一种糖修饰的叶酸衍生物制备的纳米运载颗粒及其应用的制作方法
本发明属于生物医药领域,特别涉及到一种糖修饰的叶酸衍生物制备的纳米运载颗粒及其应用。
背景技术:
叶酸受体是人体内某些肿瘤细胞表面高表达的受体,以叶酸受体为靶点进行的抗肿瘤治疗药物开发已持续多年,但至今仍未有成熟的治疗药物推向市场。现有的叶酸相关的抗肿瘤药物开发,大多是以叶酸修饰大分子peg(聚乙二醇),以此做为载体,或者做为多种材料混合载体的一部分,运载抗肿瘤药物。这种治疗策略虽然可以实现靶向叶酸受体的功能,但存在着多种不足。首先,这些载体中叶酸修饰的多是聚合物大分子,造成在分子中叶酸的含量比例较低,靶向效率低下;其次,这些大分子聚合物形成的纳米颗粒粒径不均一,运载抗肿瘤药物的效率不高;再次,这些大分子聚合物聚集之后形成的颗粒容易刺激免疫系统,造成免疫原性反应,影响治疗的效果。
综上,改善叶酸相关的抗肿瘤药物治疗思路,开发新的靶向效率高,粒径均一,运载效果好,且不易引起免疫原性反应的新的靶向叶酸受体的纳米颗运载粒,是叶酸相关的抗肿瘤治疗药物研发新方向。
技术实现要素:
针对上述问题,本发明的目的是提供一种糖修饰的叶酸衍生物制备的纳米运载颗粒,及其作为靶向抗肿瘤药物纳米运载颗粒的应用。在研究两亲性小分子纳米药物的基础上,应用与叶酸亲水性差异较大的糖类小分子,修饰叶酸,制备内部亲水性差异较大的分子,在溶液中能自组装成纳米颗粒,通过与抗肿瘤药物以配位键、氢键等非共价化学键的方式,将抗肿瘤药物包裹在纳米颗粒内部,靶向输送至肿瘤部位之后释放,发挥较好的抗肿瘤效果。
本发明的目的是通过以下技术方案实现的:
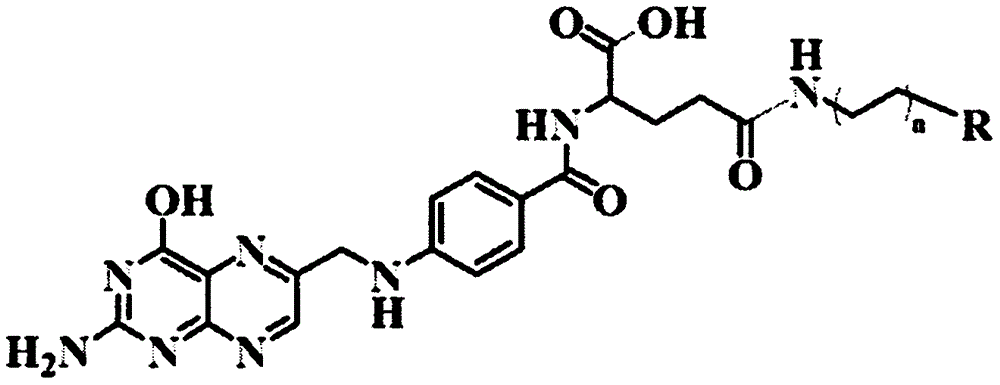
一种糖修饰的叶酸衍生物制备的纳米运载颗粒,由糖与叶酸通过碳链作为连接桥,利用化学键的方式结合形成,且糖与叶酸之间修饰的碳链长度可以为2至18个碳原子之间的任意数量。所述纳米运载颗粒的化学结构通式如图1所示。
进一步的,所述糖为还原性糖。
进一步的,所述还原性糖包括五碳糖和六碳糖,脱氧葡萄糖、葡萄糖,氨基葡萄糖,果糖、蔗糖和乳糖。
进一步的,所述化学键包括糖苷键和酯键。
所述纳米运载颗粒的合成方法,包括以下步骤:
1)制备乙酰基保护的糖分子
将干燥脱水后的糖加入无水吡啶溶液中,搅拌溶解后,冰浴下将乙酸酐缓慢滴加入溶液中,滴加完毕后撤掉冰浴,室温下搅拌24小时,然后旋蒸除去溶剂,加入乙酸乙酯溶解剩余固体,然后用柠檬酸水溶液,碳酸氢钠水溶液,饱和氯化钠水溶液各萃洗三次,之后乙酸乙酯用无水硫酸钠干燥过夜,过滤之后减压除去乙酸乙酯,用柱层析纯化,得到乙酰基保护的糖。简称化合物2;
2)生成糖分子和连接臂相连接的糖苷键
将化合物2溶解于二氯甲烷中,加入一端是溴原子的脂肪醇,冰浴下再滴加入三氟化硼乙醚,滴加完毕后撤除冰浴,室温下反应24小时,之后加入冷却过的饱和碳酸氢钠水溶液终止反应,分离出二氯甲烷层,并用二氯甲烷萃洗水相两次,合并二氯甲烷并用饱和氯化钠水溶液萃洗三次之后加入无水硫酸钠干燥过夜,过滤之后减压除去二氯甲烷,剩余油状物用硅胶柱层析纯化,洗脱液为石油醚∶乙酸乙酯,从10∶1洗脱至5∶1,得到黄色油状物,即脂肪醇和糖形成糖苷键的化合物,简称化合物3;
3)连接臂上的溴原子被叠氮基取代
将化合物3和叠氮化钠溶解于二甲基甲酰胺中,加热条件下搅拌,反应8小时后加入冰块终止反应,然后用乙酸乙酯萃取水相两次,合并有机相,并用无水硫酸钠干燥过夜,过滤之后减压除去乙酸乙酯,得到黄色油状物,即叠氮基取代溴原子的化合物,简称化合物4;
4)制备叠氮基还原的化合物
将化合物4加入乙醇中,搅拌溶解后加入林德拉催化剂和对甲苯磺酸,然后在氢气反应氛围下,室温搅拌8小时,之后过滤除去还原剂,滤液减压除去乙醇之后得到无色泡状固体,即叠氮基还原为氨基的化合物,简称化合物5;
5)制备连接臂和叶酸生成酰胺键的化合物
叶酸溶解于无水二甲基亚砜中,加入dcc,再加入化合物5,最后加入无水吡啶,避光室温反应24小时,然后减压尽可能取出多余的吡啶,残留物用甲基叔丁基醚磨洗之后得到化合物6,经柱层析后得到橙红色固体,糖被乙酰基保护的目标物,简称化合物6;
6)制备终产物
将化合物6溶解于甲醇中,然后在冰浴下滴加入甲醇钠溶液,滴加完毕后撤掉冰浴,室温下反应,然后用酸性树脂调节溶液的ph值至6,然后过滤除掉树脂,滤液减压除去之后得到粗品,用制备级高效液相色谱纯化,得到所述的纳米运载颗粒。
进一步的,步骤2)中所述的脂肪醇中脂肪链的长度为2-18个碳原子。
所述纳米运载颗粒的应用,纳米运载颗粒用于装载抗肿瘤药物,且所述抗肿瘤药物的分子量在20~2000道尔顿。
进一步的,所述纳米运载颗粒通过化学键、配位键或氢键与所述抗肿瘤药物相互作用而形成。
进一步的,所述抗肿瘤药物包括顺铂、阿糖胞苷、5-氟尿嘧啶、阿霉素和紫杉醇;所述肿瘤包括为叶酸受体高表达肿瘤,包括乳腺癌、卵巢癌和宫颈癌。本发明相比现有技术的有益效果为:
1、本发明所述的纳米运载颗粒,在最大分子量的乳糖分子修饰,连接臂碳链长度为18个碳原子时,分子量的大小不超过2000da,相比现有的大分子叶酸衍生物,在体内发挥作用时不易产生免疫原性,比较容易代谢排出体外,毒性较低;
