从PCR后的独特分子标识符频率估算PCR前的片段数的制作方法
本发明涉及校正核酸偏倚量确定方法的领域,其中所述偏倚是由扩增方法引入的。
发明背景
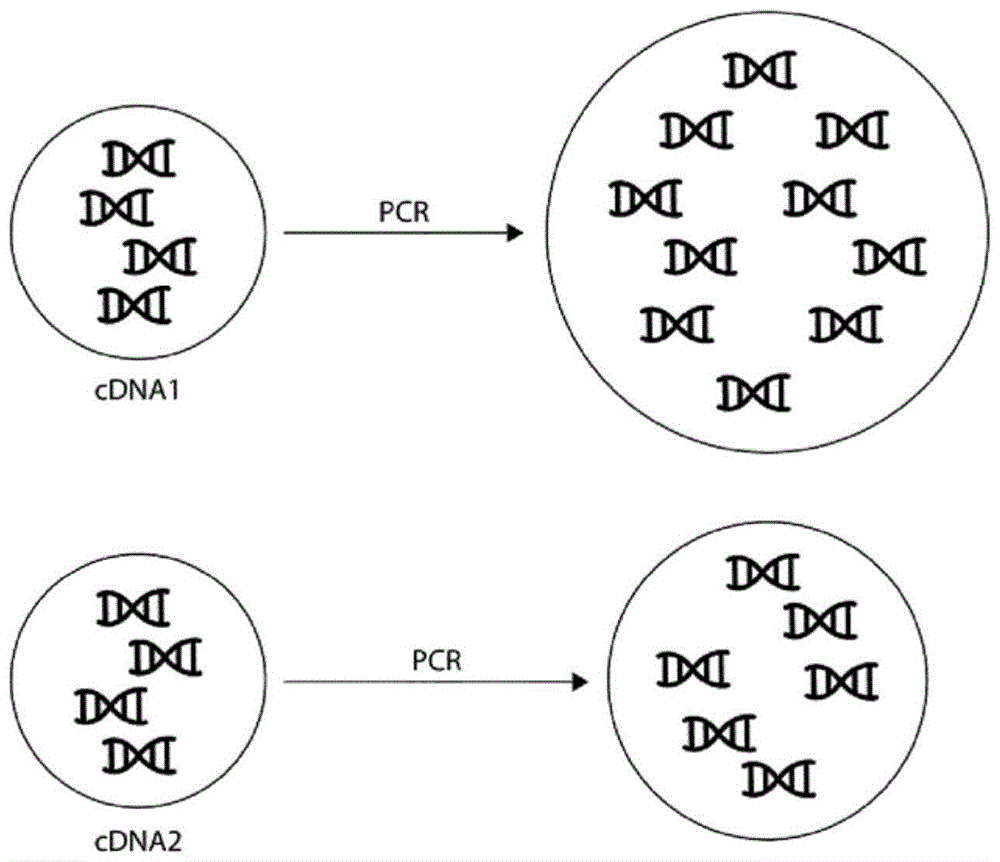
聚合酶链反应(pcr)通过进行多个由变性、退火和聚合组成的循环来扩增核酸。由于每个步骤的效率取决于进入该步骤的序列,因此pcr扩增可引入序列特异性偏倚,这意味着具有不同序列的不同核酸将以不同比例扩增。例如,对于rna-seq(rna测序,特别是片段测序或鸟枪测序),这可导致不准确的基因和异构体定量分析(图1)。
wo2017/051387a1涉及一种使用码字(codeword)并通过观察码字熵(codewordentropy)来确定核酸样品的复杂度的方法。
已有人开发出了标签,例如独特分子标识符(umi),也称分子条形码,以识别pcr复制物并减少序列特异性pcr偏倚的影响。umi是寡核苷酸,它们在每个位置上具有主要是随机的核苷酸分布,这些寡核苷酸与dna片段连接后再进入pcr扩增。如果umi均匀分布并且其数量比相同片段库大得多,则不太可能有相同的umi连接到库中两种不同片段上。在这种情况下,pcr之后库中不同umi的数量与pcr之前其片段的数量相同。因此,可以通过计数不同的umi数量而不是pcr之后每个库中的片段数量来校正不同库的pcr效率波动(图2)。但是,如果umi分布不均,例如由于连接偏倚或umi生成中核苷酸的插入不均或umi寡核苷酸内各处分布不均,这种简单的计数程序就变得不准确。umi分布不均匀与umi集大小不足具有相同的效果。在这两种情况下,相同的umi很可能与两种片段连接,从而导致低估了pcr前的片段拷贝数(图3)。
因此,需要改进的方法,能够基于扩增后的测量数来估计核酸的量和校正这种扩增偏倚。
发明概述
本发明提供了(用扩增后估值)评估样品中核酸拷贝数的方法,包括以下步骤:
a)提供包含待确定拷贝数的核酸的样品;
b)将变量标签(variablelabels)附着于这些核酸;
c)通过核酸复制程序扩增所述带标签的核酸;
d)确定扩增的带标签核酸拷贝的量,对于带不同标签的核酸拷贝,确定它们各自的量;和
e)基于步骤d)确定的量评估步骤a)的样品中的核酸拷贝数,方法是
针对样品中至少两种、优选至少四种、至少十种以上的不同种类核酸,使标签对每种核酸的附着概率近似化:对扩增后不同种类核酸的这样一种标签的量求平均值;和反复地或逐步地
(a)
优化基于步骤d)中不同标签的测得数量预期的样品中核酸拷贝数,以及根据标签附着至一种核酸拷贝的概率预期的附着至一种核酸的不同标签的数量,以及样品中核酸拷贝的预期数量或估计扩增效率的先前迭代或预设值;
或
(b)
(i)基于步骤d)确定的量,指定标签附着至一种核酸的概率,估计的扩增效率或样品中预期的核酸拷贝数,并基于在所述估计扩增效率下、每复制一个循环、核酸复制程序中扩增的带标签核酸拷贝的预期复制,对扩增的带指定标签核酸拷贝的量的概率分布建模,
(ii)根据带标签核酸拷贝的模型化概率分布,确定带标签核酸拷贝出现确定量的可能性,
(iii)通过改变估计扩增效率或样品中核酸拷贝的预期数量,使步骤(ii)的可能性最大化,
(iv)根据所述最大化模型、或根据在该最大化模型中估计的扩增效率,评估样品中的核酸拷贝数。
本发明进一步提供了一种包括计算机可读指令的计算机程序产品,该计算机可读指令用于在带标签的核酸扩增后基于本发明方法的步骤e)确定的量来计算样品中核酸拷贝的估计量。
可以组合本发明的所有实施例和方面,并且所有优选的和解释的实施例均指代本发明的所有方面。即这些方法的优选实施例也已读至计算机程序产品的优选实施例中,反之亦然。
发明详述
本发明在以上概述部分和以及在权利要求中进行了描述。用到了多种表达方式,为了便于参考和概述,结合实施例以及实施例中使用的各种定义进行说明。
“核酸拷贝数”和“多个拷贝的核酸”(“k”)是指共享相同序列的核酸分子(称为核酸拷贝)的量、丰度或浓度。如本文所用,术语“拷贝数”用于指扩增之前,即在原始样品中的量、丰度或浓度。这是经本发明方法确定的数量或拷贝数。这样的样品可以具有不同量和/或序列的不同核酸分子。这些被称为不同种类的核酸。同种类的所有核酸分子(不带标记)称为“库”。不同“库”的核酸分子是不同种类的不同核酸分子,每一种形成自己的虚拟库。“库”不需要物理分离,全部的核酸库或种类都可以在同一样品中。
“变量标签(variablelabels)”是指可以识别的不同属性或种类的标签。一个例子是随机核苷酸标签,例如随机单核苷酸(即a,t(u),g,c的混合物),随机二聚体(即aa,at,ag等与cc的混合物),随机三体、四体、五体、六体、七体、八体或更长的核苷酸。这类标签混合物具有各种不同标签,每种具体标签可能出现一次或多次。在随机核苷酸的情况下,标签混合物的组成中某个指定标签可能具有0~多个拷贝,这取决于该标签混合物库的大小。标签都附着在核酸分子上。这类附着可能效率低下,这意味着不是全部核酸分子都带有标签。标签用指示符“b”表示。
“标签附着至核酸拷贝的概率”(“pb”)将所述低效率量化为标签附着于核酸分子的概率。该量化通常是根据本发明的方法中的评估值。
“核酸复制程序”是产生核酸复制品的程序。这样的方法可以包括使引物退火和以依赖于模板的方式延伸所述引物的步骤,例如pcr。此类方法可能效率低下,这意味着不是全部的起始核酸分子都被复制(即“扩增效率”<1,其中1是理论上理想情况中每个核酸分子都被复制,0表示核酸分子都未被复制)。
“估计扩增效率”(“pd”)是所述扩增效率的估值,可在本发明的计算过程中获得。该估值是因固有的扩增偏倚原因(最大差异核酸序列)所致的、不同种类核酸之间相区别的最可能扩增效率。本发明的估值模型反映了不同种类核酸之间的这种扩增效率差异。
“扩增的带标签核酸拷贝的量”(“nb”)是指带标签的核酸拷贝扩增后可定量的量或浓度。标签种类在变量和公式描述中用下标“b”表示。该表示法是指核酸拷贝,因此关乎产生相同拷贝的一种核酸种类。值得注意的是,为了强调其重要性,一个种类(或“库”)的核酸分子可能附着有不同的标签。实际上,附着不同标签在本发明的方法中很重要,是为了确定标签附着效率、并进而监视本发明方法的扩增效率。这些不同的标签可以不加区别地附着在样品中的所有种类的核酸上。复数“量”表示可以确定不同的被扩增的带标签核酸拷贝。如所述,“核酸拷贝”是指相同的核酸种类。差异是由于各个分子之间不同的标签所致,不过,如所述,多个相同种类的标签也可以附着在这些核酸分子上。这种情况表述为“确定带不同标签的核酸拷贝各自的量”。根据确定指定种类核酸的量(也称样品中的拷贝数)的评估程序,“nb”还指所述核酸拷贝(所述种类或“库”的核酸分子)上的标签数量。相应地,表达方式“所述标签的量”(“nb”)也与表示标签的“b”一起使用。在所研究的库中,扩增的带标签核酸拷贝的量统称为“n”。它是标签“b”的各自量的总和(nb)。
