3-(4-甲氧苯基)丙脒;
其混合物,或其药学上可接受的盐。
在一种实施方式中,式(i)中r1、r2和r3独立地选自h、氰基、卤基、烷氧基、硝基、c1-6烷基和羧基。在另一实施方式中,式(i)中r4和r5独立地选自h、c1-6烷基和取代的c1-6烷基。在仍另一实施方式中,式(i)中n是2。在仍另一实施方式中,式(iia)或(iib)中m是1或2。
在一种实施方式中,所述至少一种化合物选自下述:2-氨基-4-(2-氯苯基)丁-1-醇;2-(3-氨基己基)苯酚;4-(2-氯苯基)-丁-2-胺;4-(2-氯苯基)-2-甲氨基-丁烷;4-(2-氟苯基)丁-2-胺;4-(2-溴苯基)丁-2-胺;4-(2-碘苯基)丁-2-胺;4-(2-甲氧苯基)丁-2-胺;2-(3-氨基丁基)苯酚;3-(3,4-二乙氧苯基)丙-1-胺;4-(4-氯苯基)丁-2-胺;4-(4-甲氧苯基)丁-2-胺;2-(5-氯-1-甲基-1h-吲哚-3-基)乙胺;1-(5-氟-1-甲基-1h-吲哚-3-基)丙-2-胺;1-(5-甲氧基-1-甲基-1h-吲哚-3-基)丙-2-胺;2,7-二氨基-5-(4-氟苯基)-4-氧-3,4,5,6-四氢吡啶并[2,3-d]嘧啶-6-腈;(z)-2-(1h-苯并[d]咪唑-2-基)-n′-羟基-3-(4-甲氧苯基)丙脒;其混合物,或其药学上可接受的盐。
在一种实施方式中,组合物进一步包括选自下述的药物:左旋多巴、氯氮平、溴隐亭、培高利特、普拉克索、罗匹尼罗、吡贝地尔、卡麦角林、阿扑吗啡和利舒脲,其盐和其混合物。
在一种实施方式中,将药物组合物与包括左旋多巴的第二药物组合物共同施用至患者。
在一种实施方式中,将药物组合物施用至患者给定时间期间,然后将包括左旋多巴的第二药物组合物施用至患者。
在一种实施方式中,给定时间期间从约2分钟至约24小时变化。
在一种实施方式中,患者是人。
本发明进一步包括治疗、缓解或预防患者中帕金森病的方法。该方法包括向患者施用包括治疗有效量的选自下述的至少一种化合物的药物组合物:
式(i)的化合物:
其中式(i)中:
r1、r2和r3独立地选自h、氰基、羟基、氨基、乙酰氨基、卤基、烷氧基、硝基、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基、取代的杂环基、芳基、取代的芳基、芳基-(c1-3)烷基、取代的芳基-(c1-3)烷基、羧基、烷基羧基、甲酰基、烷基-羰基、芳基-羰基和杂芳基-羰基;
r4和r5独立地选自h、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基、取代的杂环基、芳基、取代的芳基、芳基-(c1-3)烷基和取代的芳基-(c1-3)烷基;和,
n是2、3、4或5;
式(iia)或(iib)的化合物:
其中式(iia)或(iib)中:
r1和r2独立地选自h、氰基、羟基、氨基、乙酰氨基、卤基、烷氧基、硝基、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基、取代的杂环基、芳基、取代的芳基、芳基-(c1-3)烷基、取代的芳基-(c1-3)烷基、羧基、烷基羧基、甲酰基、烷基-羰基、芳基-羰基和杂芳基-羰基;
r3和r4独立地选自h、c1-6烷基、芳基、杂芳基和取代的c1-6烷基;
r5选自h、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基、取代的杂环基、芳基、取代的芳基、芳基-(c1-3)烷基和取代的芳基-(c1-3)烷基;和,
m是1、2、3或4;
2,7-二氨基-5-(4-氟苯基)-4-氧-3,4,5,6-四氢吡啶并[2,3-d]嘧啶-6-腈;
(z)-2-(1h-苯并[d]咪唑-2-基)-n′-羟基-3-(4-甲氧苯基)丙脒;
其混合物,或其药学上可接受的盐。
在一种实施方式中,所述至少一种化合物选自下述:2-氨基-4-(2-氯苯基)丁-1-醇;2-(3-氨基己基)苯酚;4-(2-氯苯基)-2-甲氨基-丁烷;4-(2-氯苯基)-丁-2-胺;4-(2-氟苯基)丁-2-胺;4-(2-溴苯基)丁-2-胺;4-(2-碘苯基)丁-2-胺;4-(2-甲氧苯基)丁-2-胺;2-(3-氨基丁基)苯酚;3-(3,4-二乙氧苯基)丙-1-胺;4-(4-氯苯基)丁-2-胺;4-(4-甲氧苯基)丁-2-胺;2-(5-氯-1-甲基-1h-吲哚-3-基)乙胺;1-(5-氟-1-甲基-1h-吲哚-3-基)丙-2-胺;1-(5-甲氧基-1-甲基-1h-吲哚-3-基)丙-2-胺;2,7-二氨基-5-(4-氟苯基)-4-氧-3,4,5,6-四氢吡啶并[2,3-d]嘧啶-6-腈;(z)-2-(1h-苯并[d]咪唑-2-基)-n′-羟基-3-(4-甲氧苯基)丙脒;其混合物,或其药学上可接受的盐。
在一种实施方式中,组合物进一步包括选自下述的至少一种药物:左旋多巴、氯氮平、溴隐亭、培高利特、普拉克索、罗匹尼罗、吡贝地尔、卡麦角林、阿扑吗啡和利舒脲,其盐和其混合物。
在一种实施方式中,患者进一步施用包括选自下述的至少一种药物的第二药物组合物:左旋多巴、氯氮平、溴隐亭、培高利特、普拉克索、罗匹尼罗、吡贝地尔、卡麦角林、阿扑吗啡和利舒脲,其盐和其混合物。
附图简述
为了说明本发明的目的,在附图中描绘了本发明的某些实施方式。但是,本发明不限于附图中描绘的实施方式的精确布置和手段。
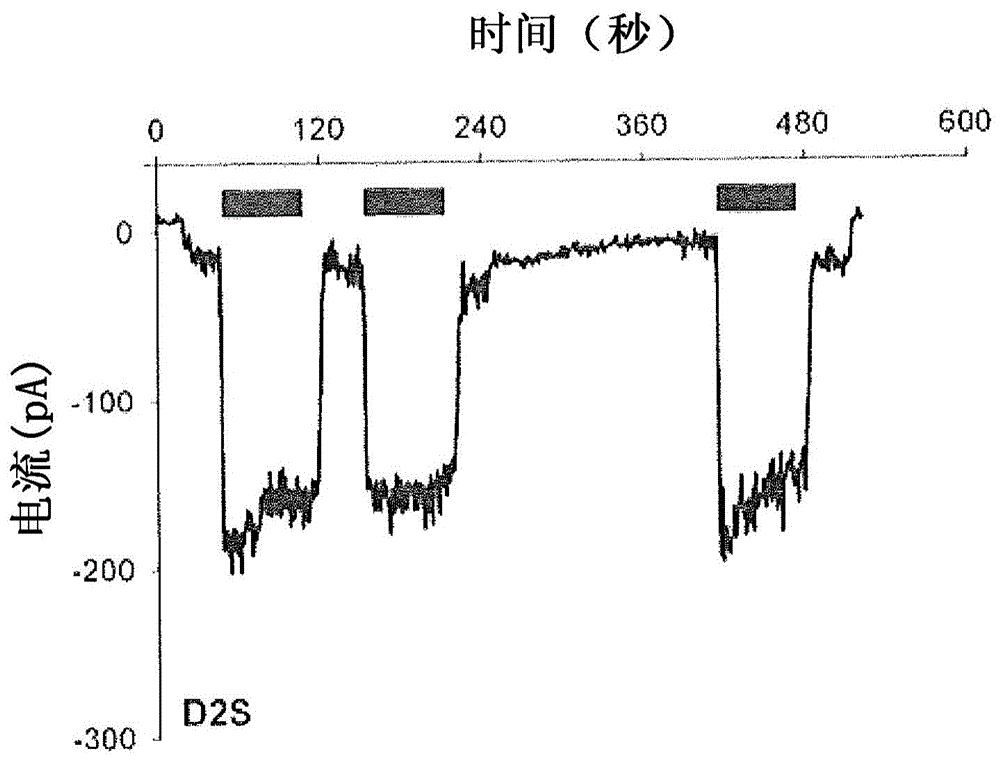
图1,包括图1a-d,图解说明了在稳定表达人d2s(图1a和1b)或d3(图1c和1d)的att-20细胞中的代表性电压钳记录(图1a和1c)和电流钳记录(图1b和1d)。对于电压钳记录(图1a和1c),将细胞保持在-65mv并将100nm多巴胺(黑条)施加1分钟。对于电流钳记录(图1b和1d),通过施加100nm喹吡罗(qp)1分钟而使自发性动作电位(spontaneousactionpotential)超极化,喹吡罗是d2样受体的激动剂。
图2,包括图2a-c,图解说明了激动剂诱导的d3受体耐受性和srt性质的调制。图解说明了来自att-d3细胞的代表性电压钳记录(图2a)和电流钳记录(图2b),所述att-d3细胞用300nmpbzi((3as,9br)-3-丙基-1,2,3a,4,5,9b-六氢苯并[e]吲哚-8-醇)或100nm喹吡罗((4ar,8ar)-5-丙基-4,4a,5,6,7,8,8a,9-八氢-1h-吡唑并[3,4-g]喹啉)处理。pbzi引起不显示耐受性或srt性质的3个响应;在相同细胞上,喹吡罗引起具有耐受性和srt的girk响应。类似地,图2c中图解的代表性电压钳记录显示300nmfauc73(正方形十字条)诱导不显示d3受体耐受性和srt的girk电流。相反,在相同的细胞中,300nmpd128907(黑条)引起耐受性和srt。细胞被保持在-65mv并且激动剂施用的持续时间为~60秒。
图3是用于设计这些新型d3激动剂的基于混合结构(hsb)的方案的流程图。
图4图解说明d3受体(seqidno:1)与β2ar(pdb号2rh1-编号为badre-2rh1;seqidno:2)的一级氨基酸序列比对。形成tm螺旋的残基以下划线形式显示。相同的残基以“*”表示并且相似度由“:”和“.”表示。
图5是结合至叠合药效团成分的d3受体的pbzi的示意图示。标注“gr”的未充填椭圆表示疏水成分,标注“pur”的未充填椭圆表示氢键供体-受体对,并且距离限制由点黑线表示。氢键相互作用由箭头表示,呈直线的离子相互作用和呈直线的π-π相互作用延伸横跨两个6元环。灰色球和轮廓线表示配体和受体之间的匹配区域。
图6,包括图6a-d,是图解说明es609(图6a)、pd128907(图6b)、pbzi(图6c)和多巴胺(图6d)的相互作用模式的一系列图示。结合位点残基根据它们的性质涂色,疏水残基为绿色(标注gr),极性残基为紫色(标注pur),和带电残基用粗体轮廓突出。灰色球和轮廓线表示配体和受体之间的匹配区域。氢键相互作用由箭头表示,呈直线的离子相互作用和呈直线的π-π相互作用延伸横跨两个六元环。使用moe程序的ligx模块产生所述图。用于筛选分子的3d药效团覆盖在pbzi结构上(图6c),空心红色圆圈表示亲水性相互作用(标注red),蓝色空心圆圈表示疏水和芳族相互作用(标注bl)并且黑色点线表示药效团成分之间的距离。
图7,包括图7a-b,是一组分子模型,图解说明hsb方法允许识别参与d3受体耐受性和srt性质的残基和构象。图7a图解说明结合至pd128907、pbzi和es609的d3受体的结构叠加,受体以美工格式表示。跨膜螺旋标为1至7并且胞外和胞内环分别标注ec1-ec3和ic1-ic3。图7b图解说明与4-(2-氯苯基)-丁-2-胺(es609)对接的多巴胺d3受体的分子模型。受体描绘为飘带,配体以空间填充模型呈现。
图8图解说明实验开发计划的流程图。
图9,包括图9a-c,图解说明电压钳实验。图9a图解说明当两个连续的100nm多巴胺(da)处理时,d3受体诱导的girk响应的代表性电压钳记录。将da溶解在具有30mm钾的胞外溶液中(以增强girk电流)。激动剂施用的持续时间为~60秒。第二girk响应与第一girk响应的比是耐受性的定量量度。图9b是条状图,其表示d3受体耐受性由100nm多巴胺(da)、喹吡罗(qp)和pd128907引起;而不是由pbzi或4-(2-氯苯基)-丁-2-胺(es609)引起。图9c是条状图,其图解说明d3受体的耐受性是激动剂依赖性的。累积数据显示稳定表达人d3多巴胺受体的att-20细胞中第二和第一激动剂诱导的girk响应的比。使用饱和浓度的多巴胺(da,100nm,n=5)、喹吡罗(100nm,n=5)、pd128907(100nm,n=5)、7oh-dpat(100nm,n=4)、7oh-pipat(100nm,n=4)、沙立佐坦(100nm,n=6)、普拉克索(300nm,n=4)、罗替高汀(100nm,n=4)、pbzi(300nm,n=10)和fauc73(300nm,n=4)引起girk响应。误差条表示士sem.*,**,p<0.05,anova,holm-sidak事后比较检验。
图10,包括图10a-b,图解说明代表性电压钳记录,其显示在att-20细胞中稳定表达的d3受体,pbzi(peun1)可与多巴胺(da)竞争并且预防耐受性和srt的发展。图10a中,100nmda引起d3受体耐受性和srt。图10b中,1000nmpbzi同时与da施用阻碍了耐受性和srt的发展。
图11,包括图11a-b,图解说明两个不同的功能分析中pbzi(图11a)和4-(2-氯苯基)-丁-2-胺(es609;图11b)的剂量响应曲线。如图11a中所图解,使用全细胞电压钳记录测量表达d3受体的att-20细胞中对pbzi的girk电流响应。该电流响应对于细胞尺寸进行标准化(使用膜电容(membrancecapacitance))。图11b图解说明通过用各种剂量的4-(2-氯苯基)-丁-2-胺活化att-20细胞中的d3受体来抑制10μm福司柯林诱导的camp水平。通过使用gehealthcare的elisa试剂盒测量camp水平。图11b中的三角形图解说明由300nm喹吡罗引起的抑制。
图12,包括图12a-b,图解说明pbzi和pd128907对balb/c小鼠移动能力的影响。如图12a所图解,pbzi诱导移动能力的剂量依赖性和单阶段抑制。水平箭头表示由10mg/kg的scpbzi(圆)诱导的活动减退,以10分钟一格(bin)绘制。1mg/kg剂量(三角形)没有显著效果。将由pbzi诱导的移动活动性相对于对照媒介(盐水)注射的小鼠的移动活动性进行标准化。由水平箭头表示的数据点显示移动活动性在统计上显著降低(p=0.002,多重newman-keuls
图13,包括图13a-b,图解说明pbzi、pd128907(诱导耐受性的d3激动剂)和对照盐水在6mg/kg左旋多巴诱导的运动障碍上的比较。施用药物10分钟后注射左旋多巴。pd128907增加了aim分数,即使在施用左旋多巴之前。pbzi延迟和减少由左旋多巴诱导的运动障碍(图13a)。如图13b所图解,曲线下方面积的积分产生总积分的aim分数,其由于施用pbzi显著减小(*p<0.05,anova,holm事后比较检验)。
图14是条状图,其图解说明左旋多巴(l-多巴)注射之后前肢运动迟缓的剂量依赖性改善,这使用前肢踏步试验测量。(*,p<0.01,anova,holm事后比较检验)。
图15图解说明体内实验的时程。
图16,包括图16a-b,图解说明由多巴胺和pbzi活化d3受体不同地调节神经放电(neuronalfiring)的发现。图16a图解说明稳定表达人d3多巴胺受体的att-20神经内分泌细胞中代表性电流钳记录。通过溶解在具有5mm钾的标准外用溶液中的100nm多巴胺(黑条)活化d3受体,在第一次应用期间超极化细胞和抑制自发动作电位,但不会至第二次和第三次应用。图16b图解说明溶解在具有5mm钾的标准外用溶液中的300nmpbzi(灰色阴影条)活化d3受体在第一次和第二次处理期间超极化细胞和抑制自发动作电位。
图17是图解说明新型d3受体激动剂es609消除耐受性和srt性质的一组图。图17a中,代表性电压钳记录显示100nmes609(十字阴影条)诱导girk电流,其在稳定表达人d3受体的att-20细胞中不显示耐受性和srt。相反,在相同的细胞中,100nm喹吡罗(qp,黑条)引起耐受性和srt。图17b中,代表性电压钳记录显示300nmpbzi(阴影条)和300nmes609(十字阴影条)在缺少外源d3受体表达的母本att-20细胞中都不诱导girk电流。图17c中,代表性电压钳记录显示100nmes609(十字阴影条)和100nm喹吡罗(黑条)在稳定表达人d2s受体的att-20细胞中诱导girk电流。es609诱导的girk电流显著小于d2s受体表达细胞中喹吡罗诱导的电流。细胞被保持在-65mv并且激动剂施用的持续时间为~60秒。图17d图解说明稳定表达人d3受体的att-20细胞中es609诱导的girk响应的累积剂量响应。黑色的填充圆是由饱和剂量的喹吡罗(qp,300nm)引起的girk响应并且显示es609是d3受体的完全激动剂。误差条表示±sem。用girk电流除以细胞电容(cellcapacitance)以针对细胞尺寸进行标准化。数据点用4参数hill方程拟合。
图18是图解说明发现pbzi和es609改善大鼠pd模型中的运动缺陷的条状图。
图19,包括图19a-b,是图解说明发现pbzi&es609改善大鼠pd模型中左旋多巴诱导的运动障碍的一对图。
图20是图解说明发现非典型的(而不是典型的)d3受体激动剂预防左旋多巴诱导的运动障碍的条状图。
图21,包括图21a-b,是图解说明pbzi和es609对海马培养物中神经元活力的作用的一组图(从培养物中2天至5天)。
图22,包括图22a-b,是图解说明pbzi&es609对用10mm过氧化氢处理4小时的海马细胞神经保护的作用的一组图。
图23,包括图23a-b,是图解说明发现es609抑制一元胺输送器摄取活性的一组图。
发明详述