2、本发明所述的纳米运载颗粒,合成方法简单,经过六步反应即可得到最终的产物,所有的化合物均经过红外、质谱和核磁共振氢谱及碳谱的确证,证实了最终合成的化合物结构正确;
3、本发明合成得到的纳米运载颗粒,在水中的溶解度比叶酸高600-2000倍,并且分子内部不同的部分存在水溶性的差异,分子之间存在着氢键相互作用,在水溶液中能够自组装成纳米颗粒,以葡萄糖修饰,连接臂碳链长度为2的化合物为例,浓度为1μmol/l的化合物的水溶液中测到的粒径为53.2nm,随着浓度的增加,粒径会逐渐变大,并且在模拟体内环境的0.5%bsa水溶液中,此化合物的自组装依然稳定存在;另外,原子力显微镜,透射电子显微镜和扫描电子显微镜这些形态学表征技术也都证实修饰和的叶酸衍生物能够自组装形成纳米颗粒;
4、本发明合成得到的纳米运载颗粒,能够通过配位键或氢键与肿瘤药物相互作用,形成具有靶向抗肿瘤功能的纳米颗粒,载药能力强、效果好;
5、本发明合成得到的纳米运载颗粒,由于分子内部碳链长度的差异,得到了一系列的化合物,这些化合物可以分别运载分子量大小不同的抗肿瘤药物,这在之前的研究中从未出现。
附图说明
图1为本发明所述糖修饰的叶酸衍生物制备的纳米运载颗粒的化学结构式;其中,n=1-9,r为脱氧葡萄糖、葡萄糖、氨基葡萄糖、果糖、乳糖、蔗糖、五碳糖或六碳糖;
图2为实施例1所述纳米运载颗粒的化学结构式;
图3为实施例1所述纳米运载颗粒的合成路线示意图;其中,(i)醋酸酐,无水吡啶;(ii),溴乙醇,二氯甲烷;(iii)叠氮化钠,二甲基甲酰胺;(iv)对甲苯磺酸,氢气,乙醇;(v)叶酸,二甲基亚砜;(vi)甲醇钠,甲醇;
图4为不同稀释倍数的实施例1所述纳米运载颗粒和叶酸饱和溶液的紫外吸收光谱图;其中,a:fa-2-dg饱和溶液稀释2000倍;b:叶酸饱和溶液稀释2倍;c:fa-2-dg饱和溶液稀释4000倍;d:叶酸饱和溶液稀释5倍;e:叶酸饱和溶液稀释10倍;
图5为实施例1所述纳米运载颗粒在水溶液中的临界胶束浓度测试结果图;其中,a:以280nm处的紫外吸光度对浓度的对数值作图;b:以280nm激发波长,442nm处的荧光发射强度对浓度的对数值作图;
图6为实施例2所述纳米运载颗粒的合成路线示意图;其中,(i)醋酸酐,无水吡啶;(ii),溴乙醇,二氯甲烷;(iii)叠氮化钠,二甲基甲酰胺;(iv)对甲苯磺酸,氢气,乙醇;(v)叶酸,二甲基亚砜;(vi)甲醇钠,甲醇;
图7为实施例2所述纳米运载颗粒的饱和溶液的稀释溶液、叶酸饱和溶液的稀释溶液的紫外吸收光谱测试结果图;
图8为所述含顺铂的靶向抗肿瘤纳米颗粒(fa-2-dg-pt)的化学结构式;
图9为所述含顺铂的靶向抗肿瘤纳米颗粒(fa-2-dg-pt)的高分辨质谱图。
具体实施方式
为了进一步阐述本发明,下面给出一系列实施例。这些实施例完全是例证性的,它们仅用来对本发明进行具体描述,不应当理解为对本发明的限制。
实施例1
本实施例提供了一种纳米运载颗粒fa-2-dg,fa-2-dg由脱氧葡萄糖和叶酸经由氨基乙醇连接形成,其中叶酸的羧基和氨基乙醇的氨基相连,形成酰胺键,氨基乙醇的羟基和脱氧葡萄糖的羟基相连,形成糖苷键。所述fa-2-dg的化学结构式如图2所示。
脱氧葡萄糖有多个羟基,在反应过程中,羟基的反应活性较高,因此需要用乙酰基保护,再进行反应。乙酰基保护的反应由脱氧葡萄糖和乙酸酐在无水的吡啶中发生。氨基乙醇的氨基在反应过程中也会有比较高的反应活性,因此在反应过程中不直接用氨基乙醇,而是选用2-溴乙醇先和乙酰脱氧葡萄糖反应,这一步的反应需要三氟化硼的催化,生成糖苷键。产物和叠氮化钠在dmf中反应之后,溴原子继续被叠氮基团取代,然后叠氮基团被对甲苯磺酸和林德尔催化剂催化还原,生成氨基苯磺酸盐,继续和叶酸反应生成酰胺键,完成糖基保护的目标物的合成,脱除糖基上的乙酰基保护,完成fa-2-dg的合成。
如图3所示,所述fa-2-dg的合成方法具体包括以下步骤:
(1)制备(4r,5s,6r)-6-(乙酰氧甲基)-四氢-2,4,5-三乙酰氧基-吡喃(化合物2)
将5.0g(30.5mmol)的2-脱氧葡萄糖溶解于25ml无水吡啶中,冰浴搅拌条件下将23.8g(233mmol)的乙酸酐滴加入溶液中,滴加完毕后撤掉冰浴,室温下搅拌16h,然后旋蒸除掉溶剂,加入100ml乙酸乙酯溶解剩余固体,然后用5%柠檬酸水溶液,5%碳酸氢钠水溶液,饱和氯化钠水溶液各萃洗三次,之后乙酸乙酯用无水硫酸钠干燥过夜。过滤之后减压除去乙酸乙酯,用柱层析法纯化,得到的产物乙酰基保护羟基的脱氧葡萄糖9.1g(27.39mmol),产率为89.9%。1h-nmr(400mhz,cdcl3):δ/ppm=5.77(dd,j=10.0,2.20hz,1h),5.08-4.97(m,2h),4.31-4.26(m,1h),4.01-4.03(m,1h),3.74-3.70(m,1h),2.35-2.29m,1h),2.09-1.90(m,12h).13c-nmr(100mhz,cdcl3):δ/ppm=170.71,170.10,169.76,168.79,91.08,72.87,70.15,68.26,61.95,34.73,20.96,20.86,20.77,20.71.hrms(m/z):calcdforc14h20o9[m+na]+:355.0999,found:355.1008。