“样品中核酸拷贝数估值”(“k”或“kest”,其中“est”表示“估计的”,强调估计的特性)涉及通过本发明方法估计评估样品中的核酸拷贝数。估值可能与样品中的实际自然量不同,但这种差异或误差比现有技术的方法大大降低。本发明方法的另一个好处是标签的偏倚(例如因标签种类较少和标记效率较低所致)大大减少了。表达方式“样品中核酸拷贝的预期数量”表示本发明的方法是预期值的细调,其中每次迭代(iteration)生成新的预期值,最终的“预期”值称为估值。ki或ki+1表示第i或i+1次迭代。扩增前带标签(即标签附着后)核酸的拷贝数在式中用小写字母“k”表示,或用“kb”表示,其中b表示标签种类。该术语通常是指,具有与样品中相同的基本核酸序列(相同的“库”)、但所附着的标签不同的核酸。换句话说,kest是将每个标签的全部核酸估值记为[kb]、由此得到的计算中所用全部标签的总和。[kb]则是根据标记概率[pb]进行标记的结果(另参见示例,公式(5)和(2)-kest和pb通过公式(3)连接;在那里,kest是公式(3)的近似解)。
“在步骤d)中检测到的不同标签的数量”(“du”)是指检测到的不同质量的区别性标签的数量,例如不同的条形码或序列。因此,它是指区别的数量或种类的数量,而不是每种具体种类的标签的数量。
“扩增的带标签核酸拷贝的预期复制”是指扩增反应中的模型化复制,由于效率低下,其在每个扩增循环中不会复制每个核酸分子。根据统计的每循环的复制数可得出概率函数。扩增(或复制)循环的次数以“c”给出。
“带标签核酸拷贝的模型化概率分布”(“p(nb)”)是指对带标签核酸在扩增过程中的结果(“nb”)进行的建模。算出扩增后达到某个量的概率。将多种量的集合概率组合在概率分布中(相对于量值)。这样的概率分布函数可以具有高斯形状或其他形状,并且可以具有与带标签核酸拷贝在该最大值的量下的最大概率相似的最大值。分布函数的形状可以具有全局最大值和一个或多个局部最大值。所述概率分布的生成取决于估计的扩增效率或样品中核酸拷贝的预期数量(这两个是可互换的),可通过改变和细调这两个参数中的一个的预期来调整模型,该参数是用于使可能性或概率最大化,以便最好地匹配带目标标签的所有核酸拷贝(不需要是样品中的所有核酸或所有标记产物)在所观察量值时的分布函数。
“扩增的核酸拷贝的确定量”(“n”)是指所研究的所有核酸拷贝的总量。注意复数“拷贝”是指不同拷贝的定性复数(qualitativeplural)。
根据此概要,本发明的方法也可以写成带有方括号中指定因子后接常规括号中指定变量指示符的形式,如下:
评估扩增后[核酸拷贝数](k)的方法,包括以下步骤:
a)提供包含待测核酸[拷贝数](k)的样品;
b)将变量标签附着至所述核酸;
c)用核酸复制程序扩增所述带标签的核酸;
d)确定[扩增的带标签核酸拷贝的量](nb),确定[带不同[标签](b)的核酸拷贝]各自的量(不同b的nb);和
e)基于确定的[步骤d)的量](nb)来提供[样品中核酸拷贝数估值](kest),这是通过如下来实现
针对样品中至少两种、优选至少四种、至少十种以上的不同种类核酸,近似化[标签附着至核酸的概率](pb):对扩增后不同种类核酸的[这样一种标签的量](nb)求平均值;和反复地或逐步地
(a)
优化基于[步骤d)中所测不同标签的数量](du)而估计或预期的[样品中核酸拷贝数估值或预期值](kest),以及根据[标签附着至核酸拷贝的概率](pb)预期的附着至一种核酸的不同标签的数量,以及[样品中核酸拷贝数预期值](kest)或[估计扩增效率](pd)的先前迭代或预设值;或
(b)
(i)基于确定的[步骤d)的量](nb),[这些标签附着至一种核酸的概率](pb),[估计扩增效率](pd)或[样品中预期的核酸拷贝数](kest),并基于在所述[估计扩增效率](pd)下、每复制一个循环、核酸复制程序中扩增的[带标签核酸拷贝](“库”)的预期复制,对[扩增的带指定标签核酸拷贝的量的概率分布](p(nb))建模,
(ii)根据[带标签核酸拷贝的模型化概率分布](p(nb)),确定[带标签核酸拷贝的确定量](nb)出现的可能性,
(iii)通过改变[估计扩增效率](pd)或[样品中核酸拷贝的预期数量](kest),使步骤(ii)的可能性最大化,
(iv)根据所述最大化模型、或根据在该最大化模型中[估计的扩增效率](pd),提供[样品中核酸拷贝数估值](kest)。
本发明的方法基于包含待确定拷贝数的核酸的样品。所述样品可以包含各种不同的核酸种类,每种种类由一个或多个分子或“拷贝”代表。例如,样品可包含1、2、3、4、5、6、7、8、9、10、20、30、50、100、150、200或更多,或这些值之间的任何范围的不同核酸种类。至少一种或多种种类的拷贝数可以是1、2、3、4、5、6、7、8、9、10、20、30、50、100、150、200、1000、10000、20000或更多个分子或这些值之间的任何范围的拷贝数。
核酸可以是全长天然核酸分子,例如mrna,microrna,rrna,trna,或基因组核酸或载体核酸,例如基因组(优选短基因组,例如病毒或细菌基因组或其分子组分),载体样质粒或转座子或人工dna构建物。通常,核酸可以是rna或dna或任何其他可以扩增的核酸类型。
优选地,核酸是起始的更大核酸的片段,例如在下一代测序(ngs)或rna-seq或鸟枪测序中产生的片段。片段化可以通过物理手段,例如剪切,或通过化学或酶促手段,例如限制酶切割来实现。片段化可以是随机的,即不具有切割的位点特异性,产生随机片段,也可以是位点特异性的,例如对所选核酸模式是特异的,如酶促限制性。随机片段化将产生许多不同种类的核酸,它们可以通过当前测序方法辅以计算机程序来处理。
优选地,通过本发明方法分析的样品的核酸(例如上述片段或其他核酸分子)具有10~10000个核苷酸(nt)的平均长度,优选15~8000或20~5000或50~4000或80~3000个核苷酸。
在步骤b)中,将变量标签附着于所述核酸。附着可通过已知的化学或酶促反应,例如连接反应或结合反应来促进。附着促进标签在整个过程中保持粘附于核酸,直至步骤d)的检测。任何适合标记核酸的标签都可以选择。在步骤c)中的扩增反应中,标签也附着在复制产物上,这一点是必须的。因此,最方便的标签是核酸标签,例如条形码或umi。但是,通过额外的努力,也可以使用其他标签如蛋白质或肽,肽核苷酸,抗体,受体,抗原,识别分子(可被结合伴侣识别者,如生物素和抗生物素蛋白,荧光标签),量子点等。优选允许高可变性的标签。同样由于这个原因,优选核苷酸标签,但并非唯一可能。