本发明涉及d3多巴胺受体(氨基酸序列为seqidno:1)的信号转导途径具有特定的耐受性和慢响应终止(slowresponsetermination)(srt)性质的发现。在展示d3多巴胺受体表达改变的神经病中,耐受性和慢响应终止(srt)性质异常表达并且可能引起所述病理。
在非限制性方面,d3受体中激动剂诱导的耐受性与受体独特的构象状态相关。该相关性表明d3受体的耐受性和srt性质是配体依赖性的并且改变耐受性特异性构象的功能选择性激动剂将消除该耐受性和srt性质。筛选已知d3受体激动剂并且确定它们的消除耐受性和srt性质的能力允许识别两种激动剂,顺式-8-oh-pbzi(pbzi)和fauc73,其虽然是d3受体的完全激动剂,但完全消除了受体的耐受性和srt性质。为了区分这些新d3受体激动剂与典型的诱导耐受性和srt的激动剂,它们统称为非典型的d3受体激动剂。基于pbzi与d3受体相互作用的药效团模型被指定为基于混合结构的(hsb)计算机筛选方法的输入,这允许识别另外的新型激动剂es609,其也消除d3受体耐受性和srt性质。
本发明因此进一步涉及不引起d3受体的耐受性和慢响应终止性质的新型功能选择性d3多巴胺受体激动剂的发现。在一个方面,这些激动剂,不是优先活化信号传导途径,而是修饰受体的信号传导性能。在另一方面,该新类型的非典型d3受体激动剂在药理学上将d3受体转化成d2受体的功能等价物。
通过研究它们活化d3受体-腺苷酸环化酶和g蛋白偶联内向整流钾通道信号传导途径的能力,表征该新类型的非典型d3受体激动剂的功能性能。该新型家族d3激动剂可用于治疗、缓解或预防遭受帕金森病或其中d3受体异常过度表达的神经病的患者中左旋多巴诱导的运动障碍(lid)。
定义
如本文所使用,下述每条术语具有与本章节中相关的含义。
除非另外定义,本文使用的所有的技术和科技术语通常具有如本发明所述领域普通技术人员所理解的相同含义。一般而言,本文所使用的命名法和细胞培养、分子遗传学、有机化学和肽化学中的实验室方法是本领域熟知的和通常使用的那些。
如本文所使用,冠词“一个(a)”和“一个(an)”指一个或多于一个(即至少一个)该冠词的语法对象。举例而言,“一个要素”意思是一个要素或多于一个要素。
如本文所使用,术语“约”将被本领域普通技术人员理解并且将基于使用其的上下文在一定程度上改变。
如本文所使用,术语“l-多巴”指左旋多巴,也称为l-3,4-二羟苯基丙氨酸或其盐。
如本文所使用,术语“喹吡罗”或“qp”指(4ar,8ar)-5-丙基-4,4a,5,6,7,8,8a,9-八氢-1h-吡唑并[3,4-g]喹诺酮或其盐。
如本文所使用,术语“gr218231”指(+)-(2r)-1,2,3,4-四氢-6-[[(4-甲氧苯基)磺酰基]甲基]-n,n-二丙基-2-萘胺或其盐。
如本文所使用,术语“pbzi”指(3as,9br)-3-丙基-1,2,3a,4,5,9b-六氢苯并[e]吲哚-8-醇或其盐。
如本文所使用,术语“氯氮平”指8-氯-11-(4-甲基哌嗪-1-基)-5h-二苯并[b,e][1,4]二氮杂
如本文所使用,术语“wst-1”指2-(4-碘苯基)-3-(4-硝基苯基)-5-(2,4-二磺苯基)-2h-四唑单钠盐或其盐。
如本文所使用,术语“pd128907”指盐酸(4ar,10br)-3,4a,4,10b-四氢-4-丙基-2h,5h-[1]苯并吡喃并-[4,3-b]-1,4-
如本文所使用,术语“7oh-dpat”指7-羟基-n,n-二丙基-2-氨基四氢萘或其盐。
如本文所使用,术语“6-ohda”指6-羟基多巴胺或其盐。
如本文所使用,术语“普拉克索”指(s)-n6-丙基-4,5,6,7-四氢-1,3-苯并噻唑-2,6-二胺或其盐。
如本文所使用,术语“罗替高汀”指(s)-6-[丙基(2-噻吩-2-基乙基)氨基]-5,6,7,8-四氢萘-1-醇或其盐。
如本文所使用,术语“es609”或“es0609”指4-(2-氯苯基)-丁-2-胺或其盐。
如本文所使用,术语“沙立佐坦”指1-[(2r)-3,4-二氢-2h-苯并吡喃-2-基]-n-([5-(4-氟苯基)吡啶-3-基]甲基)甲胺或其盐。
如本文所使用,术语“fauc73”指(4-乙炔基环己-3-炔基)二丙胺或其盐。
如本文所使用,术语“依替必利”指3-氯-5-乙基-n-{[(2s)-1-乙基吡咯烷-2-基]甲基}-6-羟基-2-甲氧苯甲酰胺或其盐
如本文所使用,术语“lid”指左旋多巴诱导的运动障碍。
如本文所使用,术语“耐受性性质”当应用于d3受体时指当通过包括多巴胺在内的典型激动剂进行重复刺激时,受体信号传导功能渐进的减少。
如本文所使用,术语“srt性质”或“慢响应终止”当应用于d3受体时指在去除激动剂之后,终止d3受体的信号传导功能所用时间增加。
如本文所使用,术语“多肽”指由氨基酸残基、相关的天然发生的结构变体和其合成的非天然发生的类似物经肽键连接组成的聚合物。例如,合成的多肽可使用自动化多肽合成仪合成。如本文所使用,术语“蛋白质”通常指大的多肽。如本文所使用,术语“肽”通常指短的多肽。本文使用常规符号表示多肽序列:多肽序列的左手端是氨基末端,和多肽序列的右手端是羧基末端。
如本文所使用,氨基酸由其全称、其对应的三字母代码或由其对应的单字母代码表示,如下面所示:
如本文所使用,术语“治疗(treament)”或“治疗(treating)”定义为应用或施用本发明的治疗剂即化合物(单独或与另一药剂结合)至患者,或应用或施用治疗剂至从患者分离的组织或细胞系(例如,用于诊断或体外应用),所述患者具有lid、lid症状或发展lid的可能性,目的是医治、治愈、减缓、减轻、改变、治疗、缓解、改善或影响lid、lid的症状或发展lid的可能性。术语“治疗(treament)”或“治疗(treating)”也定义为应用或施用本发明的治疗剂即化合物(单独或与另一药剂结合)至患者,或应用或施用治疗剂至从患者分离的组织或细胞系(例如,用于诊断或体外应用),所述患者具有帕金森病、帕金森病的症状或发展帕金森病的可能性,目的是医治、治愈、减缓、减轻、改变、治疗、缓解、改善或影响帕金森病、帕金森病的症状或发展帕金森病的可能性。基于从药物基因组学(pharmacogenomics)领域获得的知识,可对这种治疗进行具体调节或改变。
如本文所使用,术语“预防(prevent)”或“预防(prevention)”意思是如果什么都没有出现,没有不适或疾病发展,或如果已经发展了不适或疾病,没有进一步的不适或疾病发展。也考虑预防一些或所有的与该不适或疾病相关的症状的能力。
如本文所使用,术语“患者”或“对象”指人或非人哺乳动物。非人哺乳动物包括,例如,家畜和宠物,比如羊、牛、猪、犬科、猫科和鼠科哺乳动物。优选地,患者或对象是人。
如本文所使用,术语“有效量”、“药学上有效量”和“治疗有效量”指无毒的但足够量的试剂以提供期望的生物结果。该结果可以是征兆、症状或病因的减少和/或缓解,或生物系统任何其他期望的改变。在任何个案中适当的治疗量可由本领域技术人员使用常规实验确定。
如本文所使用,术语“药学上可接受的”指材料比如载体或稀释剂,其不消除化合物的生物活性或性质,并且相对无毒,即,该材料可施用至个体,而不造成不期望的生物作用或与其所包含在的组合物中的任何组分不以不利的方式相互作用。
如本文所使用,语言“药学上可接受的盐”指从药学上可接受的无毒酸包括无机酸、有机酸、溶剂化物、水合物或其笼形物制备的施用化合物的盐。这种无机酸的例子是盐酸、氢溴酸、氢碘酸、硝酸、硫酸和磷酸。适当的有机酸可选自例如脂肪族、芳族、羧酸和磺酸类有机酸,其例子为甲酸、乙酸、丙酸、丁二酸、樟脑磺酸、柠檬酸、反丁烯二酸、葡糖酸、羟乙磺酸、乳酸、苹果酸、黏酸、酒石酸、对甲苯磺酸、甘醇酸、葡萄糖醛酸、马来酸、糠酸、谷氨酸、苯甲酸、邻氨基苯甲酸、水杨酸、苯乙酸、扁桃酸、扑酸(双羟萘酸)、甲磺酸、乙磺酸、泛酸、苯磺酸(苯磺酸盐)、硬脂酸、对氨基苯磺酸、海藻酸、半乳糖醛酸等。
如本文所使用,术语“组合物”或“药物组合物”指本发明中有用的至少一种化合物与药学上可接受的载体的混合物。药物组合物利于施用化合物至患者。本领域存在多种施用化合物的技术,包括,但不限于静脉内、口服、气溶胶、肠胃外、眼用、肺部和局部施用。
如本文所使用,术语“药学上可接受的载体”意思是药学上可接受的材料、组合物或载体,比如液态或固体填料、稳定剂、分散剂、悬浮剂、稀释剂、赋形剂、增稠剂、溶剂或包封材料,其参与在患者中携带或输送本发明中有用的化合物或参与携带或输送本发明中有用的化合物至患者,以便其可执行其预期的功能。通常,这种构造从一个器官或身体的一部分携带或输送至另一器官或身体的另一部分。每种载体必须是“可接受的”,意思是与制剂中包括本发明中有用的化合物在内的其他成分相容,并且不伤害患者。可用作药学上可接受载体的材料的一些例子包括:糖,比如乳糖、葡萄糖和蔗糖;淀粉,比如玉米淀粉和马铃薯淀粉;纤维素,和其衍生物,比如羧甲基纤维素钠、乙基纤维素和醋酸纤维素;粉末状黄蓍胶;麦芽;明胶;滑石;赋形剂,比如可可脂和栓剂蜡;油,比如花生油,棉籽油,红花油,麻油,橄榄油,玉米油和大豆油;二醇类,比如丙二醇;多元醇,比如丙三醇,三梨醇,甘露醇和聚乙二醇;酯类,比如油酸乙酯和月桂酸乙酯;琼脂;缓冲剂,比如氢氧化镁和氢氧化铝;表面活性剂;海藻酸;无热源水(pyrogen-freewater);等渗盐水;林格氏溶液;乙醇;磷酸盐缓冲液;和药物制剂中使用的其他无毒相容物质。如本文所使用,“药学上可接受的载体”也包括任何和所有的包衣、抗菌剂和抗真菌剂和吸收延迟剂,以及与本发明中有用的化合物的活性相容且患者生理上可接受的类似物。补充性活性化合物也可并入组合物。“药学上可接受的载体”可进一步包括本发明中有用的化合物的药学上可接受的盐。可包括在用于本发明实践中的其他另外的成分是本领域所知的并且例如在remington’spharmaceuticalsciences(genaro,ed.,mackpublishingco.,1985,easton,pa)中描述,其通过引用并入本文。
如本文所使用,术语“烷基”本身或作为另一取代基的一部分意思是具有指定的碳原子数(即c1-6意思是1至6个碳原子)的直链或支链烃并且包括直链、支链或环状取代基,除非另外指出。例子包括甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基、戊基、新戊基、己基和环丙基甲基。最优选的是(c1-c6)烷基,尤其是乙基、甲基、异丙基、异丁基、正戊基、正己基和环丙基甲基。
如本文所使用,术语“取代的烷基”意思是如上面定义的烷基由选自下述的一个、两个或三个取代基取代:卤根、-oh、烷氧基、-nh2、-n(ch3)2、-c(=o)oh、三氟甲基、-c≡n、-c(=o)o(c1-c4)烷基、-c(=o)nh2、-so2nh2、-c(=nh)nh2和-no2,优选地包含一个或两个选自下述的取代基:卤根、-oh、烷氧基、-nh2、三氟甲基、-n(ch3)2、和-c(=o)oh,更优选地选自卤根、烷氧基和-oh。取代烷基的例子包括但不限于2,2-二氟丙基、2-羧基环戊基和3-氯丙基。
如本文所使用,除非另外指出,术语“烷氧基”,单独或结合其他术语使用,意思是具有如上面定义的指定碳原子数的烷基经氧原子与分子的其余部分连接,比如,例如,甲氧基、乙氧基、1-丙氧基、2-丙氧基(异丙氧基)和更高同系物和异构体。优选的是(c1-c3)烷氧基,尤其是乙氧基和甲氧基。
如本文所使用,除非另外指出,术语“卤基”或“卤根”,单独或作为另一取代基的一部分,意思是氟、氯、溴或碘原子,优选地,氟、氯或溴,更优选地,氟或氯。
如本文所使用,除非另外指出,术语“杂烷基”本身或结合另一术语意思是由指定数目的碳原子和一个或两个选自o、n和s的杂原子组成的稳定的直链或支链烷基,并且其中氮和硫原子可任选地被氧化以及氮杂原子可任选地季铵化。杂原子(一个或多个)可位于杂烷基的任何位置,包括杂烷基其余部分和其所连接的片段之间,以及与杂烷基中最远端的碳原子连接。例子包括:-o-ch2-ch2-ch3、-ch2-ch2-ch2-oh、-ch2-ch2-nh-ch3、-ch2-s-ch2-ch3和-ch2ch2-s(=o)-ch3。至多两个杂原子可相连,比如,例如,-ch2-nh-och3或-ch2-ch2-s-s-ch3。
如本文所使用,术语“芳族”指这样的碳环或杂环,其具有一个或多个多不饱和环且具有芳族特征,即具有(4n+2)个离开原位的π(pi)电子,其中n是整数。
如本文所使用,除非另外指出,单独或结合其他术语使用,术语“芳基”意思是这样的碳环芳族系统,其包含一个或多个环(通常一个、两个或三个环),其中这种环可以悬垂的方式连接在一起比如联苯基,或可稠合比如萘。例子包括苯基、蒽基和萘基。优选的是苯基和萘基,最优选的是苯基。
如本文所使用,术语“芳基-(c1-c3)烷基”意思是其中一个至三个碳亚烷基链连接至芳基的官能团,例如,-ch2ch2-苯基。优选的是芳基-ch2-和芳基-ch(ch3)-。术语“取代的芳基-(c1-c3)烷基”意思是其中芳基被取代的芳基-(c1-c3)烷基官能团。优选的是取代的芳基(ch2)-。类似地,术语“杂芳基-(c1-c3)烷基”意思是其中一个至三个碳亚烷基链连接至杂芳基的官能团,例如,-ch2ch2-吡啶基。优选的是杂芳基-(ch2)-。术语“取代的杂芳基-(c1-c3)烷基”意思是其中杂芳基被取代的杂芳基-(c1-c3)烷基官能团。优选的是取代的杂芳基-(ch2)-。
如本文所使用,除非另外指出,术语“杂环”或“杂环基”或“杂环的”本身或作为另一取代基的一部分意思是未取代的或取代的、稳定的单环或多环杂环环系统,其由碳原子和至少一个选自n、o和s的杂原子组成,并且其中氮和硫杂原子可任选被氧化,并且氮原子可任选地被季铵化。除非另外指出,杂环系统可连接在提供稳定结构的任何杂原子或碳原子处。杂环本质上可为芳族或非芳族的。在一种实施方式中,杂环是杂芳基。
如本文所使用,术语“杂芳基”或“杂芳族”指具有芳族特征的杂环。多环杂芳基可包括部分饱和的一个或多个环。例子包括四氢喹啉和2,3-二氢苯并呋喃基。
非芳族杂环的例子包括单环基团,比如氮丙啶、环氧乙烷、硫杂丙环、氮杂环丁烷、氧杂环丁烷、硫杂环丁烷、吡咯烷、吡咯啉、咪唑啉、吡唑烷、二氧戊环、环丁砜、2,3-二氢呋喃、2,5-二氢呋喃、四氢呋喃、四氢噻吩、哌啶、1,2,3,6-四氢吡啶、1,4-二氢吡啶、哌嗪、吗啉、硫代吗啉、吡喃、2,3-二氢吡喃、四氢吡喃、1,4-二
杂芳基的例子包括吡啶基、吡嗪基、嘧啶基(尤其是2-和4-嘧啶基)、哒嗪基、噻吩基、呋喃基、吡咯基(尤其是2-吡咯基)、咪唑基、噻唑基、
多环杂环的例子包括吲哚基(尤其是3-、4-、5-、6-和7-吲哚基)、二氢吲哚基、喹啉基、四氢喹啉基、异喹啉基(尤其是1-和5-异喹啉基)、1,2,3,4-四氢异喹啉基、噌啉基、喹喔啉基(尤其是2-和5-喹喔啉基)、喹唑啉基、酞嗪基、1,8-萘啶基、1,4-苯并二
上面提到的杂环基和杂芳基部分的列举意欲是代表性的而不是限制性的。
如本文所使用,术语“取代的”意思是原子或原子团替代氢,作为连接至其他基团的取代基。