(2)制备(2r,3s,4r)-2-(乙酰氧甲基)-6-(2-溴乙基氧基)四氢-3,4-二乙酰氧基-吡喃(化合物3)
将化合物2(5.0g,15mmol)溶解于二氯甲烷中,加入2-溴乙醇(2.3g,18mmol,1.3ml),冰浴下再滴加入三氟化硼乙醚(2.6g,18mmol,2.2ml),滴加完毕后撤除冰浴,室温下反应2h,然后加入冷却过的饱和碳酸氢钠水溶液(60ml)终止反应,分离出二氯甲烷层,并用50ml二氯甲烷萃取水相两次,合并二氯甲烷,用饱和氯化钠水溶液40ml萃洗三次之后用无水硫酸钠干燥过夜。过滤之后减压除去二氯甲烷,剩余油状物用硅胶柱层析纯化,洗脱液为石油醚∶乙酸乙酯,从10∶1洗脱至5∶1,得到化合物3(4.6g,10.4mmol,69.3%产率)为黄色油状物。1h-nmr(400mhz,cdcl3):δ/ppm=5.33-5.26(m,1h),5.01-4.95(m,2h),4.28-4.24(m,1h),4.08-4.04(m,2h),3.94-3.88(m,1h),3.82-3.76(m,1h),3.48(t,j=0.8hz,2h),2.29-2.25(m,1h),2.11-2.98(m,9h),1.85-1.76(m,1h).13c-nmr(100mhz,cdcl3):δ/ppm=170.71,170.18,169.93,97.25,69.24,68.91,68.31,67.97,62.33,34.88,30.09,20.98,20.78,20.75.hrms(m/z):calcdforc14h21bro8[m+na]+:419.0312,found:419.0324。
(3)制备(2r,3s,4r)-2-(乙酰氧甲基)-6-(2-叠氮乙氧基)四氢-3,4-二乙酰氧基-吡喃(化合物4)
将化合物3(4.5g,11.3mmol)和叠氮化钠(1.11g,16.9mmol)溶解于35ml二甲基甲酰胺中,60℃加热条件下搅拌,反应16h后加入冰水300ml终止反应,然后用乙酸乙酯(100ml)萃取两次,合并有机相,用饱和氯化钠水溶液40ml萃洗三次之后用无水硫酸钠干燥过夜。过滤之后减压除去乙酸乙酯,得到化合物4(3.9g,粗品)为黄色油状物。1h-nmr(400mhz,cdcl3):δ/ppm=5.23-5.21(m,1h),4.92(brs2h),4.23-4.19(m,1h),3.99-3.92(m,2h),3.77-3.73(m,1h),3.60-3.51(m,1h),3.36-3.35(m,2h),2.23-2.19(m,1h),2.01-2.91(m,9h),1.79-1.72(m,1h).13c-nmr(100mhz,cdcl3):δ/ppm=170.71,170.12,169.93,97.27,69.22,68.83,68.18,66.55,62.35,50.44,34.82,20.96,20.77,20.74.hrms(m/z):calcdforc14h21n3o8[m+na]+:382.1221,found:382.1211。
(4)制备(2r,3s,4r)-2-(乙酰氧甲基)-6-(2-氨基乙氧基)四氢-3,4-二乙酰氧基-吡喃(化合物5)
林德拉催化剂(0.6g,0.3mmol,10.0%纯度)和对甲基苯磺酸(0.19g,1.1mmol)先后加入到溶解有化合物4(3.0g,9.2mmol)的乙醇溶液中,然后在氢气(15psi)氛围下,室温反应16h。之后过滤,减压除去乙醇之后得到化合物5(2.82g,91.9%),为无色泡状固体。1h-nmr(400mhz,cdcl3):δ/ppm=7.71(d,j=8.0hz,1h),7.11(d,j=8.0hz,1h),6.38(brs,2h),5.28-5.19(m,1h),4.95-4.86(m,2h),4.22(m,1h),3.96-3.89(m,1h),3.77-3.66(m,2h),3.53-3.44(m,1h),3.12-3.01(m,2h),2.34(s,3h),2.21(m,1h),2.10-1.92(m,9h),1.69-1.62(m,1h).13c-nmr(100mhz,cdcl3):δ/ppm=170.77,170.29,169.81,97.39,69.07,68.83,68.18,66.55,62.35,50.44,34.82,20.96,20.77,20.74.hrms(m/z):calcdforc14h24no8[m+h]+:334.1496,found:334.1506。
(5)制备n2-{4-[(2-氨基-4-羟基蝶啶-6-甲基)氨基]苯甲酰基}-n5-{2-[(2s,4r,5s,6r)-4,5-二乙酰氧基-6-(乙酰氧基甲基)四氢吡喃]-氧乙基}-l-谷氨酰胺(化合物6)