优选地,标签是能扩增的,特别是wo2017/051387a1所述能被扩增的大分子。
在核苷酸标签的情况下,这些可以是rna或dna或任何其他核苷酸类型。它可以是与核酸分子相同的类型或另一种类型。核酸标签使用标签分子的独特属性作为识别元件。优选地,所述识别元件是核苷酸序列。核苷酸可以选自a,t(u),g,c或其任意组合,优选所有四种核苷酸类型(u优选用于rna,t优选用于dna;两者均被互补a识别)。标签可以包含1、2、3或4种不同的核苷酸类型。只有一种核苷酸类型时,识别元件实质上是标签的大小或长度。包含更多的核苷酸类型可实现许多排列,例如,对于具有4种不同核苷酸类型、长度为6个核苷酸的标签,有46(4的6次方)种排列。排列的数量是分子式核苷酸类型数的(nt长度)指数,例如43,44,45,46,47,48等或对于3种核苷酸类型而言33,34,35,36,37,38等核苷酸。从业者可以根据目标样品中预期的核酸的复杂性和多样性来选择合适数量的不同标签。例如使用2,3,4,5,6,7,8,9,10,15,20,25,30,35,40,50,60,70,80,100,120,140,160,180,200,300,400,500,750,1000,2000,5000,10000或更多,或这些值之间任何范围内的不同标签数。在一些方法,尤其是方法(a)中,优选选择不同标签的数目使得其大于预期的核酸拷贝数的数目。这在方法(b)中也是可能的,但不是必须的。方法(b)能够更好地处理较少数量的不同标签。
标签无需是分子,但也可具有标签的各种拷贝数。本发明的方法能抵消任何标签偏倚(即各种标签的浓度不均匀)以及由于标签不均匀附着核酸分子而引起的任何偏倚。优选地,各个标签种类具有1,2,3,4,5,6,7,8,9,10,50,100,1000,10000或更多的拷贝数,例如优选最高达100万(m),最高达100000或最高达10000。优选地,所有标签的总数应等于或超过所有种类的全部核酸的数量,以便对所研究的每种核酸进行标记。所有标签的总拷贝数可以至少为1000或10000或更多,例如优选最高达10000个百万(m),最高5000m,最高1000m或最高500m。
在优选的实施方案中,使用允许检测和校正标签序列错误的标签。这种纠错标签本身是本领域已知的,见krishnanetal.,electronicsletters,2011,47,236-237。这些标签可用于检测标签序列测序过程中的测序错误-在给定测序仪的误差范围内。
例如,当我们考虑两种带有acgt和tgca序列的标签时,如果这些标签在全部四个位置上都不同,那么我们可以校正这些标签中有一个核苷酸被另一个替换时的错误。例如,如果第一种标签的读取结果更改为acct,第二种标签的读取结果更改为tcca(均由于测序错误),则错误的标签与其正确版本的差距为1,而与另一种标签的正确版本的差距为3。因此,根据它们与正确标签的差距,可以将acct正确分派为acgt,将tcca正确分派为tgca。另一方面,如果第一和第二种标签都更改为acca,则与两种正确标签之间的差距将为2。因此,我们无法校正错误,而只能推断发生了一个以上的错误。结果,标签acgt和tgca都是能校正一个错误和检测到二个错误者。此例涉及取代型错误的校正(参见krishnanetal.,见上文;和bystrykhetal.,plosone,publiclibraryofscience,2012,7,1-8;所有参考文献通过引用并入本文)。所有不同标签之间核苷酸差异更大的较长标签允许校正和检测更多错误。也可能产生对取代、插入和缺失进行校正的标签(buschmannetal.,bmcbioinformatics,2013,14,272;hawkinsetal.proceedingsofthenationalacademyofsciences,nationalacademyofsciences,2018,115(27):e6217-e6226;所有参考文献均通过引用纳入本文)。优选地,这种纠错标签在本发明方法的步骤b)中使用。特别优选地,纠错标签是对取代、插入和缺失进行校正的标签。特别优选地,与之组合或替代地,纠错标签适用于校正1、2、3或更多个错误。这样的标签可能具有2n+1种序列差异,其中n是应该校正的错误数。优选地,序列差是2n+2。这种比式2n+1多出来的差异将在错误校正量之上增加另一层错误检测,如上述实例所示。
优选地,在步骤d)中,使用纠错来分配正确的标签(无测序错误)。单纯错误检测(无校正)将用于从进一步分析中删除此类标签。
步骤c)包括通过核酸复制程序,例如pcr,扩增带标签的核酸。核酸扩增程序通常遵循以下模式:对于指定模板带标签的核酸,产生(另外的)拷贝。这样的方法可以包括引物结合和延伸所述引物。引物结合的步骤可能需要接头或衔接子分子附着至引物结合区。这样做的优点是任何核酸序列都可以从头端或尾端开始扩增。其他次优选的不依赖序列的引发是随机引发。如
背景技术:
部分所述,核酸扩增方法有受困于效率低下、甚至这种效率低下引起的序列偏倚的倾向。这种偏倚通常是未知的。本发明实质上提供了一种变通方法来补偿这种未知的偏倚和低效率。
在本发明的计算步骤中,对扩增模式即通常情况下的复制进行建模和考虑。如果扩增的模式是不同的,例如每个扩增循环的三倍扩增或其他x倍扩增,那么在本发明的方法中也可以考虑。由于扩增的标准方法,如pcr,是每个循环都复制,因此本发明是关于复制而记述的。当然,本发明还考虑每个循环有另外x倍扩增。当然,通常使用超过一个的扩增循环。因此,复制方法可以总共产生许多倍的扩增。优选地,进行2,3,4,5,6,7,89,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30或更多个扩增循环。
步骤d)包括确定扩增的带标签核酸拷贝的量,针对具有不同标签的核酸拷贝确定每个量。量的确定通常包括鉴定或分离带标签的核酸拷贝,以推出混合物中核酸的量。标签(可能并且常常在分子与分子之间是不同的)同样地与核酸种类的同一性一起被鉴定出。示例方法包括序列确定,其与核酸分子的序列一起还提供其同一性,然后可以进行计数。因此,该步骤提供了扩增后核酸拷贝(连同标签)的量。现在的目标是获得扩增前的量或数,这在步骤e)中完成。