对于芳基、芳基-(c1-c3)烷基和杂环基,术语“取代的”当应用于这些基团的环时指允许取代处的任何水平的这种取代,即单-、二-、三-、四-或五-取代。取代基独立地选择,并且取代可在任何化学可及位置。在一种实施方式中,取代基的数量在1和4之间变化。在另一实施方式中,取代基的数量在1和3之间变化。在仍另一实施方式中,取代基数量在1和2之间变化。在仍另一实施方式中,取代基独立地选自c1-6烷基、-oh、c1-6烷氧基、卤基、氨基、乙酰氨基和硝基。在仍另一实施方式中,取代基独立地选自c1-6烷基、c1-6烷氧基、卤基、乙酰氨基和硝基。如本文所使用,在取代基是烷基或烷氧基的情况,碳链可为分支的、直链的或环状的,直链的是优选的。
如本文所使用的术语,“说明材料”包括可用于传达本发明化合物效用的表达的出版物、记录、图表或任何其他媒介。在一些情况下,说明材料可以是可用于实现缓解或治疗本文所述各种疾病或不适的试剂盒的一部分。任选地,或可选地,说明材料可描述缓解哺乳动物的细胞或组织中疾病或不适的一种或多种方法。试剂盒的说明材料可以例如贴在包含本发明化合物的容器上或与包含化合物的容器一起运输。可选地,说明材料可与容器分开输送,目的是接收者协作使用说明材料和化合物。例如,说明材料用于试剂盒使用;说明书用于化合物使用;或说明书用于化合物制剂的使用。
本发明的化合物
本发明中有用的化合物可使用有机合成领域熟知的技术合成。
在一个方面,本发明中有用的化合物具有式(i):
其中式(i)中:
r1、r2和r3独立地选自h、氰基、羟基、氨基、乙酰氨基、卤基、烷氧基、硝基、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基、取代的杂环基、芳基、取代的芳基、芳基-(c1-3)烷基、取代的芳基-(c1-3)烷基、羧基、烷基羧基、甲酰基、烷基-羰基、芳基-羰基和杂芳基-羰基;
r4和r5独立地选自h、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基、取代的杂环基、芳基、取代的芳基、芳基-(c1-3)烷基和取代的芳基-(c1-3)烷基;和,
n是2、3、4或5;
或其药学上可接受的盐。
在一种实施方式中,r1、r2和r3独立地选自h、氰基、羟基、氨基、乙酰氨基、卤基、烷氧基、硝基、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基、取代的杂环基、羧基、烷基羧基、甲酰基、烷基-羰基、芳基-羰基和杂芳基-羰基。在另一实施方式中,r1、r2和r3独立地选自h、氰基、羟基、氨基、乙酰氨基、卤基、烷氧基、硝基、c1-6烷基、取代的c1-6烷基、杂烷基、羧基、烷基羧基、甲酰基和烷基-羰基。在仍另一实施方式中,r1、r2和r3独立地选自h、氰基、羟基、氨基、乙酰氨基、卤基、烷氧基、硝基、c1-6烷基、取代的c1-6烷基、杂烷基和羧基。在仍另一实施方式中,r1、r2和r3独立地选自h、氰基、羟基、氨基、乙酰氨基、卤基、烷氧基、硝基、c1-6烷基和羧基。在仍另一实施方式中,r1、r2和r3独立地选自h、氰基、卤基、烷氧基、硝基、c1-6烷基和羧基。在仍另一实施方式中,r1和r2是h,和r3是氯。在仍另一实施方式中,r1、r2和r3独立地选自h、氟、氯、溴、碘、甲氧基、乙氧基、羟基、甲基、乙基或其他c1-6烷基。
在一种实施方式中,n是2、3或4。在另一实施方式中,n是2或3。在仍另一实施方式中,n是2。
在一种实施方式中,r4和r5独立地选自h、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基、取代的杂环基、芳基和取代的芳基。在另一实施方式中,r4和r5独立地选自h、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基和取代的杂环基。在仍另一实施方式中,r4和r5独立地选自h、c1-6烷基、取代的c1-6烷基和杂烷基。在仍另一实施方式中,r4和r5独立地选自h、c1-6烷基和取代的c1-6烷基。在仍另一实施方式中,r4和r5是甲基。在仍另一实施方式中,r5是h、甲基、乙基、丙-1-基、丙-2-基、羟甲基、1-羟乙基、2-羟乙基、1-羟丙-1-基、2-羟丙-1-基、3-羟丙-1-基、1-羟丙-2-基或2-羟丙-2-基。
在一种实施方式中,本发明中有用的化合物选自:
2-氨基-4-(2-氯苯基)丁-1-醇
2-(3-氨基己基)苯酚
4-(2-氯苯基)-丁-2-胺
4-(2-氯苯基)-2-甲氨基-丁烷(也称为4-(2-氯苯基)-n-甲基丁-2-胺)
4-(2-氟苯基)丁-2-胺
4-(2-溴苯基)丁-2-胺
4-(2-碘苯基)丁-2-胺
4-(2-甲氧苯基)丁-2-胺
2-(3-氨基丁基)苯酚
3-(3,4-二乙氧苯基)丙-1-胺
4-(4-氯苯基)丁-2-胺
4-(4-甲氧苯基)丁-2-胺
其混合物,或其药学上可接受的盐。
在另一方面,本发明中有用的化合物具有式(iia)或(iib):
其中式(iia)或(iib)中:
r1和r2独立地选自h、氰基、羟基、氨基、乙酰氨基、卤基、烷氧基、硝基、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基、取代的杂环基、芳基、取代的芳基、芳基-(c1-3)烷基、取代的芳基-(c1-3)烷基、羧基、烷基羧基、甲酰基、烷基-羰基、芳基-羰基和杂芳基-羰基;
r3和r4独立地选自h、c1-6烷基、芳基、杂芳基和取代的c1-6烷基;
r5选自h、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基、取代的杂环基、芳基、取代的芳基、芳基-(c1-3)烷基和取代的芳基-(c1-3)烷基;和,
m是1、2、3或4;
或其药学上可接受的盐。
在一种实施方式中,r1和r2独立地选自h、氰基、羟基、氨基、乙酰氨基、卤基、烷氧基、硝基、c1-6烷基、取代的c1-6烷基、羧基、烷基羧基、甲酰基、烷基-羰基、芳基-羰基和杂芳基-羰基。在另一实施方式中,r1和r2独立地选自h、氰基、羟基、卤基和烷氧基、c1-6烷基和取代的c1-6烷基。
在一种实施方式中,r3和r4独立地选自h和c1-6烷基。
在一种实施方式中,r5选自h、c1-6烷基和取代的c1-6烷基。
在一种实施方式中,m是1、2或3。
在一种实施方式中,本发明中有用的化合物选自
2-(5-氯-1-甲基-1h-吲哚-3-基)乙胺
1-(5-氟-1-甲基-1h-吲哚-3-基)丙-2-胺
1-(5-甲氧基-1-甲基-1h-吲哚-3-基)丙-2-胺
其混合物,或其药学上可接受的盐。
在仍其他方面,本发明中有用的化合物选自:
2,7-二氨基-5-(4-氟苯基)-4-氧-3,4,5,6-四氢吡啶并[2,3-d]嘧啶-6-腈
(z)-2-(1h-苯并[d]咪唑-2-基)-n′-羟基-3-(4-甲氧苯基)丙脒
,其混合物,或其药学上可接受的盐。
本发明的方法
在一个方面,本发明包括在遭受帕金森病的患者中治疗、缓解或预防左旋多巴诱导的运动障碍的方法。方法包括向患者施用包括治疗有效量的选自下述的至少一种化合物的药物组合物:
式(i)的化合物:
其中式(i)中:
r1、r2和r3独立地选自h、氰基、羟基、氨基、乙酰氨基、卤基、烷氧基、硝基、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基、取代的杂环基、芳基、取代的芳基、芳基-(c1-3)烷基、取代的芳基-(c1-3)烷基、羧基、烷基羧基、甲酰基、烷基-羰基、芳基-羰基和杂芳基-羰基;
r4和r5独立地选自h、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基、取代的杂环基、芳基、取代的芳基、芳基-(c1-3)烷基和取代的芳基-(c1-3)烷基;和,
n是2、3、4或5;
式(iia)或(iib)的化合物:
其中式(iia)或(iib)中:
r1和r2独立地选自h、氰基、羟基、氨基、乙酰氨基、卤基、烷氧基、硝基、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基、取代的杂环基、芳基、取代的芳基、芳基-(c1-3)烷基、取代的芳基-(c1-3)烷基、羧基、烷基羧基、甲酰基、烷基-羰基、芳基-羰基和杂芳基-羰基;
r3和r4独立地选自h、c1-6烷基、芳基、杂芳基和取代的c1-6烷基;
r5选自h、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基、取代的杂环基、芳基、取代的芳基、芳基-(c1-3)烷基和取代的芳基-(c1-3)烷基;和,
m是1、2、3或4;
2,7-二氨基-5-(4-氟苯基)-4-氧-3,4,5,6-四氢吡啶并[2,3-d]嘧啶-6-腈;
(z)-2-(1h-苯并[d]咪唑-2-基)-n′-羟基-3-(4-甲氧苯基)丙脒;
其混合物,或其药学上可接受的盐。
在一种实施方式中,r1、r2和r3独立地选自h、氰基、羟基、氨基、乙酰氨基、卤基、烷氧基、硝基、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基、取代的杂环基、羧基、烷基羧基、甲酰基、烷基-羰基、芳基-羰基和杂芳基-羰基。在另一实施方式中,r1、r2和r3独立地选自h、氰基、羟基、氨基、乙酰氨基、卤基、烷氧基、硝基、c1-6烷基、取代的c1-6烷基、杂烷基、羧基、烷基羧基、甲酰基和烷基-羰基。在仍另一实施方式中,r1、r2和r3独立地选自h、氰基、羟基、氨基、乙酰氨基、卤基、烷氧基、硝基、c1-6烷基、取代的c1-6烷基、杂烷基和羧基。在仍另一实施方式中,r1、r2和r3独立地选自h、氰基、羟基、氨基、乙酰氨基、卤基、烷氧基、硝基、c1-6烷基和羧基。在仍另一实施方式中,r1、r2和r3独立地选自h、氰基、卤基、烷氧基、硝基、c1-6烷基和羧基。在仍另一实施方式中,r1和r2是h,和r3是氯。在仍另一实施方式中,r1、r2和r3独立地选自h、氟、氯、溴、碘、甲氧基和乙氧基。
在一种实施方式中,r1和r2独立地选自h、氰基、羟基、氨基、乙酰氨基、卤基、烷氧基、硝基、c1-6烷基、取代的c1-6烷基、羧基、烷基羧基、甲酰基、烷基-羰基、芳基-羰基和杂芳基-羰基。在另一实施方式中,r1和r2独立地选自h、氰基、羟基、卤基和烷氧基、c1-6烷基和取代的c1-6烷基。
在一种实施方式中,n是2、3或4。在另一实施方式中,n是2或3。在仍另一实施方式中,n是2。
在一种实施方式中,r4和r5独立地选自h、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基、取代的杂环基、芳基和取代的芳基。在另一实施方式中,r4和r5独立地选自h、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基和取代的杂环基。在仍另一实施方式中,r4和r5独立地选自h、c1-6烷基、取代的c1-6烷基和杂烷基。在仍另一实施方式中,r4和r5独立地选自h、c1-6烷基和取代的c1-6烷基。在仍另一实施方式中,r4和r5是甲基。在仍另一实施方式中,r5是h、甲基、乙基、丙-1-基、丙-2-基、羟甲基、1-羟乙基、2-羟乙基、1-羟丙-1-基、2-羟丙-1-基、3-羟丙-1-基、1-羟丙-2-基或2-羟丙-2-基。
在一种实施方式中,r3和r4独立地选自h和c1-6烷基。
在一种实施方式中,r5选自h、c1-6烷基和取代的c1-6烷基。
在一种实施方式中,m是1、2或3。
在一种实施方式中,本发明中有用的化合物选自:2-氨基-4-(2-氯苯基)丁-1-醇;2-(3-氨基己基)苯酚;4-(2-氯苯基)-丁-2-胺;4-(2-氯苯基)-2-甲氨基-丁烷;4-(2-氟苯基)丁-2-胺;4-(2-溴苯基)丁-2-胺;4-(2-碘苯基)丁-2-胺;4-(2-甲氧苯基)丁-2-胺;2-(3-氨基丁基)苯酚;3-(3,4-二乙氧苯基)丙-1-胺;4-(4-氯苯基)丁-2-胺;4-(4-甲氧苯基)丁-2-胺;2-(5-氯-1-甲基-1h-吲哚-3-基)乙胺;1-(5-氟-1-甲基-1h-吲哚-3-基)丙-2-胺;1-(5-甲氧基-1-甲基-1h-吲哚-3-基)丙-2-胺;2,7-二氨基-5-(4-氟苯基)-4-氧-3,4,5,6-四氢吡啶并[2,3-d]嘧啶-6-腈;(z)-2-(1h-苯并[d]咪唑-2-基)-n′-羟基-3-(4-甲氧苯基)丙脒;
其混合物,或其药学上可接受的盐。
在一种实施方式中,组合物进一步包括选自下述的药物:左旋多巴、氯氮平、溴隐亭、培高利特、普拉克索、罗匹尼罗、吡贝地尔、卡麦角林、阿扑吗啡和利舒脲,其盐和其混合物。在另一实施方式中,药物组合物与包括左旋多巴的第二药物组合物共同施用至患者。在仍另一实施方式中,药物组合物施用至患者给定时间期间,然后将包括左旋多巴的第二药物组合物施用至患者。在仍另一实施方式中,给定时间期间从约2分钟至约24小时变化。在仍另一实施方式中,患者是人。
在另一方面,本发明包括治疗、缓解或预防患者中帕金森病的方法。方法包括向患者施用包括治疗有效量的选自下述的至少一种化合物的药物组合物:
式(i)的化合物:
其中式(i)中:
r1、r2和r3独立地选自h、氰基、羟基、氨基、乙酰氨基、卤基、烷氧基、硝基、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基、取代的杂环基、芳基、取代的芳基、芳基-(c1-3)烷基、取代的芳基-(c1-3)烷基、羧基、烷基羧基、甲酰基、烷基-羰基、芳基-羰基和杂芳基-羰基;
r4和r5独立地选自h、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基、取代的杂环基、芳基、取代的芳基、芳基-(c1-3)烷基和取代的芳基-(c1-3)烷基;和,
n是2、3、4或5;
式(iia)或(iib)的化合物:
其中式(iia)或(iib)中:
r1和r2独立地选自h、氰基、羟基、氨基、乙酰氨基、卤基、烷氧基、硝基、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基、取代的杂环基、芳基、取代的芳基、芳基-(c1-3)烷基、取代的芳基-(c1-3)烷基、羧基、烷基羧基、甲酰基、烷基-羰基、芳基-羰基和杂芳基-羰基;
r3和r4独立地选自h、c1-6烷基、芳基、杂芳基和取代的c1-6烷基;
r5选自h、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基、取代的杂环基、芳基、取代的芳基、芳基-(c1-3)烷基和取代的芳基-(c1-3)烷基;和,
m是1、2、3或4;
2,7-二氨基-5-(4-氟苯基)-4-氧-3,4,5,6-四氢吡啶并[2,3-d]嘧啶-6-腈;
(z)-2-(1h-苯并[d]咪唑-2-基)-n′-羟基-3-(4-甲氧苯基)丙脒;
其混合物,或其药学上可接受的盐。
在一种实施方式中,r1、r2和r3独立地选自h、氰基、羟基、氨基、乙酰氨基、卤基、烷氧基、硝基、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基、取代的杂环基、羧基、烷基羧基、甲酰基、烷基-羰基、芳基-羰基和杂芳基-羰基。在另一实施方式中,r1、r2和r3独立地选自h、氰基、羟基、氨基、乙酰氨基、卤基、烷氧基、硝基、c1-6烷基、取代的c1-6烷基、杂烷基、羧基、烷基羧基、甲酰基和烷基-羰基。在仍另一实施方式中,r1、r2和r3独立地选自h、氰基、羟基、氨基、乙酰氨基、卤基、烷氧基、硝基、c1-6烷基、取代的c1-6烷基、杂烷基和羧基。在仍另一实施方式中,r1、r2和r3独立地选自h、氰基、羟基、氨基、乙酰氨基、卤基、烷氧基、硝基、c1-6烷基和羧基。在仍另一实施方式中,r1、r2和r3独立地选自h、氰基、卤基、烷氧基、硝基、c1-6烷基和羧基。在仍另一实施方式中,r1和r2是h,和r3是氯。在仍另一实施方式中,r1、r2和r3独立地选自h、氟、氯、溴、碘、甲氧基和乙氧基。