叶酸(1.25g,2.88mmol)溶解于100ml无水二甲基亚砜中,然后加入dcc(1.5g,7.2mmol),再加入化合物5(1.75g,3.5mmol),最后加入吡啶(20g,0.25mol,21ml),避光室温反应16h,然后减压尽可能除去多余的吡啶,残留物用甲基叔丁基醚磨洗之后得到化合物6(2.4g,粗品),经柱层析纯化后得到橙色固体(1.8g,82.6%)。1h-nmr(400mhz,dmso):δ/ppm=11.44(s,1h),8.64(s,1h),7.95(m,1h),7.66(d,j=8hz,1h),7.19(s,1h),7.13(s,1h),7.07(s,1h),6.93(s,1h),6.63(t,j=8hz,1h),5.14(m,1h),4.93(s,1h),4.82(m,1h),4.48(s,1h),4.35(m,1h),4.16(m,1h),3.97-3.91(m,2h),3.56(m,1h),3.42(m,1h),3.28(m,1h),2.54(s,1h),2.26(m,1h),2.08(m,1h),2.01(s,6h),1.98(s,3h).13c-nmr(100mhz,dmso):δ/ppm=174.52,172.38,170.58,170.15,169.87,166.68,158.13,151.23,148.99,129.51,128.41,121.82,118.56,117.06,112.86,111.59,96.66,69.43,68.98,67.76,66.27,63.89,62.48,53.23,46.38,42.58,38.93,34.82,30.98,27.45,21.12,20.97,20.91.hrms(m/z):calcdforc33h40n8o13[m+h]+:757.2788,found:757.2799。
(6)制备n2-{4-[(2-氨基-4-羟基蝶啶-6-甲基)氨基]苯甲酰基}-n5-{2-[(2s,4r,5s,6r)-4,5-二羟基-6-(羟甲基)四氢吡喃]-氧乙基}-l-谷氨酰胺(目标物)
将化合物6(1.5g,2mmol)溶解于甲醇中,然后在冰浴下滴加入甲醇钠naome(3.13g,14.3mmol,25.0%purityofmeoh),滴加完毕后撤掉冰浴,室温下反应1h。然后用酸性树脂调节ph值至6,然后过滤除去树脂。滤液减压除去之后得到粗品。粗品用制备级hplc纯化,(色谱柱:ageladurashell(博纳艾吉尔,天津,中国)10μm,250×50mm;流动相:[water(10mmnh4hco3)-acn];b%:0-15.0%,20min),之后得到终产物fa-2-dg(0.94g,75%),为橙色固体。1h-nmr(400mhz,dmso):δ/ppm=8.97(s,1h),8.58(d,j=20hz,2h),7.95(d,j=10.6hz,1h),7.85(s,1h),7.68-7.46(m,4h),6.90(s,1h),6.78(s,1h),6.61(dd,j=19.0,8.0hz,2h),4.78(s,1h),4.46(s,1h),4.34(s,1h),4.20(s,1h),4.06(s,1h),3.64-3.21(m,11h),3.02-2.98(m,1h),2.19-2.11(m,2h),1.98(s,1h),1.39(t,j=12.4hz,1h).13c-nmr(100mhz,dmso):δ/ppm=172.81,166.64,162.19,156.88,155.29,151.01,148.83,129.48,128.91,128.41,121.99,111.81,97.19,73.62,72.21,68.38,65.73,65.52,61.47,54.43,46.43,38.91,38.31,33.01,32.69,29.08,27.73.hrms(m/z):calcdforc27h34n8o10[m+h]+:631.2471,found:631.2473。
(一)fa-2-dg在水中溶解度的测定:
制备fa-2-dg的饱和水溶液,然后按10,100,1000,2000,4000和8000的倍数逐级稀释,得到一系列不同浓度的溶液,待测。同时制备叶酸的饱和水溶液,按照2,5,10,20和50的倍数稀释,得到一系列不同浓度的溶液,待测。
用紫外分光光度计(uv-2600,岛津)对以上制得的样本进行测试,波长扫描范围185-500nm,扫描速度:中速,扫描间隔:1nm,为保证测试的准确性,所有样品的测试温度均为37℃,用仪器自带的uv-probe软件记录并分析数据。
fa-2-dg饱和溶液的稀释溶液和叶酸饱和溶液的稀释溶液的紫外吸收光谱测试见图4所示。图中的b、d和e谱线代表的是叶酸饱和溶液稀释2倍,5倍和10倍的紫外吸收光谱,图中的a和c代表的是fa-2-dg稀释2000倍和4000倍的紫外吸收光谱,根据紫外吸收光谱的测试结果,fa-2-dg饱和溶液稀释2000倍的溶液和叶酸饱和溶液稀释2倍的溶液谱线有部分重合,如图中的a和b所示,在最大吸收波长280nm处,fa-2-dg饱和溶液稀释2000倍的溶液吸光度稍高于叶酸饱和溶液稀释2倍的溶液。fa-2-dg饱和溶液稀释4000倍的溶液和叶酸饱和溶液稀释5倍的溶液谱线在280nm处的最大吸收波长处重合,从这个结果来看,在结构中引入脱氧葡萄糖使得叶酸在水中的溶解度提升了800倍。
(二)fa-2-dg临界胶束浓度的测定
