利用纳米孔测序数据的高原多倍体鱼类基因组注释方法与流程
本发明涉及基因组注释技术领域,涉及一种高原多倍体鱼类基因组注释方法,特别是涉及一种利用纳米孔测序数据的高原多倍体鱼类基因组注释方法。
背景技术:
一般基因组测序组装完成后需要对其蛋白编码基因进行结构注释。注释一般有三种方法:基于训练模型的从头预测、基于近缘物种编码基因的同源预测以及基于自身表达转录本的预测。将所有三种方法预测得到的结果进行整合得到这个物种的所有蛋白编码基因,这些基因通常能够得到多种预测方法的支持。如果每种方法都能得到较为理想的结果,最后整合得到的结果也会比较正确。但是各种方法适应的情况各异,不可能每种方法都能得到较好的预测结果,在整合的时候引入较差的预测结果可能会导致最终的预测结果较差。
对于一般的二倍体物种,使用二代测序组装得到的转录组即可直接进行评估。但是高原鱼类包含大量同源多倍体物种,由于加倍导致的同源基因ohnologs非常相似,因此采用二代转录组测序技术得到的短序列经常无法确定来自于两个ohnologs中的哪个,导致最终的组装结果可能存在大量的嵌合转录本。因此,对于多倍体的高原鱼类,如何获得准确的全长转录组序列用于评估成为一个需要解决的问题。现有对高原多倍体鱼类基因组进行注释的方法存在着基因预测的不确定性问题,预测成本较高,且预测结果不够可靠。
技术实现要素:
针对现有技术存在的问题,本发明提供一种利用纳米孔测序数据的高原多倍体鱼类基因组注释方法,能够提高高原多倍体鱼类基因组注释的准确性和可靠性,降低基因组注释的成本。
本发明的技术方案为:
一种利用纳米孔测序数据的高原多倍体鱼类基因组注释方法,其特征在于,包括下述步骤:
步骤1:基于纳米孔测序技术获得待注释高原多倍体鱼的全长转录组序列;
步骤2:采用n种基因组预测方法分别预测待注释高原多倍体鱼可能的蛋白编码基因;
步骤3:将步骤1中获得的全长转录组序列作为参考序列,将每种基因组预测方法预测得到的每个蛋白编码基因与参考序列进行比对,计算每个蛋白编码基因相对参考序列的重叠率、重叠相似度;
步骤4:过滤掉重叠率低于重叠率阈值且重叠相似度低于重叠相似度阈值的蛋白编码基因,对剩下的蛋白编码基因进行整合,得到最终的预测基因集。
进一步的,所述步骤1具体包括:提取待注释高原多倍体鱼各个器官组织的转录组,将各个器官组织的转录组等量混合后采用纳米孔测序技术进行全长转录组测序,获得待注释高原多倍体鱼的全长转录组序列。
进一步的,所述步骤2中,n种基因组预测方法包括从头预测方法、基于同源序列的预测方法、基于转录组数据的预测方法。
本发明的有益效果为:
本发明基于纳米孔测序技术获得待注释高原多倍体鱼的全长转录组序列,降低了高原多倍体鱼类基因组注释的成本;本发明以全长转录组序列为参考序列计算每种基因组预测方法预测得到的每个蛋白编码基因相对参考序列的重叠率、重叠相似度,并利用重叠率、重叠相似度对预测结果进行过滤,对过滤后的预测结果进行整合得到最终的预测基因集,排除了各种预测噪音的干扰,提高了高原多倍体鱼类基因组注释的准确性和可靠性。
附图说明
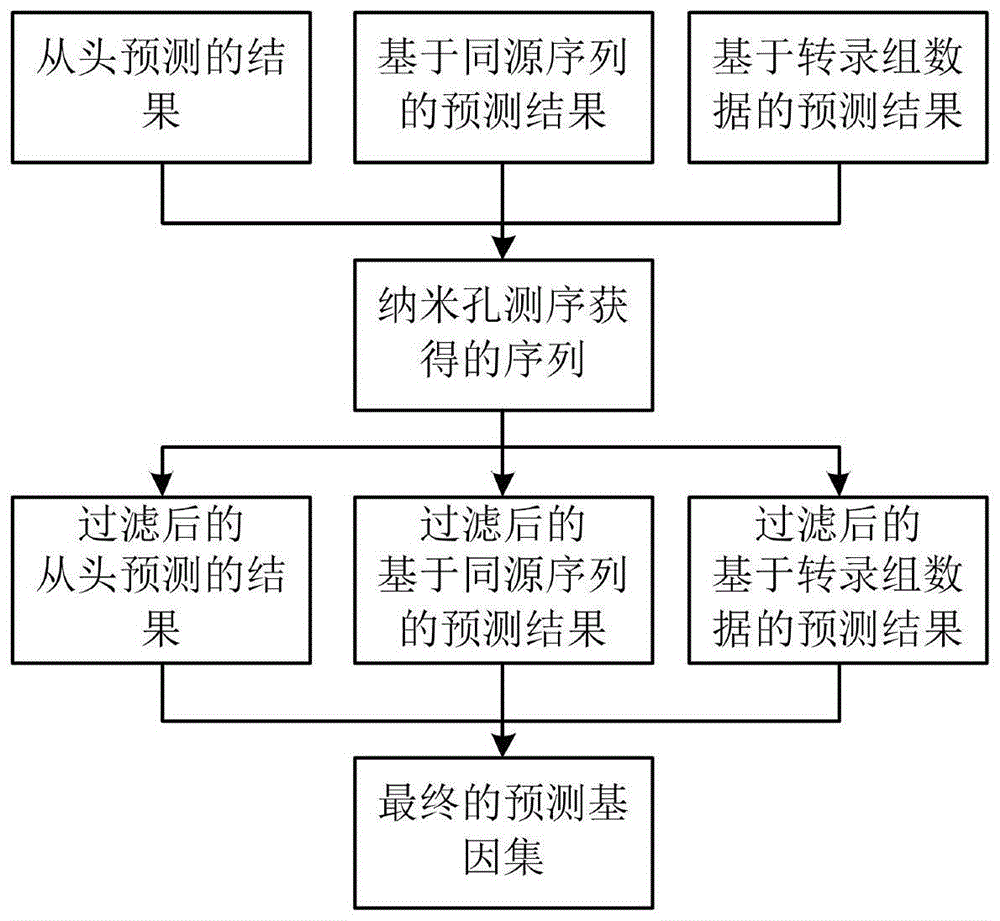
图1为具体实施方式中本发明的利用纳米孔测序数据的高原多倍体鱼类基因组注释方法的原理图。
具体实施方式
下面将结合附图和具体实施方式,对本发明作进一步描述。
如图1所示,本发明的利用纳米孔测序数据的高原多倍体鱼类基因组注释方法,包括下述步骤:
步骤1:基于纳米孔测序技术获得待注释高原多倍体鱼的全长转录组序列。