根据步骤e),本发明的方法提供了两种可选的详细方法,称为(a)和(b),分别由实施例2.1.2和2.2进一步支持。一般地,也对于方法(a)和(b),通过针对样品中至少两种、优选至少四种、至少十种以上的不同种类核酸,使标签对每种核酸的附着概率近似化:对扩增后不同种类核酸的所述标签的量求平均值,从而基于步骤d)的确定的量,提供步骤a)的样品中核酸拷贝数的估计。对所述标签的量求平均值的步骤同样是指,针对本发明方法中使用的各种标签,在不同核酸种类(或片段)上平均出的标签浓度的拷贝数。因此,可以确定特定标签多久与核酸分子连接一次–与核酸种类无关,因为它是平均的。考虑到标签对不同种类核酸的附着可能不同,因为这是随机效应,并且遵循统计波动性,这进一步受到标签的附着不均匀性和附着反应的影响。为了获得平均值,对至少2种核酸种类进行平均,优选超过2种,例如3,4,5,6,7,8,9,10,20,30,40,50或更多种不同的核酸种类或这些值之间的任何范围。然后,可以用该平均值确定标签附着概率(也称为“标签附着至核酸拷贝的概率”)或对不同标签附着至核酸的预期数目。这种概率或预期可以根据标准统计方法来计算,其中考虑了所述平均值、以及对特定核酸种类(即被平均的成员)的单个标签附着而言相对于所述平均值的偏倚。这意味着标签的量还取决于标签附着反应动力学和标签混合组合物中的任何不均匀性,可能有些标签比其他标签更多地被代表。一旦知道了扩增效率(对于指定的核酸种类),就可以用确定的扩增后的量直接计算出样品中核酸的原始数(参见实施例章节的式(27))。在不能直接得出扩增效率的情况下,可以迭代近似化所述扩增效率。由于扩增效率与核酸数量直接相关,该数可以等效地直接替换扩增效率或与扩增效率一起代入计算中。实际上,根据优选的实施方案,样品中估计的扩增效率或预期的核酸拷贝数可互换使用。它们可以根据式(27)进行互换。因此,每当本发明的方法使用“估计的扩增效率”这种表述时,该表述也指“样品中预期的核酸拷贝数”,反之亦然。
优选地,在所述核酸复制程序中所述带标签核酸的扩增效率不是100%。不同种类的核酸拷贝的扩增效率可能会不同——这是一个难题,本发明的方法要考虑并固有地补偿这一难题。通常的扩增效率为60%~99.99%,优选为65%~99%,例如70%~95%,最常见80%左右。不同种类的核酸拷贝之间的示例性平均差异高达+/-20%或高达+/-10%。
本发明提供了在迭代过程中样品中核酸拷贝数的估计。这意味着通常以重复的方式逐步提高对核酸拷贝数的临时估计或预期,直到达到对核酸拷贝数的最终估计为止。作为对逐步改进而言的替代方法,也可以采用“强力”方法,其中计算各种核酸估计值(有间隔地或逐步地覆盖整个可能或合理的范围,例如从1到步骤d)确定的全部组合的核酸种类量的总量(“n”)),并选出所有这些估计的最佳模型。当从业者对改进感到满意时(通常是在收敛之后),或者如果通过选择最佳拟合估计出足够数量的预期值,则可以停止迭代过程。收敛(convergence)是指,例如——也是从业者在满意时设置的——当迭代不改变核酸拷贝数的估计值或改变为5%以下,例如3%以下或1%以下或0.5%以下或0.3%以下时。本发明方法中使用的其他参数,例如(扩增后)确定的带标签核酸拷贝的量,也可以在决定终止迭代时用到,以符合具体的扩增模型。所述模型然后可以用于计算样品中的核酸拷贝数。本发明包括对样品中核酸拷贝的最大估计数目的迭代优化。许多最大化过程是本领域已知的,可以根据本发明来使用。可能的示例迭代过程将从0到1的扩增效率间隔平均分隔成更小的间隔。从这些更小间隔中选出一或多个,以使得在所选更小间隔的边界处的可能性高于未选的更小间隔的边界处的可能性。在每个选中的更小间隔上,执行二进制搜索[4],以查找该间隔中可能性最高的扩增效率。总体最大值是二进制搜索结果的最大值。更小间隔的数量可以是1到n,并且二进制搜索可以在1到n个更小间隔中执行。当然,许多其他最大化方法也是可能的。同样,可以使用样品中预期的核酸拷贝数代替估计的扩增效率,它们可以互换使用。在示例中,kest是(近似地)满足公式(3)的值,其中,(3)的右侧是通过(2)定义的。有许多本领域技术人员已知的用于求解诸如(3)这样的非线性方程的方法,例如非线性优化或非线性规划。一个例子是根据式(4)和(5)的定点迭代。
本发明的一个具体实施方案,称为方法(a),包括优化样品中核酸拷贝的估值或预期数目(即中间值,直到迭代后确定最终估值)。所述优化是基于并使用步骤d)中所测不同标签的数量、以及根据标签附着于核酸拷贝的概率预期的附着至核酸的不同标签的数量、以及样品中预期的核酸拷贝数或估计的扩增效率(如上述,这二个“或”不同表述形式可互换使用)的先前迭代或预设值。步骤d)中所测不同标签的数量是一个测量值,无需进一步解释(另可见术语章节和上述步骤d))。附着于核酸的不同标签的预期数目根据标签附着于核酸拷贝的概率或为了更好参照而用可变术语写成:附着于核酸的不同标签的预期数按照[标签附着于核酸拷贝的概率](pb)或“标签附着概率”,实质上是指标记效率的期望值,可以如上述计算出,是基于对扩增后不同核酸种类的该标签的数量求平均。标签附着于核酸拷贝的概率可用于计算因其相关性而附着至核酸的不同标签的预期数目,即该概率与预期值直接相关。具有统计和概率运算能力的数学家能够执行这种计算。在实施例章节中的式(2)给出示例。我们还参考上文章节对步骤e)的介绍。最终参数是样品中预期核酸拷贝数或估计的扩增效率的预设值,或者是组合模型中的两者。第一次迭代通常从预设值开始。进一步的迭代可以从先前迭代的值开始。通常,提供样品的从业人员对假定值有很好的猜测,可以在本发明的方法中使用它。其他好的起始值是基于通常或常见的扩增效率。例如,可以在步骤d)所测不同标签的数量值范围内,或者从1到扩增的核酸拷贝的确定量之间取整数,来选出样品中核酸拷贝预期数量或估值扩增效率的第一迭代的预设值。该实施例还用括号中的参数表达式和示例中为了更好参考而用到的变量数值来进行如下编写:[样品中预期的核酸拷贝数](kest)或[估计的扩增效率](pd)的第一迭代的预设值是从[步骤d)所测不同标签数](du)的值中选出、或在1到[扩增的核酸拷贝的确定量](n)之间取整数。由于该方法提供了迭代改进,因此起始值或预设值不太重要。kest值将持续提高。初始预设值偏倚很大时,需要进一步的迭代来获得与从更好的初始值或预设值开始的过程同样的最终质量。实施例2.1.2及其中所列公式进一步说明了备选方案(a)的方法,其可以根据本文所述的总体发明来使用。
发明人还提供了甚至进一步改进的方法来计算样品中核酸拷贝的数量。该方法也是迭代的,并且使用不同核酸种类的标签量的平均值或标签附着于核酸的概率。