在一种实施方式中,r1和r2独立地选自h、氰基、羟基、氨基、乙酰氨基、卤基、烷氧基、硝基、c1-6烷基、取代的c1-6烷基、羧基、烷基羧基、甲酰基、烷基-羰基、芳基-羰基和杂芳基-羰基。在另一实施方式中,r1和r2独立地选自h、氰基、羟基、卤基和烷氧基、c1-6烷基和取代的c1-6烷基。
在一种实施方式中,n是2、3或4。在另一实施方式中,n是2或3。在仍另一实施方式中,n是2。
在一种实施方式中,r4和r5独立地选自h、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基、取代的杂环基、芳基和取代的芳基。在另一实施方式中,r4和r5独立地选自h、c1-6烷基、取代的c1-6烷基、杂烷基、杂环基和取代的杂环基。在仍另一实施方式中,r4和r5独立地选自h、c1-6烷基、取代的c1-6烷基和杂烷基。在仍另一实施方式中,r4和r5独立地选自h、c1-6烷基和取代的c1-6烷基。在仍另一实施方式中,r4和r5是甲基。在仍另一实施方式中,r5是h、甲基、乙基、丙-1-基、丙-2-基、羟甲基、1-羟乙基、2-羟乙基、1-羟丙-1-基、2-羟丙-1-基、3-羟丙-1-基、1-羟丙-2-基或2-羟丙-2-基。
在一种实施方式中,r3和r4独立地选自h和c1-6烷基。
在一种实施方式中,r5选自h、c1-6烷基和取代的c1-6烷基。
在一种实施方式中,m是1、2或3。
在一种实施方式中,本发明中有用的化合物选自:2-氨基-4-(2-氯苯基)丁-1-醇;2-(3-氨基己基)苯酚;4-(2-氯苯基)-丁-2-胺;4-(2-氯苯基)-2-甲氨基-丁烷;4-(2-氟苯基)丁-2-胺;4-(2-溴苯基)丁-2-胺;4-(2-碘苯基)丁-2-胺;4-(2-甲氧苯基)丁-2-胺;2-(3-氨基丁基)苯酚;3-(3,4-二乙氧苯基)丙-1-胺;4-(4-氯苯基)丁-2-胺;4-(4-甲氧苯基)丁-2-胺;2-(5-氯-1-甲基-1h-吲哚-3-基)乙胺;1-(5-氟-1-甲基-1h-吲哚-3-基)丙-2-胺;1-(5-甲氧基-1-甲基-1h-吲哚-3-基)丙-2-胺;2,7-二氨基-5-(4-氟苯基)-4-氧-3,4,5,6-四氢吡啶并[2,3-d]嘧啶-6-腈;(z)-2-(1h-苯并[d]咪唑-2-基)-n′-羟基-3-(4-甲氧苯基)丙脒;其混合物,或其药学上可接受的盐。
在一种实施方式中,组合物进一步包括选自下述的药物:左旋多巴、氯氮平、溴隐亭、培高利特、普拉克索、罗匹尼罗、吡贝地尔、卡麦角林、阿扑吗啡和利舒脲,其盐和其混合物。在另一实施方式中,患者进一步施用包括选自下述的药物的组合物:左旋多巴、氯氮平、溴隐亭、培高利特、普拉克索、罗匹尼罗、吡贝地尔、卡麦角林、阿扑吗啡和利舒脲、其盐和其混合物。
遭受帕金森病的患者中lid的分子基础
遭受帕金森病的患者中发展lid的分子机制仍未被很好理解。研究显示,许多基因的表达在运动障碍动物中改变。尤其,在啮齿动物和灵长类中,研究已经报道了运动障碍动物的基底核中d3多巴胺受体表达特异性增加。正常表达d2多巴胺受体的区域中d3受体表达增加的功能后果是未知的。
根据功能选择性的概念,配体可有差异地激活与单个gpcr结合的信号传导途径(kenakin,2003,trendspharmacol.sci.24(7):346-354;urban等,2007,j.pharmacol.exp.ther.320(1):1-13)。对于d2受体,之前已经报道了功能选择性(gay等,2004,mol.pharmacol.66(1):97-105)。沙立佐坦——在d2样多巴胺受体具有亲和力的配体——展示了在d2l和d4.2多巴胺受体的功能选择性(kuzhikandathil&bartoszyk,2006,neuropharmacology51:873-884)。沙立佐坦是d2l-和d4.2-girk通道信号传导途径的部分激动剂,但是在d2l-和d4.2-腺苷酸环化酶途径是完全激动剂(kuzhikandathil&bartoszyk,2006,neuropharmacology51:873-884)。随后的研究揭示沙立佐坦对d2s多巴胺受体诱导耐受性和慢响应终止(srt)性质。这表明除了有差异地激活信号传导途径,某些激动剂也能调节固有的受体性质比如耐受性和慢响应终止性质。
d3多巴胺受体展示耐受性和慢响应终止(srt)性质,这将其与d2多巴胺受体区分(图1)。d3受体的耐受性性质描述当被典型的激动剂——包括多巴胺——重复刺激时,受体信号传导功能渐进的降低。srt性质描述在去除激动剂之后终止d3受体的信号传导功能所用时间延长。
已经在培养的黑质神经元中观察到d3受体耐受性和srt,并且人和小鼠d3受体在爪蟾(xenopus)卵母细胞、cho-k1细胞和att-20细胞中异种表达。耐受性和srt性质由天然激动剂多巴胺以及由至今测试的各种合成激动剂引起。d3受体的耐受性和srt性质由宽范围的激动剂浓度引起,在10、30、100和1000nm观察到。该性质在d3-girk、d3-acv和d3-map激酶信号传导途径中被证实。
d2和d3受体性质的差异产生神经放电的差别调制(图1)。这些结果表明,异常表达d3受体耐受性和srt性质的模型可导致运动障碍动物的基底核中神经放电的异常调制并且致使帕金森病中发展lid。根据该模型,如果d3受体耐受性和srt性质可被消除,运动障碍动物的基底核中由过度表达的d3受体调制神经放电与天然表达的d2受体类似并且可能预防运动障碍的表达。
多巴胺d3激动剂的初始研究
为了确定可消除d3多巴胺受体耐受性和srt的激动剂,在功能上筛选合成的偏向d3受体的激动剂。通过刺激在att-20细胞系中单独表达的人d2、d3和d4多巴胺受体,测试化合物的功能活性。在att-20细胞中,这些多巴胺受体结合并且激活g蛋白偶联内向整流钾(girk)通道并且也抑制内生腺苷酸环化酶(kuzhikandathil等,2004,mol.cell.neurosci.26:144-155;westrich&kuzhikandathil,2007,biochim.biophys.acta-mol.cellres.1773:1747-1758)。
使用全细胞电压钳记录,电生理学上测定girk通道的活化(kuzhikandathil等,2004,mol.cell.neurosci.26:144-155)。将新的配体引起的girk通道响应与喹吡罗——d2、d3和d4多巴胺受体的典型完全激动剂——比较。使用商业上可得的elisa试验,确定腺苷酸环化酶抑制(westrich&kuzhikandathil,2007,biochim.biophys.acta-mol.cellres.1773:1747-1758)。
初始功能筛选鉴定氢溴酸顺-8-羟基-3-(正丙基)-1,2,3a,4,5,9b-六氢-1h-苯并[e]吲哚(pbzi)。该化合物已经报道为d3多巴胺受体激动剂;但是,本研究显示,不像传统的d3受体激动剂,pbzi不引起耐受性和srt性质(图2)。
基干混合结构的(hsb)方案
基于混合结构的(hsb)方案(koratgere&welsh,2006,j.comp.aidedmol.des.20:789-802;图3)用于鉴定属于不引起耐受性和srt性质的新类型d3受体激动剂的另外化合物。hsb方案由5个子阶段组成(图3)。i阶段对应于构建卖家可获得的小分子的广泛电子数据库。ii阶段对应于使用混合药效团开发和筛选化合物,起始于来自md模拟的3d结构快相。iii阶段对应于使通过混合药效团筛选的化合物进行聚类、过滤、化学空间分析和分类建模以开发富集的小分子数据库。iv阶段对应于对接来自富集的数据库的分子至drd3受体,分数来自一致性评分方案。v阶段对应于测试最佳评级的化合物它们对所有多巴胺受体的活性。
开发小分子的广泛电子数据库:
由来自商业卖主的化合物组成的zinc数据库子集(irwin&soichet,2005,j.chem.inf.models45:177-82),连同包括天然产物的其他化合物、来自pdb的配体和fda批准的药物,形成近3百万化合物的完整数据库。所有的化合物以sdf格式文件获取,转化成mol2格式并且使用tripos力场进行能量最小化。进一步,针对冗余度,过滤数据库中的所有分子并且根据它们对应的卖主列表重新命名。
开发组合的配体-蛋白药效团:
产生组合的药效团(也称为混合药效团)是该方法的重要步骤。因此,定制药效团以捕获pbzi和d3受体之间相互作用的必要特征是重要的。
这种信息获自pbzi-d3受体复合物的3d结构复合物。使用部分逆激动剂与其结合的β2肾上腺素受体(b2ar)的晶体结构(2rh1)构建d3受体的同源模型(cherezov等,2007,science318(5854):1258-65)。跨膜(tm)区域和环区域与来自β肾上腺素晶体结构的对应残基具有显著的同源性。b2ar中第三细胞内环比d3受体中对应的环更长并且因此未对比对(图4)中显示的大的缺失建模。但是,显示对于srt和受体耐受性非常重要的第三细胞内环的其余部分使用来自b2ar结构的比对坐标建模。进一步,整个结构使用能量最小化和分子动力学模拟优化,生产运行2ns长。使用namd(kalé等,1999,j.comput.physics151(1):283-312)——具有charmm力场(brooks等,1983,j.comput.chem.4:187-217),进行所有的模拟。
精制的结构用于进一步的对接实验。多巴胺、pd128907和pbzi使用gold程序(ver4.0)对接(jones等,1997,j.mol.biol.267:727-48)。对接的复合物使用namd程序进行能量最小化。pbzi和d3受体之间重要的相互作用用于构建混合药效团(图5)。产生多种药效团模型,包括对于设计d3受体激动剂关键的4个药效团成分的结合,比如tm3中与天冬氨酸的盐桥以及与来自tm5和tm6的芳族氨基酸簇的疏水相互作用。进一步,tm2和tm3之间残基的小交换显示为有助于设计高选择性的d4激动剂和拮抗剂(kortagere等,2004,mol.pharmacol.66:1491-99)。将这些生化和功能证据与混合药效团的设计结合,然后用于筛选小分子数据库(其在上面描述)。
使用疏水核心和两个氢键受体-供体对的初步筛选产生满足药效团并且与d3受体对接的具有高分数的85个化合物。一个代表性化合物4-(2-氯苯基)-丁-2-胺(图6)用于体外和体内功能表征。4-(2-氯苯基)-丁-2-胺的对接与d3受体中母体pbzi化合物非常类似,产生与asp110必要的盐桥相互作用和与芳族残基比如phe345和his349的疏水芳族相互作用(图6)。
过滤、化学空间分析和聚类模块:
过滤方案:
源自基于药效团的筛选的化合物基于源自chemaxon程序的物理化学性质比如形状、logp、体积、tpsa和分子量聚类。lipinski五规则用作第一过滤器,并且基于回归的血脑屏障(bbb)渗透模型——其可滤出bbb渗透的化合物——是第二水平的过滤器(kortagere等,2008,pharm.res.8:1836-45)。bbb回归模型是如下描述的概括模型:
logbb(pred)=0.3408*logp-0.0192*tpsa+0.2503*a_nn+0.1467*a_no+0.1069*logs-0.0011*质量-0.0001*体积0.0602*#rot.键。
其中:a_nn是氮原子的数量,a_no是氧原子的数量,tpsa是拓扑极性表面积,logs是溶解度和logp是水/辛醇分配系数和疏水性的量度,和#rot.键是可旋转键的数量。
化学空间分析和聚类技术:
进行化合物的化学空间分析以分析化合物是否属于一个或多个化学类。去除与查询结构化学上不一致的离群值;但是使用宽的截断值以允许骨架迁跃(scaffoldhoping)。使用支持向量机(supportvectormachine)(svm)技术进行化合物的聚类和基于qsar的分类。基于svm的模型的方法和应用对可穿透bbb的化合物分类和对pxr活化剂分类已经实现了高精度。
因此,采用该方法基于靶标对化合物分类并且产生数据库的富集可用于避免筛选不期望的分子。本文开发的数据库中的分子不含立体异构体。在富集过程期间,化学上相同的化合物通过仅仅保留该分子的单个拷贝而去除多余的。
开发定制的评分方案:
gold程序用于初步对接。确定配体的活性位点定义为在
此外,为评估这些最佳评级的化合物对多巴胺d3受体的选择性,将它们针对其他4个多巴胺受体对接并且评分。仅仅针对多巴胺d3受体分数最佳的那些化合物用作迭代hsb方法中的前导分子。初步结果已经验证hsb方案,作为新型化合物,4-(2-氯苯基)-丁-2-胺(图7)被鉴定为不引起耐受性和srt性质的d3受体激动剂。
在基于细胞的试验中新型d3受体激动剂的功能、选择性和细胞毒性特征的评估
上面鉴定的前导化合物在基于细胞的试验中评估以确定它们激活d3受体而不诱导耐受性和srt性质的能力。通过从稳定表达各种多巴胺受体亚型的att-20神经内分泌细胞系中产生功能剂量-响应曲线,评估针对各种多巴胺受体亚型的选择性。通过测量前导化合物对代表内分泌、神经元和肝细胞类型的三种不同细胞系的增殖和细胞死亡的作用,评估细胞毒性。确定前导化合物是否进步通过临床前开发方案的每个标准显示在图8中。
d3多巴胺受体的激动:
为了确定化合物是否是d3多巴胺受体的激动剂,使用稳定表达人d3受体的att-20神经内分泌细胞系。在初始筛选中,使用全细胞电压钳记录,测量300nm前导化合物激活g蛋白偶联内向整流钾(girk)通道的能力(图1)。该方案之前用于进行全细胞电压钳和测量激动剂诱导的girk响应。将剧烈的配体诱导的girk电流与当量浓度的天然激动剂多巴胺或完全激动剂喹吡罗引起的电流比较。通过测量由前导化合物引起的非转染att-20细胞中的girk响应并且也在d2样多巴胺受体拮抗剂依替必利和d3受体选择性拮抗剂gr218231((+)-(2r)-1,2,3,4-四氢-6-[[(4-甲氧苯基)磺酰基]甲基]-n,n-二丙基-2-萘胺)的存在下,确定响应的特异性。为了进一步开发考虑,前导化合物应当产生等于或大于由当量浓度(300nm)的多巴胺或喹吡罗引起的girk响应。
d3受体耐受性和srt的消除:
为了确定化合物是否消除d3受体耐受性和srt,测量对前导化合物两次连续处理的d3受体激活的girk响应。如图9所图解,第二响应与第一响应的比对于消除耐受性的激动剂接近1,但是对于引起耐受性(例如对于多巴胺和喹吡罗)的激动剂近似为0.4。在一种实施方式中,为了进一步考虑,前导化合物应当具有在0.7和1.0之间的第二与第一girk响应的比。
在第二系列实验中,使用与girk通道的结合作为测试,测定前导化合物与100nm、300nm和1000nm多巴胺竞争以阻碍对d3受体耐受性和srt发展的能力。如图10中所图解,在该测试中,pbzi(其在lid模型中减少aim分数)能够有效地与多巴胺竞争。
