一种治疗肝细胞癌的药物组合物及其应用的制作方法
本发明属于生物工程技术领域,具体涉及一种治疗肝细胞癌的药物组合物及其应用。
背景技术:
双特异性酪氨酸-(y)-磷酸化调节激酶3(dyrk3)是哺乳动物激酶家族的五个成员之一,其包括dyrk1a,dyrk1b,dyrk2,dyrk3和dyrk4(1)。虽然该家族在催化结构域中具有高度保守的氨基酸序列,在活化环中具有tyr-x-tyr基序,但它们的n-和c-末端区域完全不同。大量研究证实了dyrk家族激酶在各种人类疾病中的重要作用,特别是在不同类型的人类肿瘤中。以前的研究报道dyrk1a和dyrk1b促进癌细胞增殖和侵袭。相反,dyrk2被鉴定为肝细胞癌(hcc)和crc中的肿瘤抑制因子。外显子组测序和阵列分析最近发现dyrk3参与了胃癌和神经胶质瘤,其在包括肝细胞癌(hcc)在内的肿瘤中的作用和作用机制仍未得到阐明。
众所周知,癌细胞的代谢重组通常由oxphos到糖酵解的变化来定义。值得注意的是,来自糖酵解和戊糖磷酸途径(ppp)的大量代谢物不仅是核苷酸和氨基酸合成的重要碳源,而且对于从头嘌呤生物合成也是必需的。新出现的证据表明,包括肿瘤在内的几种代谢紊乱中嘌呤代谢紊乱。例如,c-myc依赖性嘌呤生物合成显著抑制前列腺癌中对抗雄激素的治疗反应。嘌呤合成酶水平的升高也有助于癌症干细胞的发展,并预测胶质母细胞瘤患者的预后不良。然而,嘌呤合成途径在hcc中的作用和调节仍然没有表征。
技术实现要素:
本发明的目的在于提供一种治疗肝细胞癌的药物组合物。
本发明的另一目的在于提供一种atf4转录活性抑制剂。
本发明的技术方案之一:
一种治疗肝细胞癌的药物组合物,其有效成分包括双特异性酪氨酸-(y)-磷酸化调节激酶3,该双特异性酪氨酸-(y)-磷酸化调节激酶3对ncoa3进行磷酸化,破坏noca3与atf4的结合,引起atf4的转录活性的抑制,从而下调嘌呤代谢的发生。
在本发明的一个优选实施方案中,其有效成分为双特异性酪氨酸-(y)-磷酸化调节激酶3。
进一步优选的,所述双特异性酪氨酸-(y)-磷酸化调节激酶3在ser-1330处直接磷酸化ncoa3。
本发明的技术方案之二:
双特异性酪氨酸-(y)-磷酸化调节激酶3在制备治疗肝细胞癌的药物组合物中的应用,该双特异性酪氨酸-(y)-磷酸化调节激酶3对ncoa3进行磷酸化,破坏noca3与atf4的结合,引起atf4的转录活性的抑制,从而下调嘌呤代谢的发生。
在本发明的一个优选实施方案中,所述双特异性酪氨酸-(y)-磷酸化调节激酶3在ser-1330处直接磷酸化ncoa3。
本发明的技术方案之三:
一种atf4转录活性抑制剂,其有效成分包括双特异性酪氨酸-(y)-磷酸化调节激酶3,该双特异性酪氨酸-(y)-磷酸化调节激酶3对ncoa3进行磷酸化,破坏noca3与atf4的结合,引起atf4的转录活性的抑制。
在本发明的一个优选实施方案中,其有效成分为双特异性酪氨酸-(y)-磷酸化调节激酶3。
进一步优选的,所述双特异性酪氨酸-(y)-磷酸化调节激酶3在ser-1330处直接磷酸化ncoa3。
本发明的技术方案之四:
双特异性酪氨酸-(y)-磷酸化调节激酶3在制备atf4转录活性抑制剂中的应用,该双特异性酪氨酸-(y)-磷酸化调节激酶3对ncoa3进行磷酸化,破坏noca3与atf4的结合,引起atf4的转录活性的抑制。
在本发明的一个优选实施方案中,所述双特异性酪氨酸-(y)-磷酸化调节激酶3在ser-1330处直接磷酸化ncoa3。
本发明的有益效果是:本发明的有效成分包括双特异性酪氨酸-(y)-磷酸化调节激酶3,该双特异性酪氨酸-(y)-磷酸化调节激酶3对ncoa3进行磷酸化,破坏noca3与atf4的结合,引起atf4的转录活性的抑制,从而下调嘌呤代谢的发生,显著抑制了hcc异种移植肿瘤模型中的肿瘤生长和转移。
附图说明
图1为本发明实施例1中的实验结果图之一。
图2为本发明实施例1中的实验结果图之二。
图3为本发明实施例1中的实验结果图之三。
图4为本发明实施例1中的实验结果图之四。
图5为本发明实施例1中的实验结果图之五。
图6为本发明实施例1中的实验结果图之六。
图7为本发明实施例1中的实验结果图之七。
图8为本发明实施例1中的实验结果图之八。
具体实施方式
以下通过具体实施方式结合附图对本发明的技术方案进行进一步的说明和描述。
实施例1
一种治疗肝细胞癌的药物组合物,其有效成分包括双特异性酪氨酸-(y)-磷酸化调节激酶3,该双特异性酪氨酸-(y)-磷酸化调节激酶3对ncoa3进行磷酸化,破坏noca3与atf4的结合,引起atf4的转录活性的抑制,从而下调嘌呤代谢的发生。优选的,所述双特异性酪氨酸-(y)-磷酸化调节激酶3在ser-1330处直接磷酸化ncoa3。
本实施例通过下述方法来验证本发明的功能,具体如下:
1、材料和方法
患者和临床样本
从吉林大学第一附属医院和牡丹江医科大学附属红旗医院接受肝切除术的hcc患者(n=224)获得两组hcc组织样本和相应的相邻形态正常组织样本。所有将收集的样品在手术切除后立即置于液氮中并储存在-80℃下进行进一步分析。本研究经吉林大学伦理委员会和牡丹江医科大学附属红旗医院批准,获得了本研究所有参与者使用组织样本的书面知情同意书。根据世界卫生组织(who)标准确诊为hcc。基于国际抗癌联盟的第六版肿瘤-淋巴结-转移(tnm)分类,肿瘤分期:t1=没有血管侵犯的孤立性肿瘤;t2=单发血管侵犯,多发,5cm;t3=多个,>5cm,侵入门静脉或肝静脉的主要分支;t4=入侵胆囊以外的邻近器官,穿孔内脏腹膜。在手术前,没有患者接受过任何抗癌治疗,包括经皮消融,放射治疗或化疗栓塞。
微球体培养
提到的相关细胞(5×103细胞/孔)在含有补充有b27,20ng/mlegf和20ng/mlbfgf的培养基的超低附着24孔板中生长。媒介每隔一天更换一次。培养15天后,对直径>50μm且>150μm的球囊进行计数和分析。
体内致瘤和转移测定
所有动物实验均根据nih指南进行,以用于实验动物。4至6周龄的非肥胖/严重联合免疫缺陷(nod/scid)小鼠,获自上海生命科学研究院实验动物中心(sibs)。所有动物工作均按照国家指导方针进行,所有动物实验均经吉林大学伦理委员会和牡丹江大学批准。对于原位肝细胞癌小鼠模型,将hcclm3-plv-dyrk3和hcclm3-plv-nc以及其他指定的细胞(每只小鼠4×106个细胞)皮下注射到nod/scid小鼠的侧腹中。