方法(b)主要包括四个步骤,称为(i)至(iv)。该方法通常包括(i)为扩增的带指定标签核酸拷贝的量的概率分布建模。替代地或附加地,也可使用各个核酸种类总和的量,即所研究的所有核酸种类(“库”)被扩增的带标签核酸拷贝的量。我们参考上文术语章节。简言之,创建了扩增过程模型,其中考虑了扩增机理,该机理有赖于每循环的理论扩增率(通常为复制)、循环数和扩增效率,鉴于湿化学反应的扩增效率是未知的,对这些行为进行建模。替代地,该方法使用步骤d)确定的(带标签)核酸的量(已知的确定值),标签附着到核酸的概率(如上确定的近似值;这是与方法(a)共有的步骤),样品中的估计扩增效率或预期核酸拷贝数(如上述可互换使用,这些值进一步通过迭代来优化),并基于在所述估计扩增效率下(即,扩增过程的模型)、每复制一个循环、核酸复制程序中扩增的带标签核酸拷贝的预期复制。这样的模型是本领域公知的。扩增方法已被广泛研究,并且在(无效的)复制或其他x倍富集过程中经过已知次数的循环后的统计学扩增率。我们指向参考参考文献[1],[2]和[3]以及实施例2.2章节。优选地,扩增的带标签核酸拷贝的预期复制用高斯(gaussian)、负二项式(negativebinomial)、伽马(gamma)、狄拉克德尔塔(diracdelta)或高尔顿-沃森(galton-watson)复制概率函数或其混合来建模。优选地,随机分支模型在对扩增的带指定标签核酸拷贝的量的概率分布进行建模时用到。这样的模型考虑了复制扩增过程的性质,例如galton-watson模型。对计算时间要求低的模型例如有负二项式或单峰或多峰分布。而且,混合可用于减少计算机计算时间,诸如利用具有较低计算时间要求的此类模型完成复杂任务,诸如为带标签核酸拷贝数(“k”)较高者提供概率函数。尽管样品的所述“k”值可能是未知的,但可根据作为用p(k|k,pb)加权的p(nb|k)的总和的式(25)或(7),将“k”近似为一个变量。p(k|k,pb)由式(26)或(8)给出。对概率分布中的变量求和会将该变量从概率分布中删除。这通常用未知变量完成。根据式(25),p(nb|k)是p(nb|k)与权重p(k|k,pb)的混合。式(25)和(26)是式(8)和(7)的特例。例如,k的较大数是5以上,优选10以上,例如20,30,40,50,60或更大,或这些值之间的任何范围。针对感兴趣的各种核酸种类计算出这样的概率函数。因此,获得了多个概率分布。
方法(b)的步骤(ii)包括根据步骤(i)的带标签核酸拷贝的模型化概率分布,确定带标签核酸拷贝的确定量(见步骤d))出现的可能性。本质上,这是将模型与步骤d)的实测值进行比较。给定数据集的统计模型的可能性是众所周知的数学概念,见例如pawitan,“inalllikelihood:statisticalmodellingandinferenceusinglikelihood”,(2003),oxforduniversitypress。在实施例中,实施例2.2.1~2.2.3显示了针对不同复制模型的不同公式,尤其在实施例2.2中,可能性由公式(9)和(10)给出。通常,对于每个感兴趣的核酸分子种类,将测量值与概率分布函数进行比较。步骤d)的确定值将具有给定的概率,该概率可以(或可能不)处在所述概率分布函数的最大可能性或概率处。对于各个核酸种类,该偏移可能(并且通常)是不同的。这导致下一步:
步骤(iii)包括通过改变样品中估计的扩增效率或样品中核酸拷贝的预期数量来使步骤(ii)的可能性最大化。再次强调,“估计的扩增效率”和“预期的核酸拷贝数”可以互换使用。因此,仅一个参数需要变动。所述值是变动的,例如随机地变动或确切地变动,这是通过例如使用诸如迭代步进等其他信息或使用概率分布函数的信息而将所述值移至预期方向——例如通过上坡移动,使得确定的值向概率分布函数的最大值移动等。用于最大化过程的许多方法在本领域中是已知的,并且可以用于本发明。最后,在步骤(iv)中,根据所述步骤(iii)的最大化模型,提供了样品中核酸拷贝数的估计。最大化是例如当从业人员对样品中核酸拷贝数的提高感到满意时,或者如在上文对步骤e)的介绍中所述达到收敛时,达到一个迭代。到此结束步骤e)。
本发明还可以包括将步骤e)的结果呈现在诸如打印机输出,屏幕之类的可读介质上,或者写在诸如计算机存储设备之类的数据载体(诸如硬盘驱动器,闪存等)上。
总体上,本发明在实施例和其中所示的公式中进一步说明。这些公式可以单独用于说明本发明和进一步详述本发明的方法。例如,可以根据式(12),式(45)或式(46)来确定标签附着于核酸的概率(“pb”)。估计的扩增效率(“pd”)或样品中预期的核酸拷贝数(“k”或“kest”)可以互换使用,并可以根据公式(27)进行转换。在方法a)中,扩增的带指定标签核酸拷贝的量的概率分布(“p(nb”)可以根据式(3),优选地通过式(4)和式(5)二者来确定。在备选的b)中,扩增的带指定标签核酸拷贝的量的概率分布(“p(nb”)可以根据式(25)确定。所有这些公式应用都是本发明的优选实施方案——也是实施例,并且可以彼此组合。
本发明进一步提供一种采用本发明方法的计算机程序产品,例如,包含在计算机上执行或协助所述方法和步骤的机器代码。可以在任何种类的存储设备上提供计算机程序产品。还提供了一种系统,例如计算机设备,其被编程为辅助执行本发明方法的步骤。计算步骤通常在没有操作员帮助的情况下执行。输入和设置步骤可以由程序或系统来辅助,例如,在有需要时,通过建议一个针对数值随机步骤重复的选项。当然,所述程序或系统也可以用默认参数来执行,而无需操作员的进一步介入。特别地,本发明提供了一种计算机程序产品,包括计算机可读指令,该计算机可读指令用于或适于根据步骤e)基于带标签核酸扩增后确定的量来计算样品中核酸拷贝数的估计值。换句话说,本发明提供了一种包括指令的计算机可读介质,所述指令在被计算机执行时使计算机至少执行本发明方法的步骤e)。其他方法步骤的数据当然可以如上述用在步骤e)。
包括计算机程序产品的计算机可读存储设备适于在计算机上执行根据本发明的方法或由计算机支持根据本发明的方法。特别地,步骤e)可以在计算机上执行。甚至通常的湿化学步骤a),b),c)和/或d)也可以通过计算机来辅助,例如控制和获取来自自动或半自动序列读取器的数据。计算机程序产品或存储设备还可以配备有从样本获得短测序读数的序列生成组件,例如测序仪,优选含计算机组件的测序仪。例如,计算机可读介质可以包括但不限于磁存储设备(例如,硬盘,软盘,磁条,…),光学盘(例如,光盘(cd),数字通用盘(dvd)),...),智能卡,和闪存设备(例如卡,棒,钥匙驱动器,…)。尽管可能,为执行步骤a)到d)的所作调整属于或不属于计算机程序产品的一部分。计算机程序产品能够接收扩增的带标签核酸拷贝的确定量的输入,且带不同标签的核酸拷贝每个都有确定量——根据步骤d),这就足矣。因此,计算机程序产品适于从所述输入执行步骤e)。扩增的性质(例如复制方法,如pcr),以及优选地,扩增的循环数和可能附着的标签也被用作计算机程序产品的输入。