本实施例中,提取待注释高原多倍体鱼各个器官组织的转录组,将各个器官组织的转录组等量混合后采用纳米孔测序技术进行全长转录组测序,获得待注释高原多倍体鱼的全长转录组序列。为获得尽可能多的转录本,将来自各组织的总rna混合后逆转录成cdna,然后使用纳米孔测序得到10m以上序列,将过滤后的序列用last比对到基因组上,提取位置重叠的序列进行纠错,然后对纠错后的序列去冗余得到最终的转录组序列nanopore.fasta。这些转录本不是通过组装获得,代表了最真实的基因表达情况。
其中,纳米孔测序技术最明显的特征就是读长较长,能够很轻松地跨越整个转录本,同时其测序成本较其它三代测序技术更低,因此对于高原多倍体鱼类,相同的投入,纳米孔测序技术更容易获得相对全面的准确的表达数据。本发明采用这种技术获得高质量的全长转录组序列,提高了高原多倍体鱼类蛋白编码基因结构注释的准确性,降低了成本。
步骤2:采用n种基因组预测方法分别预测待注释高原多倍体鱼可能的蛋白编码基因。
本实施例中,n种基因组预测方法包括从头预测方法、基于同源序列的预测方法、基于转录组数据的预测方法,分别使用augustus、genewise和pasa软件对待注释高原多倍体鱼的编码基因进行预测,具体如下:
1)使用augustus进行预测
augustus--species=zebrafishgenome.fasta>augustus.gff
2)使用geneblasta+genewise进行预测
先用geneblasta提取各蛋白序列在该物种基因组中的同源区域:
genblasta_v1.0.4_linux_x86_64-pblast-qprotein.fasta-ttar_genome.fasta-pt-pgtblastn-e1e-3-r5-gt-ff-a0.5-d1000000-oprotein.gbla
再用genewise确定可能的基因结构:
genewise-gwhsp-quiet-tfor-gffprotein_seq.fastagbla_seq.fasta>genewise.gff
3)使用pasa进行预测
launch_pasa_pipeline.pl-calignassembly.config-c–r-ggenome.fasta-t-unanopore.fasta-tnanopore.fasta.clean-fnanopre.id--cpu10–alignersgmap--max_intron_length1000000
步骤3:将步骤1中获得的全长转录组序列作为参考序列,将每种基因组预测方法预测得到的每个蛋白编码基因与参考序列进行比对,计算每个蛋白编码基因相对参考序列的重叠率、重叠相似度,具体包括:
1)用步骤1中得到的nanopore.fasta序列作为reference建库
makeblastdb–dbtypenucl-innanopore.fasta-input_typefasta
2)分别提取各种方法预测得到的编码序列predict.fasta作为query,与reference比对
blastn–querypredict.fasta–dbnanopore.fasta–outcompare.bla–evalue1e-10–outfmt6
3)根据比对的重叠率、重叠相似度对预测结果进行评估、排序和筛选
步骤4:过滤掉重叠率低于重叠率阈值且重叠相似度低于重叠相似度阈值的蛋白编码基因,对剩下的蛋白编码基因进行整合,得到最终的预测基因集,具体包括:
1)切分
partition_evm_inputs.pl--genomegenome.fasta--gene_predictionsgene_predictions.gff3--protein_alignmentsprotein_alignments.gff3--transcript_alignmentstranscript_alignments.gff3--segmentsize${size}--overlapsize${overlap}--partition_listingpartitions_list.out--repeats${repeats}
2)运行
write_evm_commands.pl--genomegenome.fasta--weightsweights.txt--gene_predictionsgene_predictions.gff3--protein_alignmentsprotein_alignments.gff3--transcript_alignmentstranscript_alignments.gff3--output_file_nameevm.out--partitionspartitions_list.out--min_intron_length20--repeats${repeats}>commands.list
3)收集结果
recombine_evm_partial_outputs.pl--partitionspartitions_list.out--output_file_nameevm.outconvert_evm_outputs_to_gff3.pl--partitionspartitions_list.out--outputevm.out--genomegenome.fasta
本发明充分考虑到高原多倍体鱼类存在基因组加倍的现象,利用纳米孔测序技术测定全长转录组无须组装和通量较高的特性,构建一个能够代表绝大部分基因转录本的参考序列库作为基准,再据此评估不同预测方法的准确性程度,筛选并整合准确性程度较高的结果进行整合,大大降低了各种不利因素的影响,达到了以较低成本、较大幅度提高高原多倍体鱼类基因组注释的准确性和可靠性的目的。
显然,上述实施例仅仅是本发明的一部分实施例,而不是全部的实施例。上述实施例仅用于解释本发明,并不构成对本发明保护范围的限定。基于上述实施例,本领域技术人员在没有做出创造性劳动的前提下所获得的所有其他实施例,也即凡在本申请的精神和原理之内所作的所有修改、等同替换和改进等,均落在本发明要求的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种新型的15种人乳头瘤病毒亚型分型试剂盒和方法与流程
- 一种与水稻稻瘟病抗性基因紧密连锁的分子标记、引物及其应用的制造方法与工艺
- 一种无创高通量甲基化结肠癌诊断、研究和治疗方法与流程
- 用于检测CDKN2B基因变异的试剂盒及CDKN2B基因变异率的定量检测方法与流程
- 用于基因甲基化检测的引物和探针、采样方法、试剂盒与流程
- 一种癌症相关基因突变高灵敏度检测方法和试剂盒与流程
- 基于高通量测序法的人类多基因突变检测试剂盒的制造方法与工艺
- 一种用于检测德尔卑沙门氏菌的靶基因、特异性引物对及检测方法和试剂盒与流程
- 一种检测测序平台索引序列污染的方法及引物与流程
- 无物种限制的真核生物同时进行基因敲除和基因过表达的制造方法与工艺