确定前导化合物对各种d2样多巴胺受体的ec50:
稳定表达d2s、d2l、d3、d4.2和d4.4多巴胺受体的人同工型的att-20神经内分泌细胞系是可用的。另外,稳定表达d1多巴胺受体和5ht-1a5-羟色胺受体的att-20细胞也是可用的。此外,非转染att-20细胞表达内生促生长素抑制素和蕈毒碱受体。
作为初始步骤,从稳定表达各种多巴胺受体的细胞系分离的膜用于进行竞争性放射性配体结合测试。该方法为各种多巴胺受体亚型产生了前导化合物的ki。作为接下来的步骤,各种细胞系用于在两个功能测试中获得剂量响应曲线。
在第一测试中,全细胞电压钳记录用于测量对增加浓度的前导化合物(0.01nm至3,000nm剂量范围)的girk响应。使用hill方程拟合数据点,确定每个受体亚型的ec50值。
在第二功能测试中,评估d2样多巴胺受体抑制腺苷酸环化酶活性的能力。测量前导化合物剂量依赖性降低福司柯林诱导的camp水平的能力。通过使用hill方程拟合数据点,确定每个受体亚型的ec50值。之前,在这些细胞系中两种功能测试中已经确定了多巴胺和喹吡罗的ec50和ic50值。在一种实施方式中,为了更进一步,前导化合物的ec50或ic50值必须小于或等于多巴胺或喹吡罗在这些测试的任何一种中的值。图11图解说明pbzi和4-(2-氯苯基)-丁-2-胺对d3多巴胺受体的剂量响应曲线的例子。
前导化合物细胞毒性的评估:
为了确定前导化合物的细胞毒性,使用细胞增殖标记试剂wst-1(2-(4-碘苯基)-3-(4-硝基苯基)-5-(2,4-二磺苯基)-2h-四唑单钠盐;rocheappliedscience)。确定前导化合物对三个不同细胞系(作为神经内分泌细胞模型的att-20细胞,作为儿茶酚胺能神经元细胞模型的cad细胞和作为肝细胞模型的hepg2)的增殖的作用。三个细胞系一起提供对细胞毒性的初步筛选。
将细胞用宽浓度范围的前导化合物(0.01nm至100μm)处理24小时,接着用wst-1温育另外的4小时,并且使用分光光度计确定吸光度值。细胞毒性筛选中进行选择的标准是前导化合物应当展示大于10μm的ec50。随着前导化合物开发的进步,对化合物也进行体内admet、毒性和药物代谢动力学分析。
移动行为测试中新型d3受体激动剂作用的评估
评估本发明中有用的化合物它们在lid的大鼠内条纹6-ohda损伤模型中降低aim分数的能力。
评估balb/c小鼠中前导化合物对移动活性的作用:
靶向多巴胺d3受体的诱导耐受性和不诱导耐受性的激动剂有差异地改变移动活性。具体地,诱导耐受性的化合物(例如,pd128907,也称为盐酸(4ar,10br)-3,4a,4,10b-四氢-4-丙基-2h,5h-[1]苯并吡喃并-[4,3-b]-1,4-
初步结果(实施例5)与下述研究一致,该研究已经表明典型的d2/d3受体激动剂比如喹吡罗、pd128907或7-羟-n,n-二丙基-2-氨基四氢萘(7oh-dpat)的施用引起特点为初始抑制和随后刺激移动的两阶段移动响应。本文报道的研究显示,施用消除耐受性和srt的d3受体激动剂比如pbzi至balb/c小鼠仅仅引起活动减退。相反,pd128907对d3多巴胺受体引起耐受性和srt并且诱导活动减退和活动过度(图12)。因为pbzi显著减少大鼠lid模型中的aim分数(图13),该激动剂诱导的移动活性测试可用作成功减少lid模型中运动障碍的化合物的预报者。
在一种实施方式中,研究的目标是检验该假设,因为其可导致开发简单的行为试验,用于筛选可能有益于治疗lid的前导化合物。所以,施用三个不同剂量的前导化合物并且测定它们对移动活性的作用。基于上述基于细胞的测试的前导化合物的功能ec50值和结合亲和力确定剂量。将化合物施用至balb/c小鼠并且使用truscan自动行为监测系统(coulborninstruments)监测移动活性。在非限制性方面,消除d3受体耐受性和srt的前导化合物在移动测试中仅仅引起活动减退,与pbzi结果一致(图12)。
评估前导化合物在大鼠内条纹6-ohda损伤模型中的作用:
如果d3受体的耐受性和srt性质有助于内条纹6-羟基多巴胺(6-ohda)损伤大鼠中lid的表达,在施用左旋多巴之前,施用不诱导耐受性的d3受体激动剂,比如pbzi,应当减少或消除与运动障碍相关的症状。
帕金森病单侧内条纹6-ohda损伤的大鼠模型已经被深入表征并且广泛地用于测试神经保护和移植策略以治疗帕金森病。该大鼠帕金森病模型中黑质多巴胺神经元局部和缓慢地渐进恶化和伴随的运动缺陷发展好像模拟了遭受帕金森病的患者中的渐进恶化。尤其感兴趣的是在该动物模型中lid的发展,其与临床上见到的异常不随意运动(aim)类似。
本文使用winkler等2002描述的内条纹6-ohda大鼠帕金森病模型。具体地,使用通过在腹外侧纹状体中的三个不同位置注射6-ohda(7μg)诱导单侧内条纹损伤的大鼠。三个注射位置的坐标以mm计为:注射位置1:ap:+1.0;ml:-3.0;dv:-5.0;注射位置2:ap:-0.1;ml:-3.7;dv:-5.0;注射位置3:ap:-1.2;ml:-4.5;dv:-5.0。前-后(ap)和中侧(ml)坐标从前囱点起。背腹(dv)坐标从硬脑膜起。齿条(toothbar)位置是0。
内条纹6-ohda损伤的和假(sham)损伤的大鼠获自charlesriver实验室客户外科服务部,wilmington,massachusetts,usa。商业卖主在损伤一周后测试损伤动物的苯异丙胺诱导的旋转,然后运输。之前的研究已经表明通过三部位注射损伤的动物一周后展示苯丙胺诱导的旋转显著减少。损伤的动物在损伤对侧的肢中展示运动迟缓/运动不能症。在实验中,通过进行前肢踏步试验,首先确定左旋多巴改善对侧爪中运动迟缓/运动不能症的能力。随后,踏步试验也提供检验前导化合物是否干扰左旋多巴改善运动迟缓/运动不能症能力的方式。使用踏步试验在损伤后三周测量损伤对前肢运动迟缓/运动不能症的作用。一旦在损伤对侧的爪中确定踏步能力的明显缺陷,将增加剂量的左旋多巴(2、4和6mg/kg)与15mg/kg苄丝肼(外周多巴-脱羧酶抑制剂)一起施用并且使用踏步试验在注射后60分钟评估踏步能力。为长期施用阶段选择的左旋多巴剂量是显著改善踏步试验中分数的剂量(图14)。
损伤之后7.5周,开始长期左旋多巴治疗(6mg/kg,单个剂量每天给15mg/kg苄丝肼)。在开始长期左旋多巴治疗之后,在第1、8、10、17和24天测定踏步试验和aim分数。长期左旋多巴施用后,损伤的动物展示异常不随意运动(aim),其包括移动的、轴向的、肢和口舌组成部分。基于运动障碍症状的频率和严重性,使用分级表为aim评分。应当理解,大鼠模型中就频率和严重性而言的各种aim组分的等级顺序为:肢>口舌=轴向>>移动。该损伤模型已经用于测试pbzi对lid的作用(实施例3)。
为了确定aim分数,将大鼠放置在透明的玻璃圆筒中并且使用数字录像记录仪记录。实验不知情的记分员从注射左旋多巴之前20分钟到之后180分钟每隔20分钟观察一分钟。根据大鼠运动障碍表,确定左旋多巴诱导的aim分数。在开始长期左旋多巴治疗之后大概10天,6-ohda损伤的动物发展lid。通过在开始长期左旋多巴施用之后第10、17和24天,在左旋多巴施用之前施用pbzi,确定pbzi降低左旋多巴诱导的aim分数的能力。也在这些天进行踏步试验以评估pbzi对左旋多巴改善损伤动物中的踏步缺陷的能力。测试两种剂量的前导化合物。通过化合物对受体的亲和力和上述移动行为试验的结果,确定剂量。
实验的时程显示在图15中。3部位单侧内条纹6-ohda注射后一周,对大鼠进行苯丙胺诱导的旋转检验以确定损伤的动物。损伤后三周,对动物进行踏步试验以确定左旋多巴前的踏步缺陷。损伤后5周,在指示的天数中将增加剂量(2至6mg/kg)的左旋多巴每天仅仅施用一次并且进行踏步试验。损伤后7.5周,将左旋多巴(6mg/kg)每天施用一次,持续3.5周。在开始长期施用之后第1天和第8天,在左旋多巴注射后确定踏步试验和aim分数。在第10、17和24天,在共同注射左旋多巴和化合物后,确定踏步试验和aim分数。11周末,处死动物并且脑组织用于组织学分析:将动物的脑部分染色酪氨酸羟化酶以评估损伤的半球中多巴胺能神经元的损失。评估来自一些动物的脑部分的d2和d3受体结合和mrna表达。
联合治疗
本发明的化合物意欲结合可用于治疗帕金森病的一种或多种另外的化合物用于本发明方法。这些另外的化合物可包括本发明的化合物或已知用于治疗、预防或减少帕金森病的症状的化合物,例如,商业上可得的化合物。
在非限制例子中,本发明的化合物可结合下述药物的一种或多种使用:左旋多巴、氯氮平、溴隐亭、培高利特、普拉克索、罗匹尼罗、吡贝地尔、卡麦角林、阿扑吗啡和利舒脲,其盐和其混合物。
协同作用可例如使用合适的方法计算,比如,例如sigmoid-emax方程(holford&scheiner,19981,clin.pharmacokinet.6:429-453)、loewe相加方程(loewe&muischnek,1926,arch.exp.patholpharmacol.114:313-326)和中效方程(chou&talalay,1984,adv.enzymeregul.22:27-55)。上面提到的每个方程可应用于实验数据以产生相应的图以帮助评估药物联合的效果。与上面提到的方程相关的相应的图分别是浓度-效果曲线、等效线图曲线和联合指数曲线。
施用/剂量/制剂
施用方案可影响有效量的构成。治疗制剂可在lid出现之前或之后施用至患者。进一步,数个分开的剂量以及错开的剂量可每天或顺序施用,或剂量可持续地灌注,或可以是弹丸注射。进一步,按照治疗的紧急情况或预防情况所指示,治疗制剂的剂量可成比例地增加或降低。
可以使用已知的程序,以有效治疗患者中lid的剂量和时间期间施用本发明的组合物至患者,优选地哺乳动物,更优选地人。实现治疗效果必须的治疗化合物有效量可根据多种因素而变化,比如患者疾病或不适的状态;患者的年龄、性别和体重;和治疗化合物治疗患者中lid的能力。可调整剂量方案以提供最佳的治疗响应。例如,数个分开的剂量可每日施用或剂量可成比例地减少——按照治疗情况的紧急情况所指示的。本发明治疗化合物的有效剂量范围的非限制性例子为从约1至5,000mg/kg体重/每天。本领域技术人员能够研究相关因素并且做出关于治疗化合物有效量的决定而不用过多的实验。
可改变本发明药物组合物中活性成分的实际剂量水平以获得有效实现具体患者、组合物和施用模式期望的治疗响应而对患者无毒的活性成分量。
具体地,选择的剂量水平将依赖于因素的变化,因素包括采用的具体化合物的活性,施用时间,化合物排泄速率,治疗的持续时间,结合化合物使用的其他药物、化合物或材料,被治疗患者的年龄,性别,体重,状况,一般健康和之前的医疗史以及医学领域已知的类似因素。
具有本领域普通知识的医生,例如,内科医师或兽医,可容易地确定并且开处方需要的有效量药物组合物。例如,内科医师或兽医在开始可以以小于实现期望治疗效果所需要的水平在药物组合物中使用本发明化合物的剂量,并且逐渐增加剂量直到实现期望的效果。
在具体的实施方式中,以单位剂型配制化合物是尤其有利的,以便容易施用和剂量的均匀。如本文所使用,单位剂型指适合作为被治疗患者单次剂量的实体上分散的单位;包含预定量的治疗性化合物的每个单位计算为与必要的药学载体结合产生期望的治疗效果。本发明的单位剂型由下述规定并且依赖于下述:(a)治疗性化合物的独特性质和将要实现的具体治疗效果,和(b)混合/配制用于治疗患者中lid的这种治疗性化合物的领域中固有的限制。
在一种实施方式中,本发明的组合物使用一种或多种药学上可接受的赋形剂或载体配制。在一种实施方式中,本发明的药物组合物包括治疗有效量的本发明化合物和药学上可接受的载体。
载体可以是溶剂或分散介质,其包括,例如,水、乙醇、多元醇(例如,甘油、丙二醇和液态聚乙二醇和类似多元醇)、其合适的混合物和植物油。可保持适当的流动性,例如,通过使用包衣比如卵磷脂、在分散的情况下通过保持必要的颗粒尺寸和通过使用表面活性剂。可通过各种抗菌和抗真菌剂,例如,对羟基苯甲酸酯类、氯丁醇、苯酚、抗坏血酸、硫柳汞和类似物,实现防止微生物作用。在许多情况下,在组合物中优选包括等渗剂,例如,糖、氯化钠或多元醇比如甘露醇和三梨醇。可通过在组合物中包括延迟吸收的试剂,例如单硬脂酸铝或明胶,可实现延长可注射组合物的吸收。在一种实施方式中,药学上可接受的载体不单独为dmso。
在一种实施方式中,本发明的组合物以范围从每天1次至5次或更多次的剂量施用至患者。在另一实施方式中,本发明的组合物以下述范围剂量施用至患者,所述范围包括,但不限于每天一次、每两天一次、每三天一次至一周一次和每两周一次。对本领域技术人员显而易见的是施用本发明的各种组合的组合物的频率将在个体间变化,其取决于许多因素,包括但不限于年龄、被治疗的疾病或不适、性别、总体健康和其他因素。因此,本发明不应解释为限于任何具体的剂量方案并且施用至任何患者的精确剂量和组合物将由主治医师考虑关于患者的所有其他因素而确定。
本发明的化合物施用的范围可为从约1μg至约10,000mg,约20μg至约9,500mg,约40μg至约9,000mg,约75μg至约8,500mg,约150μg至约7,500mg,约200μg至约7,000mg,约3050μg至约6,000mg,约500μg至约5,000mg,约750μg至约4,000mg,约1mg至约3,000mg,约10mg至约2,500mg,约20mg至约2,000mg,约25mg至约1,500mg,约50mg至约1,000mg,约75mg至约900mg,约100mg至约800mg,约250mg至约750mg,约300mg至约600mg,约400mg至约500mg,以及其间的任何和所有的全部或部分增量。
在一些实施方式中,本发明化合物的剂量为从约1mg和约2,500mg。在一些实施方式中,在本文所描述的组合物中使用的本发明化合物的剂量小于约10,000mg,或小于约8,000mg,或小于约6,000mg,或小于约5,000mg,或小于约3,000mg,或小于约2,000mg,或小于约1,000mg,或小于约500mg,或小于约200mg,或小于约50mg。类似地,在一些实施方式中,本文所述的第二化合物(即,用于治疗帕金森病的药物)的剂量小于约1,000mg,或小于约800mg,或小于约600mg,或小于约500mg,或小于约400mg,或小于约300mg,或小于约200mg,或小于约100mg,或小于约50mg,或小于约40mg,或小于约30mg,或小于约25mg,或小于约20mg,或小于约15mg,或小于约10mg,或小于约5mg,或小于约2mg,或小于约1mg,或小于约0.5mg,以及其间任何和所有的全部或部分增量。
在一种实施方式中,本发明涉及包装的药物组合物,其包括容纳单独或结合第二药剂的治疗有效量的本发明化合物的容器;和使用化合物治疗、预防或减少患者中lid的一种或多种症状的说明书。
制剂可以以与常规赋形剂即药学上可接受的有机或无机载体物质——其适于口服、肠胃外、鼻、静脉内、皮下、肠或本领域已知的任何其他合适的施用模式——的混合物应用。药物制剂可被灭菌并且如果需要,可与辅助剂混合,例如润滑剂、防腐剂、稳定剂、湿润剂、乳化剂、影响渗透压缓冲液的盐、着色剂、调料和/或芳香物质和类似物。在需要的时候,它们也可结合其他活性剂,例如,其他止痛剂。