因此,本发明还涉及在计算机上执行步骤e),特别是在计算机已经接收到所述输入之后。
所述计算机程序产品可以适于将步骤e)的结果呈现(包括写入)或展示在诸如打印机输出、屏幕之类的可读介质上,或者写在诸如计算机驱动设备(像硬盘,闪存等)之类的数据载体上,如上述。同样,在计算机上执行步骤e)的方法优选还包括在这种可读介质上呈现步骤e)的这种结果的步骤。
本发明通过以下附图和实施例进一步举例说明,而不限于本发明的这些实施方案。在具体实施方案中,本发明提供的方法对于不均匀分布的多组标签(例如条形码,umi)或对于标签太少不足以进行标签计数的标签组,可以产生高精度pre-pcr(或其他扩增)核酸数的估计数。为此,我们首先估算了扩增前标签的分布(图4),随后将其用于估算进行pcr之前核酸的数量。首先,我们研究了标签计数的一种改良方法,该方法使用扩增后观察到的独特标签数作为输入,但也考虑了扩增前标签的分布。其余方法依赖于扩增后的标签频率作为输入,并使用统计模型来描述扩增过程(图5)。例如,我们用pcr程序的泊松(poisson),二项式(binomial)和高尔顿-沃森模型生成的合成数据来评估我们的方法。
附图
图1:序列特异性pcr偏倚。相同cdna片段的两个相同大小的库(标签为cdna1和cdna2)通过pcr扩增。pcr后,它们的大小不同。这可能导致不准确的基因和异构体定量。
图2:umi帮助识别pcr生成的片段拷贝。在pcr之前,将umi连接至cdna片段。如果一组umi足够大,则相同的umi将不会与一种cdna片段的两个拷贝连接。在这种情况下,pcr之后不同umi的数量与pcr之前片段拷贝的数量相同。
图3:正确估计pcr前库大小需要大量均匀分布的umi。如果umi集太小或分布不均匀,则相同umi可以连接至两种不同片段拷贝。对pcr后的不同umi进行计数会导致低估pcr前的拷贝数。
图4:根据每个库中的pcr后umi分布估算umi的pcr前分布。为此,在所有片段库中对umi计数的pcr后频率进行平均。
图5:估计一个库中片段的pcr前数量。模型依赖型方法使用pcr之后的umi计数作为输入。模型非依赖型方法使用pcr后不同umi的数量作为输入。两种方法都使用估计的pcr前umi分布作为输入。
图6:概率分布p(n|k,pd,d)(实线)比对通过负二项式(虚线)和正态分布(点线)得出的近似值。每组显示k=1,...,5的分布。从左到右的峰按升序对应于k。
图7:umi概率pb的概率密度函数。在每个umi位置(pa,pc,pg,pt)取样至狄利克雷(dirichlet)分布,α=5。umi概率pb是其核苷酸概率的乘积。垂直的橙色线位于x轴的1/b处,pb值表示均匀的umi分布
图8:umi计数的准确性。数据来自二项式模型。umi分布的类型(均匀/不均匀)和umi数b在图的标题中给出。预复制片段的估计数k的相对误差在y轴上,真实数在x轴上。
图9:经频率校正的umi计数的准确性。数据来自二项式模型。umi分布的类型(均匀/不均匀)和umi数b在图的标题中给出。预复制片段的估计数k的相对误差在y轴上,真实数在x轴上。
图10:泊松-正态(poisson-normal)法的准确性。数据来自poisson模型,umi数b=64。umi分布的类型(均匀/不均匀)和效率pd在图的标题中给出。预复制片段的估计数k的相对误差在y轴上,真实数在x轴上。
图11:泊松-正态分布法的准确性。数据来自poisson模型,umi数b=256。umi分布的类型(均匀/不均匀)和效率pd在图的标题中给出。预复制片段的估计数k的相对误差在y轴上,真实数在x轴上。
图12:二项式-正态方法的准确性。数据来自二项式模型,umi数b=64。umi分布的类型(均匀/不均匀)和效率pd在图的标题中给出。预复制片段的估计数k的相对误差在y轴上,真实数在x轴上。
图13:二项式-正态方法的准确性。数据来自二项式模型,umi数b=256。umi分布的类型(均匀/不均匀)和效率pd在图的标题中给出。预复制片段的估计数k的相对误差在y轴上,真实数在x轴上。
图14:gw多组分混合的准确性。数据来自galton-watson模型,umi数b=64。umi分布的类型(均匀/不均匀)和效率pd在图的标题中给出。预复制片段的估计数k的相对误差在y轴上,真实数在x轴上。
图15:gw多组分混合的准确性。数据来自galton-watson模型,umi数b=256。umi分布的类型(均匀/不均匀)和效率pd在图的标题中给出。预复制片段的估计数k的相对误差在y轴上,真实数在x轴上。
实施例方法
本节中用于估计pcr之前片段拷贝数的方法分为两类。第一类见第2.1节,是基于观察到的不同umi的数量。由于此数量在pcr之前和之后都是相同的,因此这些方法与pcr过程无关。第二类方法见第2.2节,是基于pcr之后的umi计数。由于后者受pcr影响,因此这些方法依赖于pcr过程的统计模型。2.1和2.2节中的方法要求了解pcr之前的umi分布。这可以从描述umi每个位置插入的核苷酸的说明中得出,或从pcr后可得的数据中得出。由于pcr之前的umi分布不仅受核苷酸插入频率的影响,而且还受连接偏倚的影响,因此第二种方法似乎是优选的。相应的方法在2.3节中讨论。
1.预备
在下文中,我们用b表示umi的总数,用b=1,...,b表示umi标签。umib的pcr前概率用pb表示,pcr前完整的umi分布用
我们还设定
因此,k和n是pcr之前和之后库f的大小。请注意,在pcr之前和之后的计数符号中,我们不会区分不同的库。在下文中将始终理解,除非另有明确说明,否则方法将应用于单个库。
2.1根据观察到的不同umi数量进行估计
2.1.1计数观察到的不同umi
从umi计数
仅在极不可能将相同umi与两种不同片段拷贝连接时,此方法才会产生合理的估值。这需要b的数量远远超过k的,并且umi必须均匀分布。如果k足够大,以至于
2.1.2对观察到的不同umi进行频率校正计数
与其简单地计数观察到的不同umi的数量,不如通过从具有预复制umi分布
对于均匀分布的umi,在[1]和[2]中也给出了上述公式,在那里将其用于估计umi计数与实际的pcr前片段拷贝数的偏差。在这里,我们使用(2)通过以下要求来估计k
式(3)对于总体
该迭代过程收敛到(5)的固定点,此点求解(3)。因此,ki收敛于式(3)中的kest。如前所述,该方法需要n足够小,以至于
2.2根据umi计数估算