4周后,切除皮下肿瘤并切成1mm3立方体,然后植入小鼠肝脏的左肺叶(每组10只)。对于肿瘤转移小鼠模型,将200μlpbs中的2×106个细胞注射到小鼠的尾静脉中(每组10个)。通过生物发光对肿瘤形成和转移进行成像。将d-荧光素(milliporesigma,usa)以100mg/kg腹膜内注射到小鼠中,并且每周用xenogenivisspectrumimagingsystem(caliperlifesciences,usa)检测生物发光。6周后,处死小鼠。分离肺用于检查转移性肿瘤的数量,并测量肝肿瘤的重量。然后制备肝脏和肺用于苏木精和伊红(he)染色或蛋白质印迹分析。
统计分析
student′st检验用于比较两组之间的参数。单因素方差分析用于比较大于2的组之间的参数,并且使用双向anova来比较生长曲线的参数。通过pearson相关分析评估两种给定蛋白质的表达的相关性。使用社会科学统计软件20.0(spss)。所有统计测试都是双面的。
2、结果
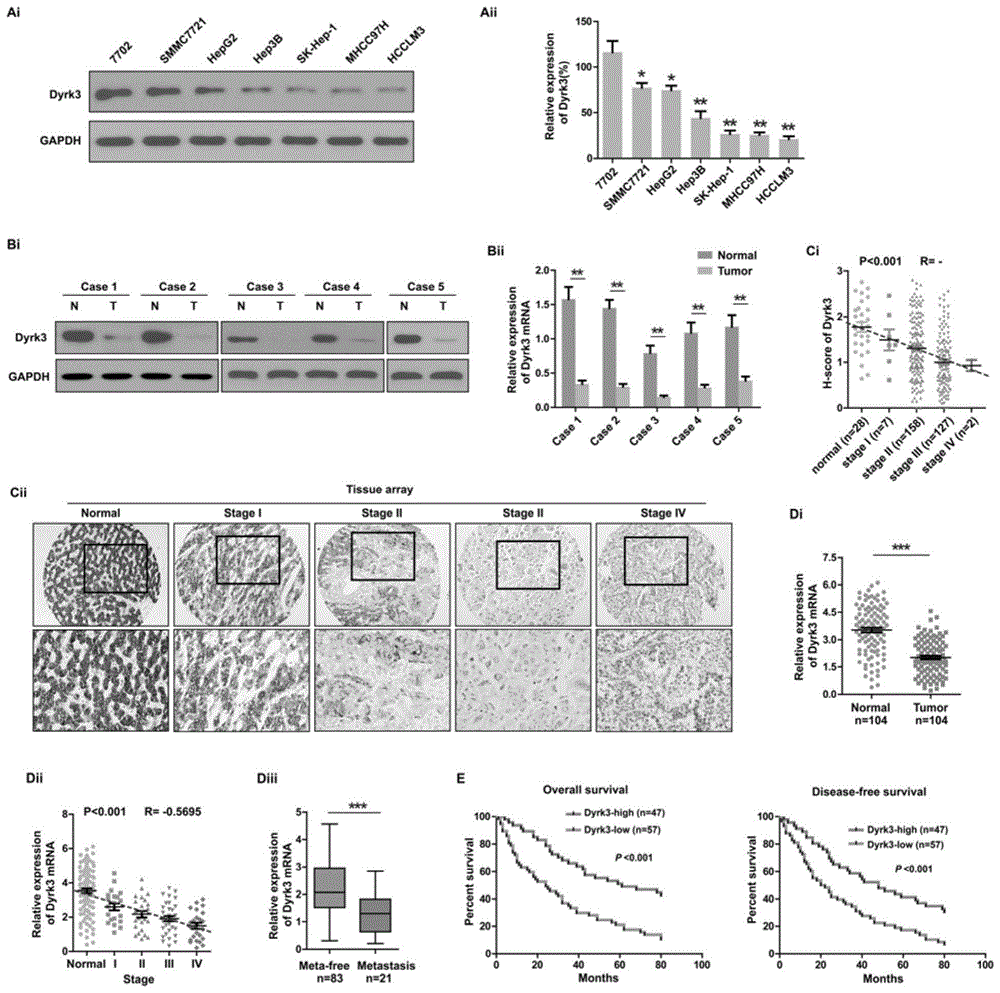
dyrk3表达在hcc组织中显著下调,这预示着不良预后
为了探讨dyrk3在hcc发展中的生物学功能,本实施例首先确定了dyrk3在hcc细胞系以及正常肝细胞系hl-7702中的表达。与对照相比,蛋白质印迹和qrt-pcr测定揭示了所有hcc细胞系中dyrk3蛋白和mrna水平的急剧降低(图1ai和aii)。类似地,与相应的相邻正常组织相比,5个hcc患者样本中dyrk3蛋白和mrna的水平显著下调(图1bi和bii)。为了确认较大样本组中dyrk3表达降低并将其与临床表型相关联,本实施例对由294名患者组成的hcc组织阵列进行了免疫组织化学染色(ihc)。本实施例的数据揭示了dyrk3与匹配的正常组织相比较低的表达;此外,表达dyrk3的细胞百分比与hcc恶性程度呈负相关(图1ci和1cii)。
为了阐明dyrk3在hcc中下调的临床意义,本实施例分析了104例hcc患者标本中临床病理特征与dyrk3表达的关系。正如预期的那样,dyrk3水平在另一个患者队列中的hcc组织中下调(图1di)并且与肿瘤分化呈正相关(补充表1),但与晚期tnm分期(图1dii)和转移(图1diii)呈负相关。kaplan-meier分析表明,较高水平的dyrk3与总体存活(os)和无病存活(dfs)显著相关(图1e)。多变量分析显示肿瘤大小和dyrk3表达是os的独立预测因子。
这些发现表明,dyrk3缺失可能在hcc进展中起关键作用,并且是该疾病的有价值的生物标志物。
dyrk3过表达显著抑制hcc的生长和转移
观察到dyrk3低表达与hcc患者生存率低的关联,本实施例开始在功能上表征dyrk3对hcc的影响。为此,本实施例在hcclm3和sk-hep1细胞系中稳定表达dyrk3(补充图1a)。dyrk3过表达不仅显著抑制了贴壁依赖性和不依赖贴壁的生长(图2ai和aii),而且还降低了hcc细胞的生长速率(图2aiii)。
然后,本实施例使用异种移植和原位癌模型检查了dyrk3对hcc生长的影响。与体外发现一致,与对照组相比,具有dyrk3过表达的hcc细胞产生较小的肿瘤(图2b)。然而,hepg2细胞中的dyrk3沉默显著促进体内hcc生长。