术语“容器”包括用于容纳药物组合物的任何贮器。例如,在一种实施方式中,容器是包含药物组合物的包装。在其他实施方式中,容器不是包含药物组合物的包装,即,容器是贮器,比如盒子或小瓶,其包含包装的药物组合物或未包装的药物组合物和药物组合物的使用说明书。而且,包装技术是本领域熟知的。应当理解,药物组合物的使用说明书可包含在包含药物组合物的包装上,并且这样说明书与包装产品形成改进的功能关系。但是,应当理解,说明书可包含与化合物进行其期望功能例如治疗、预防或减轻患者中lid的能力相关的信息。
本发明的任何组合物的施用途径包括口、鼻、直肠、阴道内、肠胃外、含服、舌下或局部。用于本发明的化合物可配制用于以任何合适的途径施用,比如口服或肠胃外,例如,经皮的、经粘膜的(例如,舌下、舌、(经)含服、(经)尿道、阴道(例如,经阴道和阴道周围)、鼻(内)和(经)直肠)、膀胱内、肺内、十二指肠内、胃内、鞘内、皮下、肌内、真皮内、动脉内、静脉内、气管内、吸入和局部施用。
合适的组合物和剂型包括,例如,片剂、胶囊、小胶囊、丸剂、软胶囊(gelcap)、锭剂、分散剂、悬浮液、溶液、糖浆、颗粒、滴剂(bead)、经皮贴片、凝胶、粉末、粒剂、乳浆剂、糖锭、乳剂、膏剂、膏药、洗液、圆片、栓剂、用于鼻或口服施用的液体喷雾剂、用于吸入的干粉末或气溶胶化的制剂、用于膀胱内施用的组合物和制剂以及类似形式。应当理解,本发明中有用的制剂和组合物不限于本文所描述的具体制剂和组合物。
口服施用
对于口服应用,尤其合适的是片剂、锭剂、液体、滴剂、栓剂、或胶囊、小胶囊和软胶囊。意欲口服使用的组合物可根据本领域已知的任何方法制备并且这种组合物可包含选自适合制造片剂的惰性的、无毒的药学上赋形剂的一种或多种试剂。这种赋形剂包括例如惰性稀释剂比如乳糖;粒化剂和崩解剂比如玉米淀粉;结合剂比如淀粉;和润滑剂比如硬质酸镁。片剂可没有包衣或它们可通过已知的技术包衣,为了精美或延迟活性成分的释放。口服使用的制剂也可以以硬明胶胶囊提供,其中活性成分与惰性稀释剂混合。
对于口服施用,本发明的化合物可为片剂或胶囊的形式,其通过常规手段用下述制备:药学上可接受的赋形剂,比如结合剂(例如,聚乙烯吡咯烷酮、羟丙基纤维素或羟丙基甲基纤维素);填料(例如,玉米淀粉、乳糖、微晶纤维素或磷酸钙);润滑剂(例如,硬酯酸镁、滑石或二氧化硅);崩解剂(例如,淀粉乙醇酸钠钠);或湿润剂(例如,月桂基硫酸钠)。如果需要,片剂可使用合适的方法和包衣材料包衣,比如从colorcon,westpoint,pa.可得的opadrytm膜包衣系统(例如,opadrytmoy型、oyc型、organicentericoy-p型、aqueousentericoy-a型、oy-pm型和opadrytmwhite,32k18400)。口服施用的液体制剂可为溶液、糖浆或悬浮液的形式。液体制剂可通过常规手段用下述药学上可接受的添加剂制备:比如悬浮剂(例如,三梨醇糖浆、甲基纤维素或氢化可食用脂肪);乳化剂(例如,卵磷脂或金合欢);非水性媒介(例如,杏仁油、油酯类或乙醇);和防腐剂(例如,对羟基苯甲酸甲酯或丙酯,或山梨酸)。
药学领域熟知粒化技术用于改变活性成分的起始粉末或其他颗粒状材料。粉末通常与粘合剂材料混合成较大永久性自由流动团块或颗粒,称为“成粒作用”。例如,使用溶剂的“湿”成粒方法一般的特点在于粉末与粘合剂材料混合并且用水或有机溶剂在导致形成湿颗粒状物质的条件下混合,然后溶剂必须从其蒸发。
熔融成粒作用通常在于使用在室温下为固体或半固体的材料(即具有相对低的软化点或熔点范围),以促进粉末状或其他材料的成粒,其必须是在缺乏添加的水或其他液体溶剂的情况下。当加热至在熔点范围内的温度时,低熔点固体液化以起到粘合剂或粒化媒介的作用。液化的固体本身扩散在与其接触的粉末状材料的表面,并且当冷却时,形成固体颗粒状物质,其中初始材料结合在一起。然后所得熔化颗粒可提供至压片机或形成胶囊,用于制备口服剂型。通过形成固体分散物或固溶体,熔化颗粒改善了活性成分(即药物)的溶解速率和生物利用度。
美国专利号5,169,645公开了具有改善的流动性能的直接可压缩的含蜡颗粒剂。当蜡与某些流动改善添加剂混合在熔融物中,随后混合物冷却和成粒,获得颗粒剂。在某些实施方式中,仅蜡本身熔化在蜡(一种或多种)和添加剂(一种或多种)的熔融组合中,并且在其他情况下蜡(一种或多种)和添加剂(一种或多种)都熔化。
本发明也包括多层片剂,其包括提供延迟释放本发明一种或多种化合物的层,以及提供立即释放用于治疗帕金森病的药物的进一步层。使用蜡/ph敏感性聚合物混合物,可获得其中包裹活性成分的胃不可溶组合物,确保其延迟释放。
肠胃外施用
对于肠胃外施用,本发明的化合物可配制用于注射或灌注,例如,静脉内、肌内或皮下注射或灌注,或用于以弹丸剂量施用和/或持续灌注。可使用在油性或水性媒介中的任选地包含其他配制剂如悬浮剂、稳定剂和/或分散剂的悬浮液、溶液或乳剂。
另外的施用形式
本发明另外的剂型包括如下述中描述的剂型:美国专利号:6,340,475、6,488,962、6,451,808、5,972,389、5,582,837和5,007,790。本发明另外的剂型也包括如下述美国专利申请中描述的剂型:20030147952、20030104062、20030104053、20030044466、20030039688和20020051820。本发明另外的剂型也包括如下述pct申请中描述的剂型:wo03/35041、wo03/35040、wo03/35029、wo03/35177、wo03/35039、wo02/96404、wo02/32416、wo01/97783、wo01/56544、wo01/32217、wo98/55107、wo98/11879、wo97/47285、wo93/18755和wo90/11757。
控释制剂和药物输送系统
在某些实施方式中,本发明的制剂可为,但不限于,短期,快速开始(rapid-offset),以及受控制剂,例如,持续释放、延迟释放和脉冲释放。
术语持续释放以其常规意思使用,指药物制剂在延长的时间期间内提供逐渐的药物释放,并且尽管不是必须,其可在延长的时间期间导致药物基本上恒定的血液水平。该时间期间可以长如一个月或更长,并且应当是比以弹丸形式施用的相同量药剂更长的释放。
对于持续释放,化合物可用为化合物提供持续释放性能的合适的聚合物或疏水材料配制。这样,使用本发明方法的化合物可以以微粒形式施用,例如,通过注射,或通过植入以晶片或圆片形式施用。
在本发明优选的实施方式中,使用持续释放制剂,本发明的化合物单独或与另一药剂结合施用至患者。
术语延迟释放在本文中以其常规的意思使用,指药物制剂在药物施用之后的延迟后提供药物的初始释放,并且尽管不是必须,其可包括从约10分钟至多达约12小时的延迟。
术语脉冲释放在本文中以其常规的意思使用,指药物制剂以在药物施用之后产生该药物脉冲型血浆特征的方式提供该药物的释放。
术语立即释放在本文中以其常规的意思使用,指药物制剂在药物施用之后立即提供药物的释放。
如本文所使用,短期指在药物施用之后的多达并且包括约8小时、约7小时、约6小时、约5小时、约4小时、约3小时、约2小时、约1小时、约40分钟、约20分钟、或约10分钟以及其任何或所有的全部或部分增量的任何时间期间。
如本文所使用,快速开始指在药物施用之后的多达和包括约8小时、约7小时、约6小时、约5小时、约4小时、约3小时、约2小时、约1小时、约40分钟、约20分钟、或约10分钟,以其任何或所有的全部或部分增量的任何时间期间。
剂量
本发明化合物的治疗有效量或剂量取决于患者的年龄、性别和体重,患者目前的医学情况和被治疗患者中帕金森病的进展。根据这些和其他因素,本领域技术人员能够确定适当的剂量。
本发明化合物的合适剂量范围可为每天约0.01mg至约5,000mg,比如每天约0.1mg至约1,000mg,例如,约1mg至约500mg,比如约5mg至约250mg。剂量可以以单个剂量或以多剂量施用,例如每天1次至4次或更多次。当使用多剂量时,每个剂量的量可相同或不同。例如,每天1mg的剂量可施用为两个0.5mg剂量,剂量之间约12小时间隔。
应当理解,在非限制例子中,每天施用的化合物量可每天、每隔天、每两天、每三天、每四天或每五天施用。例如,对于每隔天施用,每天5mg剂量可在星期一开始,随后的第一次每天5mg剂量在星期三施用,随后的第二次每天5mg剂量在星期五施用,等等。
本发明方法中使用的化合物可配制为单位剂型。术语“单位剂型”指物理上不连续的单位,适合作为经历治疗的患者的单剂量,每个单位包含预定量的计算为产生期望治疗效果的活性材料,任选地与合适的药物载体相连。单位剂型可用于单次日剂量或多次日剂量之一(例如,每天约1次至4次或更多次)。当使用多次日剂量时,对于每次剂量,单位剂型可以相同或不同。
本领域技术人员仅仅使用常规实验将认识到或能够确定本文描述的具体过程、实施方式、权利要求和实施例的各种等价物。考虑这种等价物在本发明的范围内并且由所附的权利要求所覆盖。例如,应当理解,采用本领域认识到的选择且仅仅使用常规实验,改变反应条件包括但不限于反应时间、反应大小/体积和实验试剂比如溶剂、催化剂、压力、环境条件如氮气氛和还原/氧化剂在本申请的范围内。
应当理解,在本文无论何处提供值和范围,这些值和范围包括的所有值和范围意味着包括在本发明的范围内。而且,落在这些范围内的所有值以及值范围的上限或下限也被本申请考虑。
下述实施例进一步说明本发明的方面。但是,它们绝不是本文阐释的本发明教导或公开的限制。
实施例
现在参考下述实施例描述本发明。仅仅为了说明目的提供这些实施例,并且本发明不限于这些实施例,而是包括由于本文提供的教导而显见的所有变型。
材料:
除非另外指出,所有的起始材料和树脂获得自商业供应商并且不纯化即使用。
将喹吡罗(sigma-aldrich,st.louis,mo),pd128907(tocris,ellisville,mo),7oh-dpat(tocris),普拉克索(tocris),罗替高汀(tocris),4-(2-氯苯基)-丁-2-胺(es609;asinex,moscow,russia)和氢溴酸顺-8-羟基-3-(正丙基)-1,2,3a,4,5,9b-六氢-1h-苯并[e]吲哚(pbzi;sigma-aldrich)溶解在水中并且以指示的浓度使用。将沙立佐坦(merckkgaa,gibbstown,nj),fauc73(sigma-aldrich)和7oh-pipat(tocris)溶解在dmso中。将10mm多巴胺(sigma-aldrich)原料新鲜溶解在100mm抗坏血酸中并且以100nm的终浓度使用。
细胞培养:
att-20小鼠垂体细胞在具有5%fbs、10%热失活马血清、2mm谷氨酰胺和50μg/ml庆大霉素(invitrogen,carlsbad,ca)的ham’sf10培养基中生长。稳定表达人d2s、d2l、d3和d4.2和受体的att-20细胞保持在补充500μg/mlg418(invitrogen)的f10培养基中。为了进行电生理表征,将细胞铺板在涂布40μg/ml聚l-赖氨酸(sigma-aldrich)的玻璃盖玻片上。以前已经报道了稳定表达各种人多巴胺受体的att-20细胞的产生和表征(kuzhikandathil&oxford,2000,j.gen.physiol.115:697-706;kuzhikandathil等,1998,mol.cellneurosci.12:390-402;kuzhikandathil&bartoszyk,2006,neuropharm.51:873-884;westrich&kuzhikandathil,2007,biochim.biophys.acta-mcr1773:1747-1758;kuzhikandathil等,2004,mol.cellneurosci.26:144-155;westrich等,2010,biochem.pharmacol.79:897-907)。
camp的测量:
使用campbiotrak酶免疫测试(eia)试剂盒(gehealthcare,piscataway,nj,usa)评估环状amp(camp)水平,如之前描述的(kuzhikandathil&bartoszyk,2006,neuropharm.51:873-884)。每个处理样品的camp水平测量三次,并且整个实验独立重复三次。
电生理学:
通过全细胞膜片钳技术(whole-cellpatchclamptechnique)以电压钳和电流钳模式测量激动剂激活的电流,如之前描述的(kuzhikandathil&oxford,2000,j.gen.physiol.115:697-706;kuzhikandathil等,1998,mol.cellneurosci.12:390-402;kuzhikandathil&bartoszyk,2006,neuropharm.51:873-884;westrich&kuzhikandathil,2007,biochim.biophys.acta-mcr1773:1747-1758;kuzhikandathil等,2004,mol.cellneurosci.26:144-155;westrich等,2010,biochem.pharmacol.79:897-907)。经多管微滴定管阵列将药物溶液输送至细胞。将电流响应对于细胞电容进行标准化,以考虑细胞尺寸的变化。
统计学:
用
计算模拟(hsb方法):
用于设计g蛋白偶联受体的小分子抑制剂的hsb方案已经被描述(kortagere&welsh,2006,j.compu.taidedmo.ldes.20(12):789-8026)。简言之,该方法包括创建源自商业卖主的小分子的集中库。数据库中所有的分子转化成unity格式并且使用sybyl(sybyl8.0,triposinternational)中的unity模块筛选。
为了基于d3受体结合位点中pbzi的相互作用制造3d药效团,使用与部分逆激动剂(pdb号:2rh1)的复合体中β肾上腺素(β2-ar)受体(cherezov等,2007,science318(5854):1258-1265)的晶体结构作为模板,用同源建模程序modeller(ver9.4)(sali等,1993,j.mol.biol.234(3):779-815;westrich等,2010,biochem.pharmacol.79:897-907:kortagere等,2011,biochem.pharmacol.81(1):157-163)创建d3受体的同源模型。pbzi由于其三环结构具有相对刚性的构象。使用moe中采用的随机搜索方法的构象分析用于获得从胺基延伸的烷基尾部区域的最佳构象。基于能量对获得的所有构象进行聚类并且从最多的(populated)的聚类选择代表性的成员以使用moe中采用的am1半经验量子化学方法进一步优化。类似的程序在该研究中用于其他配体。优化的构象用于进一步的对接实验。使用对接程序gold(ver4.1)(jones等,1995,j.mol.biol.245(1):43-53)将pbzi与优化的d3结构的3d结构对接。进行20个独立的试验并且使用goldscore(jones等,1995,j.mol.biol.245(1):43-53)、化学得分(chemscore)(eldridge等,1997,j.comput.aidedmol.de.s11(5):425-445)和定制的评分方案(kortagere&welsh,2006,j.comput.aidedmol.des.20(12):789-802)给对接复合物评分。