与上一节相比,以下方法利用了pcr后umi计数的全向量
这里,
我们在本节中开发三种复制模型。poisson和二项式模型产生了单代后代,而galton-watson过程模型产生多代。后者是pcr过程的更准确描述,因此相应的方法应在真实数据上产生更好的结果。原则上,可以通过最大化概率来估计片段拷贝的原始数k和效率pd
但是,对于较大k值,(8)中的求和数变得过高。替代地,我们通过最大化以下两者之一来估算k和pd
或
这里,p(nb|k,pb,1-pb,pd)是当umi集合只由b和另一
2.2.1poisson复制
对于poisson模型,从具有复制效率λ的k片段生成n的概率通过以下poisson概率得出
p(n|k,λ)=pkλ(n-k)(11)
其可以示出,在这种情况下,(10)中的条件概率p(n|n,k,pb,λ)具有如下给出的均值μ(n,k,pb,λ)和方差σ2(n,k,pb,λ)
μ(n,k,pb,λ)=pbn(12)
和
因(12)和(13)与效率λ无关,因此此参数将从下面的符号中删除。对于每个umib,我们用(12)和(13)作为正态分布的均值和方差。然后,可以找到使(10)最大化的k值作为以下二次多项式的正根。
k2-k((n+1)-n2v)-n(n-1)=0(14)
其中
由于其推导和通过正态分布对p(n|n,k,pb)建模,因此该方法称为poisson-正态(normal)或p-normal。p-normal方法与复制率λ无关,并且因为可以高效地计算(14)和(15)而非常快。
2.2.2二项式复制
上一节中的poisson复制允许从单个片段生成无限数量的片段。然而,实际上,只能生成有限数量的片段。因此,二项式模型是对poisson模型的明智替代。对于二项式模型,复制概率由下式给出:
其中,m是从单个片段生成的最大片段数,pd是在单次进行复制片段的尝试中成功的概率。如前述,我们设置qd=1-pd。如果每个片段可以在每个pcr循环中复制一次,则在c个循环后
m=2c-1(17)
此外,如果pd(1)是一个片段在一个循环后被复制的概率,则c个循环后(16)中的成功概率pd(c)如下得出
随着m增加,概率(16)收敛至(11),且λ=mpd。因此,渐近地,poisson和二项式复制模型变得等效。如前述,概率p(n|n,k,m,pb,pd)的均值由(12)给出。而另一方面方差σ2(n,k,m,pb,pd)由以下给出
表达式(19)同样与效率pd无关,并且在m→∞时收敛为(13)。如前述,如果用(12)和(19)作为(10)中正态分布的均值和方差,则可以找到使(10)最大化的k值作为二次多项式的根。在这种情况下,该多项式由以下给出
k2(m+1)-k(m(n-v+1)+2n-1)-(n(mn-m-n-2)+v)=0(20)
其中的v,如前述,由(15)给出。这种估计k的方法称为二项式-正态分布法或b-normal。
2.2.3galton-watson复制
pcr的迭代性质通过分支过程建模比poisson或二项式复制模型更适合。这里,我们选择galton-watson法。故本节中开发的方法将以galton-watson或gw作为前缀。后代分布由p(2|1)=pd和p(1|1)=1-pd给出。因此,在该模型中,每个片段都可以在一个循环内以概率pd产生新的后代。如果在循环开始时看到k片段,则在循环结束时看到n片段的概率是二项分布,即
如果循环数c大于1,则概率p(n|k,pd,c)最好通过其概率生成函数pgf(p(n|k,pd,c))(x)(也称为普通生成函数)来描述。例如,我们有[3]的第10章,
pgf(p(n|1,pd,1))(x)=qdx+pdx2(22)
和
其中
在下文中,我们将以p(n|k,pb,pd,c)模型研究单峰和多峰分布,并利用以下事实:
及
为了简化对使(9)最大化的k的搜索,我们进一步要求pd满足
单峰分布
可看出,(25)及(24)中galton-watson复制模型的均值和方差由下式给出
μ(k,pb,pd,c)=(1+pd)cpbk(28)
和
通过将参数分布的均值和方差设置为(28)和(29),我们为galton-watson复制过程定义了单峰模型。在我们的实验中,我们使用正态(normal),伽马和负二项式(negativebinomial)分布,分别通过以下给出均值和方差参数
和
多峰分布
我们在上一节中用模型进行的实验表明,galton-watson复制模型的分布不适合用单峰模式描述。因此,在本节中,我们分别对(25)中混合物的每种成分进行建模。我们利用这样一个事实,即,如果pcr之前有k次看到b,则在pcr进行n次之后看到b的概率具有以下均值和方差
μ(k,pd,c)=(1+pd)ck(33)
和
σ2(k,pd,c)=qdk(1+pd)c-1((1+pd)c-1)(34)
delta分布和单峰分布的混合
式(33)和(34)表明,对于k=0来说,分布p(n|k,pd,c)等于克罗内克δ函数(kroneckerdelta)δ0。因此,在小k的情形下,p(n|k,pd,c)在n=0处有一个明显的峰值。因此,与其将p(n|k,pd,c)建模成单峰分布,更明智的做法是对n=0和n>0这两种情况分别建模。这可以通过将p(n|k,pb,pd,c)写成混合体来完成
p(n|k,pb,pd,c)=p(n=0|k,pb,pd,c)δ0+p(n>0|k,pb,pd,c)p(n|k,pb,pd,c,n>0)
(35)
对于由两个权重为w1,w2、均值为μ1,μ2和标准误差为σ1,σ2的组分组成的混合体,根据混合体的整体均值μ和标准误差σ的公式,我们有
因此,μ2,σ2可源自μ,σ,μ1,σ1。若第一组分对应于n=0、第二组分对应于n≠0,则w1由以下给出
p(n=0|k,pb)=p(k=0|k,pb)=(1-pb)k(38)
因为μ1和σ1由μ(δ0)=σ2(δ0)=0给出,对于μ2,我们有
且对于
如前述,我们可以用参数概率分布来近似化p(n|k,pb,pd,c,n>0),这是通过将其均值和方差设置为(39)和(40)来实现。为此,我们再次使用正态(30),伽玛(31)和负二项式(32)分布。
多组分混合体。即使delta分布和单峰分布的混合体正确地描述了小k和大k时的情况,我们仍在实验中发现了中等范围k的系统偏差。这是由于以下事实:在此范围内,总和(25)由小k的项主导,负二项式或gamma分布给出的近似值p(n|k,pd,c)不准确。这可见于图6,该图还显示了近似值的不准确率随效率pd的提高而增加。实际上,对于pd接近1的情形,这些图显示出类似分形的结构,其中整体形状以较小的比例重复。