transwell测定表明,与对照相比,dyrk3转染的hcc细胞的侵袭能力较弱(图2c),而在hcc细胞中沉默dyrk3具有相反的作用。在体内,通过尾静脉注射hcclm3-dyrk3的裸鼠发生较小且较少的肺转移(图2di和2dii),而hepg2-shdyrk3细胞与对照相比具有更大且更多的肺转移,通过组织学分析进一步证实。
本实施例还分析了它们的球形成能力。过表达dyrk3的hcc细胞产生的球体数量少于其亲本细胞产生的球体数量(图2e)。相反,dyrk3耗尽的hcc细胞产生的球体数量大于亲本细胞产生的球体数量。毫不奇怪,两种关键干细胞转录因子sox2和oct4的水平在dyrk3过表达球体中显著下调(图2f)。
总之,这些发现表明dyrk3显著抑制hcc的生长和转移。
dyrk3缺失通过激活嘌呤合成途径促进hcc细胞增殖和侵袭
考虑到改变的代谢活动在肝脏肿瘤发生和转移中的关键作用,本实施例探讨了是否沉默dyrk3重编程的肿瘤相关代谢途径。为此,本实施例进行了基于质谱(ms)的hepg2细胞的代谢分析,所述hepg2细胞表达针对dyrk3的shcontrol或shrna。有趣的是,敲除dyrk3显著增加了核糖-5p(r5p),嘌呤核苷酸及其中间体(如imp,腺苷,鸟嘌呤和黄嘌呤)的细胞内库(图3a-b),表明dyrk3敲低可能促进从头嘌呤合成途径。
为了证实这一假设,本实施例再次使用lc-ms/ms测量稳定同位素标记的谷氨酰胺(酰胺-15n)的相对通量,其可以在两个不同的位置掺入嘌呤环中。如所预期的,沉默dyrk3显著增加双标记的15n-嘌呤中间体(imp,amp和gmp)的量(图3ci)。当测量从13c-甘氨酸到嘌呤中间体的通量时,获得了类似的结果(图3cii)。同样地,dyrk3损失显著增强了14c-甘氨酸通量进入hepg2细胞中的dna和rna(图3ciii)。值得注意的是,dyrk3的敲低显著上调了嘌呤合成途径所需的关键限速酶(图3d)。因此,本实施例得出结论,dyrk3敲低显著促进hcc细胞中的从头嘌呤合成途径。
重要的是,dyrk3的过表达显著抑制了hcc细胞的增殖和侵袭,而在培养基中补充嘌呤或在hcc细胞中过量表达ppat显著挽救了dyrk3过表达诱导的增殖和侵袭缺陷(图3e-f),表明dyrk3缺失通过激活嘌呤合成途径促进hcc细胞增殖和侵袭。
dyrk3缺失通过激活atf4促进嘌呤合成和hcc发展
为了阐明dyrk3损失驱动的嘌呤合成在hcc中的潜在机制,筛选了几个与嘌呤代谢有关的众所周知的转录因子:小眼相关转录因子(mitf),激活转录因子4(atf4)和c-myc。在三种转录因子中,仅耗尽atf4强烈地降低了dyrk3缺陷型hcc细胞中嘌呤合成的增加(图4ai,aii,aiii和图3b)。相反,沉默c-myc或mitf未显示显著效果(图4bi,bii,biii)。此外,atf4的消耗显著逆转了dyrk3缺陷型hcc细胞中参与嘌呤合成途径的几种限速基因的mrna水平的增加(图4c)。有趣的是,dyrk3敲低不影响atf4mrna丰度,但促进atf4核转位和polii与ppat启动子结合(图4di,dii和diii),表明dyrk3的缺失显著增强atf4转录活性。