制备fa-2-dg的水溶液,浓度为19nmol/l,47.5nmol/l,95nmol/l,190nmol/l,475nmol/l,950nmol/l,1.9μmol/l,4.75μmol/l,9.5μmol/l,19μmol/l,47.5μmol/l,63μmol/l。在紫外最大吸收波长280nm处测试各个浓度溶液的吸光度。测试温度37℃,用仪器自带的uv-probe软件记录吸光度数据。以溶液的吸光度对浓度的对数值作图,得到吸光度随浓度变化的趋势,从而找到临界胶束浓度。
在紫外测试的基础上,由于fa-2-dg有荧光性质,在280nm激发光照射下,在442nm处发射荧光,测试荧光发射的强度,以荧光发射强度对浓度的对数值作图,得到荧光强度随浓度变化的趋势,从而找到临界胶束浓度。
应用紫外吸收光谱和荧光发射光谱测定不同浓度下的样品的紫外吸光度和荧光发射强度,分别对样品浓度的对数值做图,得到如图5所示的结果。图中a为紫外吸收的测定结果,可以看到有明显的两条趋势线,线段的交叉点在x轴上的值为-5.2左右,换算成浓度约为6.31μmol/l,图中b为荧光发射光谱的测定结果,也有两条与紫外吸收光谱测得结果一致的趋势线,线段的交叉点在x轴上的值为-5.1左右,换算成浓度值约为7.94μmol/l,根据两种方法计算的临界胶束浓度,求算平均值,可得fa-2-dg在水溶液中的临界胶束浓度为7.12μmol/l。
由上述测定得到的结果可知,fa-2-dg在水溶液中的临界胶束浓度为7.12μmol/l,因此为评估fa-2-dg聚集之后的颗粒的纳米粒径分布,用动态光散射的方法测定fa-2-dg在水溶液中的粒径,同时为了评估浓度对组装的影响,特配制三个不同浓度的溶液,分别为8μmol/l,14μmol/l,和47μmol/l的三个溶液,测定它们的粒径大小,在8μmol/l的浓度条件下,fa-2-dg组装的颗粒粒径为72.5nm,当浓度为14μmol/l时,粒径增大到146.8nm,浓度为47μmol/l时,粒径进一步增大,达到269.1nm,说明化合物的聚集程度与浓度呈正相关,浓度越大,聚集的程度越严重,聚集的颗粒也越大。0.5%的bsa对fa-2-dg的组装并未产生影响,浓度为9.5μmol/l的fa-2-dg水溶液中的粒径为82.1nm,当把溶剂换成0.5%bsa水溶液时,粒径并没有发生明显的变化,为83.8nm。
(三)fa-2-dg的红外光谱测定
采用傅里叶变换红外光谱仪对fa-2-dg的红外吸收进行测定,同时分别测定叶酸和脱氧葡萄糖以及叶酸混合脱氧葡萄糖的红外光谱,判断结构中引入脱氧葡萄糖之后分子之间的相互作用。测试仪器型号为美国thermofisher公司生产的nicoletis5型傅里叶变换红外光谱仪进行测试,选择衰减全反射(attenuatedtotalreflection,atr)的模式测试。
(四)fa-2-dg纳米形态表征测定
分别利用原子力显微镜、透射电子显微镜和扫描电子显微镜测定fa-2-dg及fa-2-dg在溶液中的纳米粒度表征。
原子力显微镜的结果显示,fa-2-dg形成规则的纳米线状结构,在纳米线的末端,有粒径约为20nm的纳米颗粒形成。透射电子显微镜下fa-2-dg可以看到很多纳米线绕在一起成团状,这些纳米线的宽度约为5-20nm不等。fa-2-dg从扫描电子显微镜观察到成比较薄的层状结构,在边缘断裂处还有少许的卷曲,推测是由纳米线结合在一起形成的纳米薄膜结构,由于薄膜的韧性较差,可以看到薄膜断裂形成的小块膜状结构。
实施例2
本实施例提供了一种纳米运载颗粒——葡萄糖修饰的叶酸衍生物,所述叶酸衍生物由葡萄糖和叶酸经两个碳原子的连接臂氨基乙醇连接之后形成,是一种纳米运载颗粒。葡萄糖的1位羟基和氨基乙醇的羟基形成糖苷键,叶酸的羧基和氨基乙醇的氨基形成酰胺键,这样形成的结构在体外能保持很好的稳定性,在体内能被广泛存在的酰胺酶和糖苷酶水解,快速代谢排出体外,不易造成蓄积。
如图6所示,以葡萄糖和连接臂为2个碳原子的氨基乙醇为例,所述叶酸衍生物的合成包括以下步骤:
(1)制备6-(乙酰氧甲基)-四氢-2,3,4,5-四乙酰氧基-吡喃(化合物2)
将5.0g(27.8mmol)的葡萄糖溶解于25ml无水吡啶中,冰浴搅拌条件下将23.8g(233mmol)的乙酸酐滴加入溶液中,滴加完毕后撤掉冰浴,室温下搅拌16h,然后旋蒸除掉溶剂,加入100ml乙酸乙酯溶解剩余固体,然后用5%柠檬酸水溶液,5%碳酸氢钠水溶液,饱和氯化钠水溶液各萃洗三次,之后乙酸乙酯用无水硫酸钠干燥过夜。过滤之后减压除去乙酸乙酯,用柱层析法纯化,得到的产物为乙酰基保护羟基的葡萄糖10.1g(27.4mmol),产率为89.9%。1h-nmr(500mhz,cdcl3):δ/ppm=6.41(d,j=10hz,1h),5.32(m,1h),5.16(m,1h),4.78(m,1h),4.57(m,1h),4.27(dd,j=8.0hz,j=1.6hz,1h),3.95(dd,j=8.0hz,j=1.6hz,1h),1.9-2.3(m,15h);13c-nmr(125mhz,cdcl3):δ/ppm=170.41,170.20,169.82,169.53,168.58,91.32,71.02,70.35,69.73,68.31,58.64,21.16,20.97,20.73,20.54,20.51.hrms(m/z):calcdforc16h22o11[m+na]+:413.1167,found:413.1158。