为获得结合激动剂的模型更理想的构象,使用schrodinger套装程序的desmond模块(kevin等,2006,proceedingsoftheacm/ieeeconferenceonsupercomputing(sc06),newyork,ny,ieee),将模型浸入清亮水-popc脂质双层-水模型膜系统。将模型膜与β2-ar晶体结构预比对,以采用其对于d3受体-pbzi模型的取向。选择双层组合物的默认条件,包括na+和cl-离子的条件,并且使用默认的所有原子opls力场进行整个模拟。随后是由预松弛、最小化、加热、平衡和3ns的生产运行的常规程序组成的四步方案,以充分优化模型(chien等,2010,science330(6007):1091-1115)。在整个模拟期间,使用低水平的约束,保持pbzi与来自tm3和tm5的残基的相互作用。但是,所有的约束在生产运行期间被去除以使配体在蛋白环境中完全松弛。pbzi和d3受体之间的关键相互作用,即与asp110的盐桥、与ser192的氢相互作用、与his349的芳环相互作用、与val111和来自tm6的其他芳族残基的疏水相互作用用于构建4点混合药效团,如图6c中所示。筛选卖主可获得的小分子的电子库以识别对应药效团的命中。
最后,使用lipinski五规则(lipinski等,2001,adv.drugdeliv.rev.46(1-3):3-26)、血脑屏障(bbb)渗透(kortagere等,2008,pharm.res.25(8):1836-1845)和对孕烷异型生物质受体的脱靶筛选(kortagere等,2009,pharm.res.26(4):1001-1011)以及herg通道(chekmarev等,2008,chem.res.toxicol.21(6):1304-1314)筛选命中。使用gold程序将所得290个命中与d3受体的结合位点对接并且使用各种评分函数进行评分,如之前对pbzi对接所描述的。15个最佳等级命中获自卖主并且在功能上评价。
实施例1:化合物的体外表征
就它们与多巴胺受体的相互作用,表征pbzi和4-(2-氯苯基)-丁-2-胺。两种化合物经d3多巴胺受体激活g蛋白偶联内向整流钾(girk)通道(图2)并且抑制腺苷酸环化酶活性(图11)。两种化合物的体外功能数据显示在表1中。最感兴趣的是观察到不像传统的d2/d3受体激动剂,pbzi和4-(2-氯苯基)-丁-2-胺消除了d3r的耐受性和srt性质(,图2)。这两种化合物代表新类型的d3多巴胺受体激动剂,其消除d3受体的耐受性和srt性质同时是完全激动剂。
表1:
在att-20稳定细胞系中pbzi&4-(2-氯苯基)-丁-2-胺对d2样受体信号传导功能作用的表征
nd:未测定
实施例2:配体依赖性d3受体耐受性和srt性质
图9a图解说明当受体被内生激动剂多巴胺刺激时d3多巴胺受体诱导的天然girk通道的激活。d3受体的耐受性质量化为第二与第一激动剂诱导的响应之比。在d3受体-腺苷酸环化酶和d3受体-有丝分裂原活化的蛋白激酶途径中也观察到d3受体耐受性质(kuzhikandathil&bartoszyk,2006,neuropharm.51:873-884;westrich&kuzhikandathil,2007,biochim.biophys.acta-mcr1773:1747-1758)。另外,图9a显示d3受体srt性质,其是在去除激动剂之后,激动剂诱导的响应的延迟终止。耐受性d3受体采用不同的构象(westrich等,2010,biochem.pharmacol.79:897-907),这表明耐受性和srt性质可能通过改变该不同构象状态的功能选择性激动剂调整。为了确认可能调节d3受体耐受性和srt性质的激动剂,使用d3受体-girk通道信号传导途径作为试验,筛选10个不同的激动剂它们诱导耐受性和srt的能力。选择的激动剂包括内生配体多巴胺——展示对d3选择性的配体——和临床上用于治疗帕金森病的化合物。图9c中的结果表明,尽管大部分激动剂诱导耐受性,但两个激动剂,顺-8oh-pbzi和fauc73,消除了耐受性性质。有趣地,pd128907诱导增强的耐受性,其明显不同于其他激动剂。
图2a和2c说明稳定表达人d3多巴胺受体并且用诱导耐受性或不诱导耐受性的激动剂处理的att-20细胞的代表性电压钳记录。pbzi(图2a)和fauc73(图2c)消除了耐受性和srt性质二者;但是,在相同的细胞中,喹吡罗(图2a)或pd128907(图2c)诱导了严重的耐受性和srt。母体att-20细胞中的对照实验以及用d2/d3拮抗剂依替必利(100nm)预处理显示pbzi和fauc73的激动作用对d3受体是特异性的。此外,消除耐受性和srt的能力不是浓度依赖性的——以100nm至10μm的剂量测试的pbzi不诱导耐受性。这些结果一起表明d3受体耐受性和srt性质是配体依赖性的。在两个消除耐受性和srt的化合物中,pbzi是水溶性的并且在体外和体内更广泛地被表征。
实施例3:化合物的体内表征:体内行为响应
为确定新型d3多巴胺受体激动剂的体内行为作用,比较pbzi和典型的诱导耐受性和srt的d3多巴胺受体激动剂pd128907(盐酸(4ar,10br)-3,4a,4,10b-四氢-4-丙基-2h,5h-[1]苯并吡喃-[4,3-b]-1,4-
实施例4:化合物的体内表征:内条纹6-ohda大鼠帕金森病模型
为了进一步评估新型d3受体激动剂的治疗可能性,测试纹状体中d3受体的上调有助于发展帕金森病中左旋多巴诱导的运动障碍(lid)的假设。已经广泛表征帕金森病的单侧内条纹6-ohda损伤的大鼠模型并且大量用于测试神经保护和移植策略以治疗帕金森病(winkler等,2002,neurobiol.dis.10(2):165-86)。该大鼠帕金森病模型中黑质多巴胺神经元和伴随的运动缺陷发展的部分和缓慢渐进恶化好像模拟遭受帕金森病的患者中渐进的恶化(winkler等,2002,neurobiol.dis.10(2):165-86)。尤其感兴趣的是在该动物模型中发展lid,其中运动与临床上见到的异常不随意运动(aim)类似。
winkler和合作者描述的内条纹6-ohda大鼠帕金森病模型用于测试pbzi和es0609对lid的作用(winkler等,2002,neurobiol.dis.10(2):165-86)。为确定aim分数,将大鼠放置在透明的玻璃圆筒中并且使用数字录像记录仪记录。大概在开始长期左旋多巴治疗的10天之后,6-ohda损伤的动物发展lid。通过施用新型d3受体激动剂10分钟然后在开始长期左旋多巴施用之后第10、17和24天施用左旋多巴,确定pbzi或4-(2-氯苯基)-丁-2-胺降低左旋多巴诱导的aim分数的能力。在这些天也进行踏步试验以评估这些新激动剂对左旋多巴改善损伤的动物中踏步缺陷的能力。
实施例5:化合物的体内表征:运动障碍的减少
在大鼠模型中测试诱导耐受性的(pd128907)和不诱导耐受性的d3受体激动剂(pbzi或4-(2-氯苯基)-丁-2-胺)以减少与长期左旋多巴治疗相关的运动障碍的能力。在左旋多巴治疗之前10分钟施用对照(盐水)、pd128907(0.1mg/kg,sc)、pbzi(17mg/kg,sc)或4-(2-氯苯基)-丁-2-胺(20mg/kg)。图13a图解说明对照、pd128907(诱导耐受性的d2/d3激动剂)和pbzi(不诱导耐受性的d3/d3激动剂)对在17天(长期左旋多巴阶段)损伤动物的总aim分数(包括移动、轴、肢和口舌)的影响。有趣地,即使在左旋多巴施用之前,施用pd128907至损伤的动物本身诱导运动障碍。这与lid也可由在帕金森病的治疗中使用的多巴胺受体激动剂引起的临床报道一致。
图13a的结果表明pbzi本身不诱导运动障碍,并且延迟左旋多巴诱导的运动障碍的出现。通过计算图13b中曲线下方的面积获得的累积数据说明,pbzi使总积分的aim分数显著减少(~300%)。在pbzi和左旋多巴共同施用期间获得的踏步试验数据表明,pbzi不削弱左旋多巴对运动迟缓/运动不能症的有益效果。这些结果表明,pbzi和其类似物是作为治疗帕金森病中lid的新疗法有期望的临床前开发候选物。
实施例6:化合物的体内表征:小鼠中的移动行为
评估balb/c小鼠中pbzi对移动行为的作用。该实验的具体目标是:(a)确定pbzi是否诱导balb/c小鼠中移动的降低;(b)评估pbzi在较长试验时段中的作用;和(c)确定在balb/c小鼠中pd128907是否引起两阶段移动作用。
总共12个刚成年雄性balb/c小鼠获自charlesriver,inc.(wilmington,ma),并且以四组放入标准笼子中。动物维持在12小时光/12小时暗循环并且允许无限制地获取食物和水。在动物的光阶段进行实验。基于评估balb/c小鼠中多巴胺能化合物和运动活性之间的关系的之前工作选择小鼠的品系。小鼠接受pbzi的单个注射(0、1或10mg/kg,sc),并且其后立即分别放入检验台(truscanarena,coulbouminstrumentsinc.),法线照明2小时。对于pd128907,单独组的balb/c小鼠接受盐水的皮下注射(n=4)或0.05或0.4mg/kgpd128907(每个n=4)。注射药物的动物的移动(“行进距离”)分数在10分钟间隔内暴跌,并且为了比较目的对于注射盐水的动物进行标准化(图12a)。在三个分开的场合进行实验。作为剂量x时间相互作用的函数,移动显著变化(f(2,4)=5.40,p=.002)。包括该相互作用的newman-keuls倍(α=.05)主效应显示,与其他组小鼠比较,20分钟内接受10mg/kgpbzi的小鼠的移动显著减少(大概90%)。进一步重要地,在其他时间间隔中没有显著的组间区别。该作用在balb/c小鼠中注射后持续40分钟。
相反,pd128907,如之前就其他小鼠品系所报道,在balb/c小鼠中引起两阶段作用。在注射后前40分钟观察到活动减退,随后在注射后70至110分钟期间观察到活动过度(图12b)。在0.05mg/kg或更高的剂量下观察到pd128907的两阶段作用。尽管较低剂量的pd128907(0.05mg/kg)引起较短时间(20分钟)的活动减退,但是其引起相同持续时间和程度的活动过度。引起活动减退需要的剂量的差异可能是由于d3多巴胺受体对pbzi(ki=22nm)和pd128907(ki=2.3nm)的结合亲和力不同。利用pbzi和pd128907二者,注射后120分钟移动活性回到对照盐水水平。这些结果清楚地表明诱导d3受体耐受性的激动剂(pd128907)引起两阶段移动响应。但是,不诱导d3受体耐受性的激动剂(pbzi)引起单阶段活动减退移动响应。
实施例7:d3受体信号传导性能对细胞功能和信号传导途径的作用
稳定表达的d3受体结合至att-20神经内分泌细胞中的内生girk通道并且调节自发性动作电位和分泌(kuzhikandathil&oxford,1999,j.neurosci.19(5):1698-1707;kuzhikandathil&oxford,2000,j.gen.physiol.115:697-706;kuzhikandathil等,1998,mol.cellneurosci.12:390-402)。通过多巴胺活化稳定表达的人d3多巴胺受体在第一应用期间抑制自发性动作电位,但当随后应用时不抑制(图16a)。相反,通过消除耐受性和srt的激动剂pbzi活化d3受体在第一应用期间和随后的应用期间抑制自发性动作电位(图16b)。该结果表明,通过诱导耐受性的d3受体激动剂调节神经放电非常不同于不诱导耐受性的那些。后一类型的激动剂将d3受体转化成d2受体的功能等价物。
实施例8:通过使用d3受体-pbzi药效团模型识别不诱导耐受性和srt的新型激动剂。
图9c、2a和2c的结果显示pd128907诱导严重的耐受性和srt;有趣地,不诱导耐受性和srt的pbzi与pd128907共有少许核心结构成分(图6)。pd128907和pbzi诱导耐受性和srt能力的完全不同表明,对接在d3受体同源模型中的这些化合物的比较性建模研究可能产生信息以开发药效团模型来筛选消除耐受性和srt性质的另外化合物。
为进一步理解由pd128907和pbzi诱导的信号传导性能的不同,将它们与d3受体的结合位点对接。对接由可质子化的胺与asp110的盐桥相互作用、与跨膜(tm)5中保守的丝氨酸残基的氢键相互作用和与来自tm6和tm7残基的芳族相互作用限定。结合配体的复合物被最小化并且进一步使用分子动力学(md)模拟优化。优化的复合物的结构叠加产生
为了验证pbzi与d3受体的这些相互作用是否重要,设计了并入疏水成分、盐桥相互作用和芳族-π相互作用的三维药效团。使用该三维药效团,使用hsb方法筛选三百万个化合物库,以筛选可模拟pbzi药效团特征的小分子。来自筛选的命中接着进行过滤方案,其包括lipinski药物样性能、孕烷异生物质受体活化和更重要的血脑屏障(bbb)渗透。从过滤方案得到的290个命中对接至d3受体的结合位点并且使用各种评分方案评分。评分方案定制为仅仅对与asp110形成盐桥相互作用并且与由tm5、tm6和tm7形成的芳族聚类具有有利的相互作用的那些分子评级。
通过hsb计算机筛选识别的15个化合物被评估它们活化d3受体并且诱导girk响应、耐受性和srt的能力。功能研究确定了一种新型d3受体激动剂es609(4-(2-氯苯基)-丁-2-胺),其不诱导耐受性和srt。图17显示es609的代表性迹线和累积数据,并且表明,如在pbzi和fauc73的情况,es609也消除d3受体耐受性和srt性质(图17a)。对照实验显示es609在母本att-20细胞中不引起girk电流(图17b)并且在稳定表达d3受体的att-20细胞中诱导的电流通过用拮抗剂依替必利预处理而被阻止。对接实验确认es609符合pbzi的相互作用模式,与his349具有有利的π-叠加相互作用和与asp110具有盐桥(图6a)。使用md模拟研究进一步优化d3受体-es609复合物显示es609引起与pbzi类似的构象。结合es609的d3受体结构叠合至结合pbzi的d3受体结构,rmsd为
实施例9:pbzi和es609的功能表征
为了比较新型d3受体激动剂的功能作用,在稳定表达人d3、d2s、d2l或d4.2的att-20细胞中测试pbzi(表2)和es609(表3)。通过评估pbzi和es609抑制腺苷酸环化酶或活化与这些“d2样”多巴胺受体连接的girk通道的能力确定功效。对于d3受体,pbzi和es609抑制腺苷酸环化酶和活化girk通道的ec50值范围为0.2nm至30nm。通过比较由饱和浓度(300nm)的完全激动剂喹吡罗引起的响应,结果显示pbzi和es609是腺苷酸环化酶和girk通道二者测试中d3受体的完全激动剂。相反,两种化合物是d2s多巴胺受体的部分激动剂。对于d2l多巴胺受体,pbzi在两个测试中为完全激动剂;相反,es609在腺苷酸环化酶测试中不引起任何响应并且在girk通道测试中是部分激动剂。对于d4.2多巴胺受体,pbzi不引起响应并且es609在腺苷酸环化酶测试中是部分激动剂。在girk通道测试中,pbzi对于d4.2受体是部分激动剂,而es609不引起响应。这些结果一起表明pbzi和es609二者对于d3受体是完全激动剂,并且对于其他d2样多巴胺受体是部分激动剂或不引起响应。