为了适应(25)中混合体组分的复杂结构,我们研究了一种替代模型,其中p(n|k,pb,pd,c)是两个以上分布的混合体,其均值和方差由(33)和(34)给出。对于k=0,我们再次使用kroneckerdeltaδ0。对于k>0,我们最初拟合负二项式分布。但是,我们发现这在小k的情形下拟合较差。因此,我们将k分为小、中和大值的范围。对于小值,我们借助(24)精确计算每个k确切的p(n|k,pd,c),对于中等值,我们对每个k使用正态分布且均值和方差由(33)和(34)给出,对于大k的部分总和,我们使用单个负二项式分布。我们为k的范围选择了固定边界。然而,原则上,可以自适应性选择这些边界以最小化模型与真实分布之间的误差。在我们的实验中,小k的范围从k=0到k=15,中等k的范围从k=16到k=49,大k的范围从k=50开始。
截断概率分布
应注意,如果p(n|k,pd,c)是在n<k时由分布f(n)且f(n)>0建模的,则f(n)必须限制在n≥k的范围内。这意味着
其中cdff(n)是的f(n)累积函数。当然,这也意味着p(n|k,pd,c)的均值和方差与非限制性f(n)的均值和方差不一致。通常,如果一个分布pdf(n)是通过将另一分布f(n)限制在n≥k范围内而给出的,则pdf(n)=cf(n)且c=1/(1-cdff(k-1))、且非限制性f(n)的均值和方差必须等于
和
如果f(n)定义在非负整数上,例如负二项式或伽马分布的情形,则计算(42)和(43)很容易。但是,我们发现截断作用较小,仅在大k时用于多组分混合体的单个负二项式分布。
2.3根据数据估算umi频率
由于(12)适用poisson、二项式和galton-watson复制模型,这表明对于从这些模型生成的数据,pb可以根据nb的分布进行估计。重写(12)如下
表明,
这得出相对于b的概率分布。即使(44)中的期望值与n和k无关,但
这减少了小库对pb估算的影响。
3实验
3.1生成合成数据
在我们的实验中,我们使用了umib=64和b=256,它们是均匀和非均匀分布的。对于非均匀数据,umi每个位置上核苷酸的分布的取样来自dirichlet分布,即
其中pa,pc,pg,pt是观察到核苷酸a,c,g和t的概率。总的来说,umib的概率由下式给出:
其中nt(b,i)是条形码b第i位的核苷酸,乘积延伸至b的所有位置。我们在(47)中设定α=5。b=64和b=256的对应概率密度函数pb在图7中给出。这显示了围绕1/b(垂直线)的pb可变性及其概率密度函数的不对称形状。因此,取样的umi分布
λ=(1+pd_gw)c(49)
和
其中m=2c-1。在下文中,我们使用pd_gw而不是λ和pd_bin,并以pd来简单表示pd_gw。
3.2结果
umib=64和b=256的umi计数结果可见于图8,频率校正后的umi计数结果见图9。与其他方法相比,两种版本的umi计数都与复制模型无关,因此与复制效率无关。由于这一原因,我们仅包括效率为0.5的二项式复制模型的结果。其他复制模型和效率的结果相同。图8显示,仅当umi数b远大于k时,umi计数才产生良好的k估值。当b=64,仅在k=10时估值是很准确的。当b=256,对k=100的估计也算合理。但是,对于大k值,umi计数将k低估至少50%。图8进一步显示,对于不均匀的数据,umi计数得出准确度略有降低的结果。另一方面,图9的频率校正计数产生更准确的结果,在更大的k范围内无偏差。但是,对大k,一旦观察到的不同umi数
图10和11中给出了b=64和b=256时的poisson-正态分布法结果,其定义为(14)和(15)中二次方程的正根。这些结果表明,p-normal方法的准确性在很大程度上与效率pd无关。但是,不均匀性似乎在小k的情形下具有负面影响。与频率校正计数相比,p-normal在k的整个范围内都能产生良好的结果。图12和13给出了二项式-正态分布法的结果。这表明poisson-正态分布法和二项式-正态分布法之间的差异很小。如第2.2.2节所述,这是由于poisson和二项式复制模型的渐近等效性引起的。poisson-正态分布法和二项式-正态分布法之间的细微差异仅适用于小k的情形。
图14和15包含gw-multimix方法的结果,该方法在所有gw方法中具有最高的准确性。图14和15显示,准确性随效率pd提高而略有提高。另一方面,不均匀性对结果影响很小。gw-multimix方法可在k的整个范围内产生有效结果,并且比poisson-正态分布法和binomial-正态分布法更准确,尤其是对于小k情形而言。在b=256umi的情形中,gw-multimix方法具有相当高的效率准确性,远远超过了所有其他方法。鉴于此方法的优越性,可能值得对poisson和二项式复制模型使用多成分混合进行p(n|n,k,pb)建模。
总之,我们的实验表明,我们的方法可以准确估算出复制前片段的数量k。这些估值在很大程度上与umi的复制效率pd和复制前分布
参考文献
[1]glennk.fu,jinghu,pei-huawang,andstephenp.a.fodor.countingindividualdnamoleculesbythestochasticattachmentofdiverselabels.proceedingsofthenationalacademyofsciences,108(22):9026–9031,2011.
[2]glennk.fu,weihongxu,juliewilhelmy,michaeln.mindrinos,ronaldw.davis,wenzhongxiao,andstephenp.a.fodor.molecularindexingenablesquantitativetargetedrnasequencingandrevealspoorefficienciesinstandardlibrarypreparations.proceedingsofthenationalacademyofsciences,111(5):1891–1896,2014.
[3]charlesm.grinsteadandlauriej.snell.grinsteadandsnell’sintroductiontoprobability.americanmathematicalsociety,versiondated4july2006edition,2006.
[4]donalde.knuth.theartofcomputerprogramming:sortingandsearching.addison-wesley,secondedition,chapter6,1998.
- 还没有人留言评论。精彩留言会获得点赞!