(2)制备2-(乙酰氧甲基)-6-(2-溴乙基氧基)四氢-3,4,5-三乙酰氧基-吡喃(化合物3)
将化合物2(5.0g,12.8mmol)溶解于二氯甲烷中,加入2-溴乙醇(2.3g,18mmol,1.3ml),冰浴下再滴加入三氟化硼乙醚(2.6g,18mmol,2.2ml),滴加完毕后撤除冰浴,室温下反应2h,然后加入冷却过的饱和碳酸氢钠水溶液(60ml)终止反应,分离出二氯甲烷层,并用50ml二氯甲烷萃取水相两次,合并二氯甲烷,用饱和氯化钠水溶液40ml萃洗三次之后用无水硫酸钠干燥过夜。过滤之后减压除去二氯甲烷,剩余油状物用硅胶柱层析纯化,洗脱液为石油醚∶乙酸乙酯,从10∶1洗脱至5∶1,得到化合物3(5.1g,10.8mmol,84.9%产率)为黄色油状物。1h-nmr(500mhz,cdcl3):δ/ppm=5.62(d,j=10hz,1h),5.19(m,1h),5.02(m,1h),4.78(m,1h),4.61(m,1h),4.29(dd,j=8.0hz,j=1.6hz,1h),3.98(dd,j=8.0hz,j=1.6hz,1h),3.41(t,j=6.6hz,2h),1.91-2.32(m,12h),1.67(t,j=6.6hz,2h);13c-nmr(125mhz,cdcl3):δ/ppm=170.20,169.82,169.53,168.58,98.73,71.12,70.54,69.28,68.16,64.17,63.15,29.46,21.07,20.89,20.81,20.63.hrms(m/z):calcdforc16h23bro10[m+na]+:477.0469,found:477.0461。
(3)制备2-(乙酰氧甲基)-6-(2-叠氮乙氧基)四氢-3,4,5-三乙酰氧基-吡喃(化合物4)
将化合物3(5.0g,11.0mmol)和叠氮化钠(1.1g,16.9mmol)溶解于35ml二甲基甲酰胺中,60℃加热条件下搅拌,反应16h后加入冰水300ml终止反应,然后用乙酸乙酯(100ml)萃取两次,合并有机相,用饱和氯化钠水溶液40ml萃洗三次之后用无水硫酸钠干燥过夜。过滤之后减压除去乙酸乙酯,得到化合物4(3.9g,粗品)为黄色油状物。1h-nmr(500mhz,cdcl3):δ/ppm=5.58(d,j=10hz,1h),5.27(m,1h),4.96(m,1h),4.74(m,1h),4.66(m,1h),4.31(dd,j=8.0hz,j=1.6hz,1h),4.03(dd,j=8.0hz,j=1.6hz,1h),3.56(t,j=6.6hz,2h),1.91-2.32(m,12h),1.72(t,j=6.6hz,2h);13c-nmr(125mhz,cdcl3):δ/ppm=170.23,169.87,169.56,168.51,100.51,71.14,70.49,69.31,68.09,64.13,63.02,47.32,21.12,20.93,20.93,20.73.hrms(m/z):calcdforc16h23n310[m+na]+:440.1371,found:440.1376。
(4)制备2-(乙酰氧甲基)-6-(2-氨基乙氧基)四氢-3,4,5-三乙酰氧基-吡喃(化合物5)
林德拉催化剂(0.6g,0.3mmol,10.0%纯度)和对甲基苯磺酸(0.19g,1.1mmol)先后加入到溶解有化合物4(3.0g,7.2mmol)的乙醇溶液中,然后在氢气(15psi)氛围下,室温反应16h。之后过滤,减压除去乙醇之后得到化合物5(2.32g,82.4%),为无色泡状固体。1h-nmr(500mhz,cdcl3):δ/ppm=7.81(d,j=7.2hz,2h),7.61(br,3h),7.38(d,j=7.2hz,2h),5.72(d,j=10hz,1h),5.32(m,1h),4.97(m,1h),4.73(m,1h),4.61(m,1h),4.33(dd,j=8.0hz,j=1.6hz,1h),4.06(dd,j=8.0hz,j=1.6hz,1h),3.59(t,j=6.6hz,2h),2.71(t,j=6.6hz,2h),2.41(s,3h),1.91-2.32(m,12h);13c-nmr(125mhz,cdcl3):δ/ppm=170.25,169.81,169.67,168.52,147.61,143.25,129.31,126.53,100.51,71.17,70.53,69.35,68.02,66.31,62.71,43.09,25.01,21.15,20.98,20.93,20.71.hrms(m/z):calcdforc16h25n10[m+h]+:392.3691,found:392.3697。
(5)制备n2-{4-[(2-氨基-4-羟基蝶啶-6-甲基)氨基]苯甲酰基}-n5-{2-3,4,5-三乙酰氧基-6-(乙酰氧基甲基)四氢吡喃]-氧乙基}-l-谷氨酰胺(化合物6)