与atf4在从头嘌呤合成中的作用一致,atf4耗竭阻断了dyrk3损失诱导的hcc细胞增殖和侵袭(图4e和f)。因此,atf4是hcc中dyrk3下游的重要效应物,表明dyrk3缺失通过其对atf4转录活性的调节至少部分地促进了从头嘌呤合成和hcc发展。
dyrk3在s1330直接磷酸化ncoa3
为了研究dyrk3如何激活atf4,对来自hek293t的细胞裂解物的flag标记的dyrk3进行了lc-ms/ms分析。有趣的是,ncoa3,一种众所周知的atf4共激活因子,被鉴定为dyrk3相互作用蛋白。
免疫共沉淀测定揭示了异位表达的ncoa3和dyrk3之间的相互作用(图5a)。类似地,内源性ncoa3也与内源性dyrk3相关(图5b),表明dyrk3和ncoa3之间的结合不是dna转染的人工产物。if分析进一步证实flag-ncoa3与ha-dyrk3共定位,主要在hek293细胞的细胞质中(图5c)。有趣的是,先前显示激活ncoa3转录活性的egf刺激破坏了它们的共定位(图5c)。总之,这些数据表明ncoa3直接与哺乳动物细胞中的dyrk3相互作用。
鉴于dyrk3是激酶并且存在两个在ncoa3中保守的最佳dyrk3识别基序(图5d),检查了dyrk3是否直接磷酸化ncoa3并调节其活性。当加入体外磷酸化反应时,发现野生型(wt)-dyrk3非催化失活的dyrk3-k218m磷酸化重组ncoa3(图5e)。然而,当ncoa3-s1330a不是t1166a突变体与wt-dyrk3一起温育时,未观察到磷酸化(图5e)。与这些作用一致,wt-dyrk3在hcc细胞中的过表达显著增加了磷酸化ncoa3的量(图5f)。但添加星形孢菌素(sts),即ser/thr激酶抑制剂,完全消除了dyrk3在s1330处的ncoa3磷酸化(图5f)。因此,ncoa3是dyrk3的新磷酸化底物。
dyrk3在ser-1330(s1330)处的ncoa3磷酸化显著抑制atf4诱导的嘌呤代谢酶和hcc进展的表达
为了阐明dyrk3在s1330中ncoa3磷酸化的功能意义,首先评估了这种修饰对ncoa3内在活性的影响。为此,在内源性ncoa3耗尽的sk-hep1细胞中转染wt-pbind-ncoa3,磷酸化缺陷型pbind-ncoa3-s1330a突变体或磷酸化模拟pbind-ncoa3-s1330e突变体。用葡萄糖或egf处理这些细胞,之前已显示其激活ncoa3的转录活性。如图6a所示,与wt-ncoa3相比,pbind-ncoa3-s1330a突变体的活性进一步增加,而ncoa3-s1330e突变体对葡萄糖或egf诱导的信号传导显著无效,表明dyrk3依赖性ncoa3。磷酸化显著抑制ncoa3驱动的转录反应。
由于ncoa3是已建立的atf4相互作用因子,本实施例研究了dyrk3在s1330处的ncoa3磷酸化是否改变了它们的结合亲和力。值得注意的是,在存在25mm葡萄糖或响应egf信号传导的情况下,与wt-ncoa3相比,atf4与pncoa3-s1330a的相互作用强烈增加,而ncoa3-s1330e在ncoa3敲低的hcc细胞中的重新表达显著降低它与atf4的关联(图6bi和bii)。