表2:
稳定表达单个多巴胺受体亚型的att-20细胞中顺-8oh-pbzi诱导的腺苷酸环化酶的抑制和girk通道的活化
*,p<0.05,统计学上显著,studentt检验。
表3:
稳定表达单个多巴胺受体亚型的att-20细胞中es609诱导的腺苷酸环化酶的抑制和girk通道的活化
*,p<0.05,统计学上显著,“学生”t检验。
本文描述的结果显示d3受体耐受性和srt性质是配体依赖性的并且帮助识别不诱导这些性能的新型非典型的d3受体激动剂。识别了水溶性化合物pbzi,其结构上与pd128907相似但是不诱导耐受性和srt。之前的结合研究已经显示,对于d2样受体,pbzi的ki对于d3为27nm,对于d2s为1800nm,以及对于d4.2为280nm(scheideler等,1997,eur.j.pharmacol.339(2-3):261-270)。对d1样受体和多种其他神经递质受体、离子通道和运载蛋白的结合被忽略。因此在受体结合测试中,pbzi展示d3受体选择性。这与功能研究的结果一致,其显示pbzi对于d3受体是完全激动剂并且对d2s多巴胺受体是部分激动剂(表2)。对d2s受体的部分激动作用也与之前的体外结果一致。体内,施用pbzi的动物在具有高d3受体表达的区域,内侧额叶前皮质和伏核的壳区域,显示c-fos表达的特异性增加。之前未确定pbzi对d2i受体的作用。本文描述的结果显示pbzi对d2l多巴胺受体是完全激动剂(表2)。分别基于d2s和d2l受体的突触前和突触后区域,预测pbzi主要具有突触后作用。这也被之前体内研究支持(fink-jensen等,1998,eur.j.pharmacol.342(2-3):153-161)。
典型的d2样受体激动剂pd128907对d3受体是选择性的,对d3的ki是0.4nm,对于d2s是202nm并且对于d4.2是114nm(scheideler等,1997,eur.j.pharmacol.339(2-3):261-270);但是,本文描述的研究显示其诱导严重的耐受性和srt。有趣地,pd128907的化学结构与pbzi类似。尽管pd128907和pbzi具有与d3受体完全激动剂类似的核心结构和功能,但是它们具有对耐受性和srt性质显著不同的作用。本文描述的结果表明pbzi和pd128907之间的功能差异是由于d3受体中这些激动剂诱导的不同的构象。这些构象变化的大部分限制在最接近结合位点的区域和胞外环(ec)2环区域。结合pbzi和pd128907的结构的比较表明最大的位移出现在tm4,其与ec2的大移动相关。另外,在细胞内环(ic)2中也观察到之前显示对于介导耐受性质重要的构象改变(westrich等,2010,biochem.pharmacol.79:897-907)。在结合pd128907的形式中,其他重要的构象变化包括沿着tm6螺旋长度向下的位移和tm3螺旋中第一转角的展开。
在该研究中,通过小心地监测在计算机模型中通过pbzi和pd128907的结合产生的相互作用的性质和构象作用,设计展示选择性并且消除耐受性和srt性质的新型非典型的d3受体激动剂,es609。hsb方法可用于识别另外的非典型的d3受体激动剂。基于pbzi与d3受体相互作用的性质设计es609。假设是通过在配体上引入吸电子基团,增加与芳核的π-π相互作用强度。因此在es609中邻位的卤基非常适合该设计,连同靠近可质子化的胺增加疏水性(图6d)。后者增强与tm3上疏水基团的相互作用,与fauc73与tm3中的残基的相互作用类似。his349可在促进d2l受体中偏向配体的信号传导中起主要作用。这些结果与增强与his349和其他芳核成员的π-π相互作用可有利地有助于pbzi、fauc73和es609的非典型性能的发现一致。调整研究以理解ec2和tm4在促进功能选择性中的具体作用并且进一步理解这些非典型激动剂在与d3受体的复合物中完整的构效关系。功能上,es609和pbzi对于激活连接d3受体的信号传导途径具有类似的效能。但是,与pbzi相反,当在两个不同的信号传导途径中测试时,es609是部分激动剂或对其他d2样受体(d2s、d2l和d4.2)不引起响应。es609展示的选择性表明其结构可用作设计未来选择性d3受体激动剂的模板。由其ec50限定的es609的功能亲和力与我们研究的两个信号传导途径中的其他d2样多巴胺受体激动剂类似。此外,其小的分子量和水溶解度使其理想地适合体内研究。假设pbzi和es609在体外具有类似的信号传导性能,预期它们在体内具有类似作用。
这里描述的新型的非典型d3受体激动剂代表功能选择性概念的新变化。常规地,功能选择性归因于不同的配体在与相同受体结合的不同的信号传导途径中引起不同响应的能力。配体不同程度地活化与相同受体结合的不同路径的能力是由于配体在受体中造成的不同构象。假设d3受体耐受性和srt性质由不同的构象状态决定,则预测改变这些构象状态的配体的识别调节这两种性质。与该预测一致,在该研究中识别了改变两种d3受体性质的配体。本文描述的结果表示,功能选择性概念可不限于与相同受体结合的不同通路的选择性激活,而是也可扩展至包括在单个途径中调节信号传导性质的配体。如果这些性质涉及多个信号传导途径,如本文的情况,异常表达或这些具体性能的改变可能是各种不适的病理学基础。在该背景下,这些受体性质,以d3受体的耐受性和srt性质为例,可能代表新的药物靶标。本文描述的研究允许识别特异性靶向这两种d3受体信号传导性质的新类型激动剂。
已经提出具有左旋多巴诱导的运动障碍的动物纹状体中d3受体耐受性和srt性质的异常表达促进了运动障碍行为。通过消除耐受性和srt的非典型d3受体激动剂可能改善左旋多巴诱导的运动障碍症状。在耐受性和srt性质的背景下发生的d3受体表达的改变可能提供对在神经学疾病中观察到的一些行为表型的解释,比如神经分裂症、精神错乱、长期可卡因使用、紧张和抑郁。在许多这些疾病中d3受体异常表达改变可能影响d3/d2受体表达的比,导致观察到的病理。这里描述的新类型非典型的d3受体激动剂,通过将d3受体信号传导转化成d2受体的功能等价物,可能提供新型治疗方法以治疗这些疾病。
本文引用的每篇和所有的专利、专利申请和出版物的内容通过引用以其整体并入本文。
尽管已经参考具体的实施方式公开了本发明,但显然,在不背离本发明真正精神和范围的情况下,本领域技术人员可设计本发明的其他实施方式和变型。所附权利要求意欲被解释为包括所有的这种实施方式和等价变型。
序列表
<110>德雷克塞尔大学
罗特格斯新泽西州立大学
s.克尔塔格勒
e.v.库兹坎达尔
<120>治疗帕金森病中运动障碍的新型d3多巴胺受体激动剂
<130>46528-6035-00-wo.601019
<150>us61/372,733
<151>2010-08-11
<160>2
<170>patentin版本3.5
<210>1
<211>400
<212>prt
<213>智人
<400>1
metalaserleuserglnleuserserhisleuasntyrthrcysgly
151015
alagluasnserthrglyalaserglnalaargprohisalatyrtyr
202530
alaleusertyrcysalaleuileleualailevalpheglyasngly
354045
leuvalcysmetalavalleulysgluargalaleuglnthrthrthr
505560
asntyrleuvalvalserleualavalalaaspleuleuvalalathr
65707580
leuvalmetprotrpvalvaltyrleugluvalthrglyglyvaltrp
859095
asnpheserargilecyscysaspvalphevalthrleuaspvalmet
100105110
metcysthralaserileleuasnleucysalaileserileasparg
115120125
tyrthralavalvalmetprovalhistyrglnhisglythrglygln
130135140
sersercysargargvalalaleumetilethralavaltrpvalleu
145150155160
alaphealavalsercysproleuleupheglypheasnthrthrgly
165170175
aspprothrvalcysserileserasnproaspphevaliletyrser
180185190
servalvalserphetyrleupropheglyvalthrvalleuvaltyr
195200205
alaargiletyrvalvalleulysglnargargarglysargileleu
210215220
thrargglnasnserglncysasnservalargproglypheprogln
225230235240
glnthrleuserproaspproalahisleugluleulysargtyrtyr
245250255
serilecysglnaspthralaleuglyglyproglypheglngluarg
260265270
glyglygluleulysarggluglulysthrargasnserleuserpro
275280285
thrilealaprolysleuserleugluvalarglysleuserasngly
290295300
argleuserthrserleulysleuglyproleuglnproargglyval
305310315320
proleuargglulyslysalathrglnmetvalalailevalleugly
325330335
alapheilevalcystrpleuprophepheleuthrhisvalleuasn
340345350
thrhiscysglnthrcyshisvalserprogluleutyrseralathr
355360365
thrtrpleuglytyrvalasnseralaleuasnprovaliletyrthr
370375380
thrpheasnilegluphearglysalapheleulysileleusercys
385390395400
<210>2
<211>500
<212>prt
<213>智人
<400>2
asptyrlysaspaspaspalametglyglnproglyasnglyserala
151015
pheleuleualaproasnargserhisalaproasphisaspvalthr
202530
glnglnargaspgluvaltrpvalvalglymetglyilevalmetser
354045
leuilevalleualailevalpheglyasnvalleuvalilethrala
505560
ilealalysphegluargleuglnthrvalthrasntyrpheilethr
65707580
serleualacysalaaspleuvalmetglyleualavalvalprophe
859095
glyalaalahisileleumetlysmettrpthrpheglyasnphetrp
100105110
cysgluphetrpthrserileaspvalleucysvalthralaserile
115120125
gluthrleucysvalilealavalaspargtyrphealailethrser
130135140
prophelystyrglnserleuleuthrlysasnlysalaargvalile
145150155160
ileleumetvaltrpilevalserglyleuthrserpheleuproile
165170175
glnmethistrptyrargalathrhisglnglualaileasncystyr
180185190
alaglugluthrcyscysaspphephethrasnglnalatyralaile
195200205
alaserserilevalserphetyrvalproleuvalilemetvalphe
210215220
valtyrserargvalpheglnglualalysargglnleuasnilephe
225230235240
glumetleuargileaspgluglyleuargleulysiletyrlysasp
245250255
thrgluglytyrtyrthrileglyileglyhisleuleuthrlysser
260265270
proserleuasnalaalalyssergluleuasplysalaileglyarg
275280285
asnthrasnglyvalilethrlysaspglualaglulysleupheasn
290295300
glnaspvalaspalaalavalargglyileleuargasnalalysleu
305310315320
lysprovaltyraspserleuaspalavalargargalaalaleuile
325330335
asnmetvalpheglnmetglygluthrglyvalalaglyphethrasn
340345350
serleuargmetleuglnglnlysargtrpaspglualaalavalasn
355360365
leualalysserargtrptyrasnglnthrproasnargalalysarg
370375380
valilethrthrpheargthrglythrtrpaspalatyrlysphecys
385390395400
leulysgluhislysalaleulysthrleuglyileilemetglythr
405410415
phethrleucystrpleuprophepheilevalasnilevalhisval
420425430
ileglnaspasnleuilearglysgluvaltyrileleuleuasntrp
435440445
ileglytyrvalasnserglypheasnproleuiletyrcysargser
450455460
proasppheargilealapheglngluleuleucysleuargargser
465470475480
serleulysalatyrglyasnglytyrserserasnglyasnthrgly
485490495
gluglnsergly
500
- 还没有人留言评论。精彩留言会获得点赞!