叶酸(1.25g,2.88mmol)溶解于100ml无水二甲基亚砜中,然后加入dcc(1.5g,7.2mmol),再加入化合物5(1.75g,4.5mmol),最后加入吡啶(20g,0.25mol,21ml),避光室温反应16h,然后减压尽可能除去多余的吡啶,残留物用甲基叔丁基醚磨洗之后得到化合物6(2.4g,粗品),经柱层析纯化后得到橙色固体(1.8g,82.6%)。1h-nmr(500mhz,dmso):δ/ppm=8.56(s,1h),8.21(s,1h),7.71(d,j=7.2hz,2h),6.61(d,j=7.2hz,2h),5.52(d,j=10hz,1h),5.01(m,1h),4.19(m,1h),4.53(m,1h),4.47(m,1h),4.42(t,j=6.8hz,1h),4.33(dd,j=8.0hz,j=1.6hz,1h),4.28(s,2h),4.06(dd,j=8.0hz,j=1.6hz,1h),3.59(t,j=6.6hz,2h),3.31(t,j=6.6hz,2h),2.21(m,2h),2.15(m,2h),1.91-2.12(m,12h);13c-nmr(125mhz,dmso):δ/ppm=175.06,173.12,172.04,170.15,169.91,169.69,168.57,167.05,155.12,151.24,149.42,147.61,144.87,144.53,129.25,128.31,123.53,113.51,100.61,71.17,70.53,69.35,67.02,64.61,62.05,54.62,47.09,39.74,30.84,26.93,21.15,20.98,20.93,20.71.hrms(m/z):calcdforc35h42n8o15[m+h]+:815.2756,found:815.2751。
(6)制备n2-{4-[(2-氨基-4-羟基蝶啶-6-甲基)氨基]苯甲酰基}-n5-{2-3,4,5-三羟基-6-(乙酰氧基甲基)四氢吡喃]-氧乙基}-l-谷氨酰胺(目标物)
将化合物6(1.5g,1.84mmol)溶解于甲醇中,然后在冰浴下滴加入甲醇钠naome(3.13g,14.3mmol,25.0%purityofmeoh),滴加完毕后撤掉冰浴,室温下反应1h。然后用酸性树脂调节ph值至6,然后过滤除去树脂。滤液减压除去之后得到粗品。粗品用制备级hplc纯化,(色谱柱:ageladurashell(博纳艾吉尔,天津,中国)10μm,250×50mm;流动相:[water(10mmnh4hco3)-acn];b%:0-15.0%,20min),之后得到终产物fa-2-dg(0.94g,75%),为橙色固体。1h-nmr(500mhz,dmso):δ/ppm=8.58(s,1h),8.21(s,1h),7.71(d,j=7.2hz,2h),6.61(d,j=7.2hz,2h),5.03(m,1h),4.42(t,j=6.8hz,1h),4.32(s,2h),3.83(dd,j=8.0hz,j=1.6hz,1h),3.79(m,1h),3.72(m,1h),3.63(t,j=6.6hz,2h),3.53(dd,j=8.0hz,j=1.6hz,1h),3.41(m,1h),3.38(m,2h),2.19(m,2h),2.11(m,2h);13c-nmr(125mhz,dmso):δ/ppm=174.86,172.62,171.04,167.55,155.21,151.24,149.45,147.91,144.97,144.53,128.25,127.31,123.53,113.51,104.61,77.17,74.05,73.38,71.34,64.82,62.27,54.57,47.89,39.68,30.75,26.86.hrms(m/z):calcdforc27h34n8o11[m+h]+:647.2339,found:647.2346。
(一)叶酸衍生物在水中溶解度的测定:
制备葡萄糖修饰的叶酸的衍生物的饱和水溶液,然后按照10,100,1000,2000,3000,4000和8000的倍数逐级稀释,得到一系列不同浓度的溶液,待测。同时制备叶酸的饱和水溶液,按照2,5,10,20和50的倍数稀释,得到一系列不同浓度的溶液,待测。
用紫外分光光度计(uv-2600,岛津)对以上制得的样本进行测试,波长扫描范围185-500nm,扫描速度:中速,扫描间隔:1nm,为保证测试的准确性,所有样品的测试温度均为37℃,用仪器自带的uv-probe软件记录并分析数据。
葡萄糖修饰的叶酸衍生物的饱和溶液的稀释溶液和叶酸饱和溶液的稀释溶液的紫外吸收光谱测试见图7所示。图中a、b、d分别为衍生物饱和溶液稀释1000、2000和3000倍之后测试的紫外吸收光谱图,c、e、f分别为叶酸饱和溶液稀释2、5和10倍之后测试得到的紫外吸收光谱图。根据图3所示的结果,图中d和e两条曲线接近重合,d代表衍生物饱和溶液稀释3000倍之后测得的光谱吸收曲线,e代表叶酸饱和溶液稀释5倍之后测得的光谱吸收曲线,由于衍生物的分子结构和叶酸的分子结构中,产生紫外吸收的结构部分是一样的,都是结构中的碟啶和苯环部分,说明衍生物的饱和溶液稀释600倍之后与叶酸的吸光度相当,也就是说衍生物的水溶性比叶酸强600倍。
(二)糖修饰的叶酸衍生物的纳米形态表征