由于ncoa3是已建立的atf4共激活因子,本实施例进行chip和定量pcr测定以测量s1330处ncoa3磷酸化对atf4和ncoa3在上文定义的关键嘌呤合成靶基因的启动子上的染色质占据的影响。如所预期的,s1330e突变体的重新表达显著降低了这些关键代谢基因的启动子上的atf4-ncoa3复合物占据率,而ncoa3-s1330a具有相反的效果(图6c)。毫不奇怪,s1330处的ncoa3磷酸化显著抑制嘌呤合成途径中关键代谢酶的转录(图6d)。
为了研究dyrk3在s1330处的ncoa3磷酸化是否会影响体内hcc的生长,本实施例在sk-hep1细胞中稳定表达shrna抗性wt,ncoa3-s1330a或s1330e突变体,同时敲除内源性ncoa3蛋白,并将这些细胞植入裸鼠。观察到外源性wt-ncoa3或磷酸化缺陷型s1330a构建体在ncoa3耗尽细胞中的拯救表达完全恢复了肝肿瘤的生长,而s1330e突变体在移植肿瘤细胞后显著抵抗肿瘤发生(图6e)。在体外实验中也观察到类似的效果(图6f)。
总之,这些数据表明s1330处ncoa3的dyrk3依赖性磷酸化破坏了其与atf4的相互作用,从而抑制了嘌呤合成途径和hcc进展中关键代谢酶的转录。
atf4与dyrk3启动子结合并抑制hcc中的dyrk3转录
由于atf4是转录因子,并且来自dyrk3启动子序列的分析揭示了从位置+41到+49的一个经典atf4结合位点(图7a),本实施例假设atf4可能调节dyrk3启动子的转录活性。有趣的是,增加量的atf4质粒与dyrk3启动子-荧光素酶报告基因的共转染诱导荧光素酶活性的剂量依赖性下调(图7b)。为了确定+41至+49区域内的atf4结合位点是否对于该调节是必需的,本实施例在dyrk3启动子中突变该推定的atf4结合基序。本实施例观察到,与wt-dyrk3荧光素酶报告基因相比,atf4质粒与dyrk3-突变体荧光素酶报告基因的共转染显示出近3倍的荧光素酶活性(图7b),表明atf4抑制dyrk3的启动子的转录活性。
chip数据进一步证实内源性atf4在体内与dyrk3启动子的该区域结合(图7c)。一致地,atf4水平通过与smmc7721和hepg2细胞中的dyrk3启动子结合而以剂量依赖性方式显著抑制dyrk3蛋白水平(图7d)(图7e)。此外,atf4质粒的转染抑制了pdyrk3近端启动子的活性,而atf4表达的敲低显著增加了启动子活性(图7f)。这些数据强烈支持本实施例的假设,即atf4在体内与dyrk3启动子的推定区域结合并抑制其转录,从而发挥负反馈环(图8d)。
hcc患者组织中dyrk3,ps1330-ncoa3,atf4和嘌呤代谢相关酶的临床相关性为了进一步验证本实施例在hcc细胞系和小鼠模型中的观察结果,使用ihc检测dyrk3,p-s1330-ncoa3,atf4和ppat的蛋白质水平,这是120个人hcc样品中的嘌呤代谢速率限制酶。ihc测定显示dyrk3和atf4或ppat表达之间显著的负相关(图8a)。然而,dyrk3与ps1330-ncoa3水平显著相关(图8a和补充表5)。pearson分析加强了上述发现(图8b)。kaplan-meier分析显示hcc中atf4高表达或dyrk3和ps1330-ncoa3低表达与差的os显著相关(图8c),其进一步由另一组独立的人hcc样品证实(数据未显示)。因此,这些蛋白质的表达水平是总体存活的独立预后因素。总之,本实施例的数据突出了dyrk3/ncoa3/atf4反馈回路在人类hcc临床侵袭性中的关键作用。
3、讨论
本实施例报道dyrk3在hcc组织和细胞中下调,与晚期tnm分期呈负相关。dyrk3过表达显著抑制hcc细胞增殖,侵袭和癌症干细胞活性,表明dyrk3在hcc中充当肿瘤抑制因子。本实施例确定了dyrk3作为hcc嘌呤代谢中重要的检查点调节剂的关键作用。dyrk3通过在s1330直接磷酸化转录共激活因子ncoa3来抑制嘌呤合成途径,其破坏ncoa3与atf4的相互作用并抑制atf4的活性。ncoa3-s1330e突变体重新引入或atf4敲低显著逆转dyrk3损失增强的嘌呤合成途径和hcc进展。有趣的是,dyrk3的启动子活性受到atf4的负调节,表明双负反馈环。重要的是,dyrk3的水平不仅与人类hcc标本中的atf4表达呈负相关,而且与hcc患者的总体存活率呈正相关。因此,本实施例认为dyrk3可能是这一肝癌病例的有希望的靶点。
本实施例提供体外和体内证据表明dyrk3是hcc中的肿瘤抑制因子。支持本实施例的研究结果,显示dyrk3的下调对人脑胶质瘤的预后不良(4);反复突变而非wt-dyrk3也参与早期胃癌的发生。然而,在高侵袭性肺癌细胞株和来自肾细胞癌患者的样品中观察到dyrk3的表达增加,暗示dyrk3可能是这些癌症中的致癌基因。支持这种可能性,wippichf等最近观察到dyrk3作用于pras40,一种mtorc1抑制蛋白(1)。他们发现dyrk3磷酸化pras40,释放mtorc1并促进其活性(1)。总之,这些数据表明dyrk3可能会根据细胞环境或特定的突变效应促进或阻碍肿瘤的发展。显然,定义dyrk3的抗肿瘤作用或促肿瘤发生作用的生物决定因素的知识是一个仍然存在的挑战。因此,需要进一步的工作,包括人源肿瘤样品和动物模型,以揭示dyrk3在癌症中的细胞类型特异性和复杂活性。
本实施例的项目也报道了另一个有趣的发现。本实施例发现dyrk3缺失不会改变atf4的mrna和蛋白水平,但通过增强atf4与其共激活因子ncoa3的结合来激活atf4。值得注意的是,这种活化与其响应细胞应激的真核起始因子2α(eif2α)磷酸化下游的规范诱导无关(数据未显示)。这使人想起mtorc1,它也激活atf4而不依赖于其建立的eif2α调节机制(8)。然而,与本实施例的发现不同,mtorc1上调了atf4蛋白水平(8)。有趣的是,已经确定mtorc1和pfkfb4响应生长信号或高葡萄糖通过atf4促进嘌呤合成(8,10)。相比之下,本实施例观察到dyrk3下调嘌呤代谢。因此,dyrk3信号传导可以作为分子制动器通过抑制atf4活性来限制嘌呤合成途径过度激活。