分别利用原子力显微镜、透射电子显微镜和扫描电子显微镜测定糖修饰之后的叶酸衍生物在固体状态下的纳米形态,应用纳米粒度仪测试叶酸衍生物在液体状态下的纳米形态。
以葡萄糖修饰,以长度为2个碳原子的氨基乙醇为连接臂的叶酸衍生物为例,叶酸衍生物成规则的纳米线状结构,纳米线的端点能看到聚集成球形的纳米颗粒,粒径约为30nm。透射电子显微镜下观察到衍生物呈现出较多的纳米线绕在一起的形态,纳米线的宽度在5-20nm,聚集成的团状结构的直径约为300nm左右。从扫描电镜的结果看,衍生物呈现出较零散的纳米薄片,薄片的厚度约5nm,呈方形或圆形,圆形的薄片直径约40nm,方形的薄片边长约为35nm。
实施例3
本实施例利用实施例1所制备的纳米运载颗粒,运载抗肿瘤药物顺铂,得到一种靶向抗肿瘤纳米颗粒,通过配位键连接实施例1所述的纳米运载颗粒和顺铂。制备fa-2-dg和顺铂的配合物,将相同浓度的顺铂和fa-2-dg的水溶液混合后室温搅拌,两小时后得到顺铂和fa-2-dg形成了如图8所示的配位化合物(fa-2-dg-pt)。将混合溶液注入hplc-ms分析,证实了配合物生成。质谱结果见图9。
原子力显微镜,透射电子显微镜和扫描电子显微镜这些形态学表征技术都证实fa-2-dg本身能够自组装形成纳米颗粒,和顺铂形成配位键,载药之后得到fa-2-dg-pt的也可以自组装形成纳米结构。
(一)对fa-2-dg-pt进行体外抗肿瘤活性评价:
1)受试样品
实施例1制备的fa-2-dg,本实施例制备的fa-2-dg-pt和脱氧葡萄糖用pbs配制成所需浓度,顺铂为阳性对照品,用pbs配制成所需浓度。
2)细胞株
人卵巢癌细胞系skov-3,人乳腺癌细胞系mcf-7,人宫颈癌细胞系hela,3株肿瘤细胞均购自中科院上海细胞库。
3)试验方法
分别将生长状态良好、处于对数生长期的skov-3、mcf-7和hela细胞按照2×105个/ml的密度接种于96孔板,每孔200μl。3株细胞按预设的浓度梯度加入待测、经灭菌处理的化合物溶液,每孔25μl,对照孔加入等体积的pbs。在37℃、5%co2培养箱中培养48小时,每孔加入25μl浓度为5mg/ml的mtt溶液,继续置于37℃、5%co2培养箱中培养4小时。离心4分钟(3000rpm/min)。小心吸出上清液,每孔加入100μldmso溶解紫色残留物(甲瓒),平板振荡10分钟使沉淀全部溶解,于570nm酶标仪上测定o.d.值(吸光度),波长570nm。
按照公式“相对生存率=(d含药-d空白)/(d对照-d空白)×100%”计算每一个样品浓度下的样品对肿瘤细胞的抑制率。
4)实验结果
本实验各个化合物的体外抗肿瘤效果如表1所示。
表1不同化合物体外抗肿瘤细胞增殖的ic50值(μm)
a)2-dg代表2-脱氧葡萄糖,ic50的浓度为mm。
(二)对fa-2-dg-pt进行体内抗肿瘤评价
1)实验材料
受试化合物:2-dg,fa-2-dg,fa-2-dg-pt;
阳性对照:顺铂;
实验动物:6周龄的balb/c裸鼠,体重约为18g;
肿瘤细胞:skov-3人卵巢癌细胞,密度为每毫升2.5×108个细胞。
2)剂量设置
按照临床治疗剂量设置顺铂的给药剂量为1.5mg/kg,按此剂量设置fa-2-dg-pt中铂的含量与顺铂的剂量一致,即装载顺铂的剂量为1.5mg/kg;fa-2-dg的给药剂量与fa-2-dg-pt中载体的摩尔浓度一致,为6.34mg/kg;2-dg的剂量与fa-2-dg的摩尔浓度一致,为1.63mg/kg。所有药物均使用生理盐水配制。
3)给药方案
按照给药剂量,每只小鼠给药0.2ml,腹腔给药,在实验开始第一天给药,从第二次开始每隔两天给药,共给药6次,实验持续17天。
4)动物模型的建立
选用6周龄的balb/c裸鼠,spf级,体重约为18g,动物饲养在屏障系统内静置一日后,在背部右侧上方接种肿瘤细胞,细胞密度为每毫升2.5×108个细胞,接种之前细胞可用低密度的matrixgel基质胶按1∶1的比例混合,以保证接种成功率。接种量为0.2ml/只,细胞接种数量为每只动物5×107个细胞。接种时选用1.0ml规格的无菌注射器吸取肿瘤细胞,左手抓取动物,用医用酒精在小鼠右侧背部靠近前肢的部位消毒,然后右手拿起吸好瘤液的注射器进行注射,先将针尖刺入皮肤,挑起并继续刺入,左右稍晃动针尖,然后打入细胞液,针尖稍转动后轻轻拔出。以和小鼠头尾平行的方向为长l,以和小鼠头尾垂直的方向为宽w,计算小鼠的肿瘤体积的公式为:
tumorvolume=l×w^2/2
肿瘤接种约两周后,选取肿瘤体积在120mm3左右的动物随机分组,分组之后静养一天给药,给药方式为腹腔注射。
5)抑瘤率的计算
本实验采用肿瘤体积和瘤重两种考察指标,在实验第17天,用游标卡尺测量肿瘤的体积,然后断颈处死动物,手术取出肿瘤及内脏,用天平称取肿瘤重量。
肿瘤体积的增长率为考察抑瘤率的第一个指标,增长率计算方法为小鼠第17天的肿瘤体积除以第一天的肿瘤体积,体积增长倍数小的抑制肿瘤增长的效果较好。
肿瘤重量抑制率为第二个考察指标,计算方法为:
瘤重抑制率%=(1-给药组瘤重/空白组瘤重)×100%。
统计方法:本实验数据统计均采用t检验和方差分析。
6)实验结果
本实验各个给药组的体内抗肿瘤活性如表2所示。
表2.不同化合物体内抗肿瘤活性
- 还没有人留言评论。精彩留言会获得点赞!