作为细胞生长和生物体发育的主要调节因子,ncoa3(也称为src-3和aib1)的关键作用已在许多癌症中广泛描述(10)。因此,ncoa3是前瞻性癌症治疗的理想靶标。然而,ncoa3位于许多细胞内信号传导途径的关系中,并且在多种重要的生理作用中起着关键的“平台”共激活因子的作用(15)。因此,直接抑制ncoa3本身以抑制肿瘤发展将不是一个好的策略。鉴于失调的激酶信号过度激活ncoa3是许多肿瘤的标志(10),尤其是ncoa3的磷酸化通常会改变其转录活性,蛋白质稳定性和亚细胞定位(16),因此,针对ncoa3的翻译后修饰可能是一种可行的方法。降低肿瘤的风险。支持这一观点的先前研究报道,c-abl,非典型蛋白激酶c(apkc)和pfkfb4是已知在y1357激活ncoa3的三种蛋白激酶,1031-1130区域的8个ser/thr残基和ser857(10,16,17)。
与c-abl,apkc和pfkfb4不同,本实施例通过s1330的直接磷酸化将dyrk3鉴定为癌症中ncoa3活性的负调节剂。本实施例推测该位点的磷酸化可能会通过降低该区域的灵活性来增加ncoa3的结构稳定性。因此,磷酸化的ncoa3对于atf4是不可接近的,这导致嘌呤代谢和hcc进展的抑制。当然,这个假设应该通过更复杂的ncoa3生化或/和结构分析来验证。虽然p38和gsk3的ncoa3磷酸化增加了其降解(16),但本实施例不能排除除dyrk3之外的其他未识别激酶也可能负调节ncoa3活性的可能性。本实施例的主要数据暗示grk5可能是调节ncoa3活性的新型负激酶。进一步的研究无疑将丰富人们对ncoa3在癌症中的调控机制的认识。
atf4通常在癌症(包括hcc)的发病机制中失调,并且代表癌细胞依赖性的丰富来源。然而,与ncoa3类似,作为必需的tf,atf4也在生理条件中起重要作用(8)。因此,应该注意如何针对这个tf。此外,tf的dna结合结构域和激活区域通常是药物开发的挑战性靶标。这些障碍促使人们普遍关注通过间接手段瞄准tf,例如瞄准其核心指标。本实施例的研究通过将dyrk3或ncoa3磷酸化作为抑制促进hcc发展的atf4的策略来指出更广泛的潜力。本实施例预计,为了揭示atf4与hcc细胞中激酶依赖性之间的相关性而扩大的努力将扩大这一概念,以扩展到其他无法治愈的恶性肿瘤。
该研究表明靶向dyrk3/ncoa3/atf4信号轴可能在hcc中具有治疗意义。由于ncoa3和atf4在其他恶性肿瘤中均失调,因此dyrk3信号传导在其他癌症环境中也很重要。尽管本实施例目前在该模型系统中的研究结果不能简单地推断到其他癌症背景而没有经过仔细验证,但本实施例的结果确实提供了进一步优化和评估强效和选择性小分子的基本原理,这些小分子将dyrk3活性作为临床前hcc模型中的治疗剂。
总之,该研究支持以下模型:在基础生理条件下,dyrk3在s1330处直接磷酸化ncoa3,其减少ncoa3与atf4的结合并引起atf4转录活性的抑制。因此,嘌呤合成途径被阻断,这抑制了hcc的发展。在生长信号或高葡萄糖刺激下,dyrk3水平被atf4或其他因子下调。因此,未磷酸化的ncoa3结合并激活atf4,其显著促进嘌呤合成途径和hcc进展。因此,靶向dyrk3/ncoa3/atf4反馈环可以为hcc患者的诊断和治疗提供有用的策略。
以上所述,仅为本发明的较佳实施例而已,故不能依此限定本发明实施的范围,即依本发明专利范围及说明书内容所作的等效变化与修饰,皆应仍属本发明涵盖的范围内。
- 还没有人留言评论。精彩留言会获得点赞!