一种磷酸钙纳米颗粒及其制备方法和应用与流程
本发明涉及药物载体技术领域,具体涉及一种磷酸钙纳米颗粒及其制备方法和应用。
背景技术:
肿瘤的化疗目前存在生物利用度低,非特异性靶向,并且治疗过程中容易出现多种耐药等缺陷。因此,在癌症治疗中许多纳米药物递送系统正在进行临床前开发。近年来,无机纳米磷酸钙药物递送平台已经成为肿瘤学中潜在的化学治疗系统。
磷酸钙(capo)是一种天然且生物相容的材料,它对酸的敏感性使得包裹的药物可以从酸性内体释放到细胞质中从而发挥功效。磷酸钙可以通过钙离子和磷酸根离子之间的反应来合成。但是,这样制备的磷酸钙的大小和稳定性很难控制,并且表面带负电,作为生物载药工具容易被机体的内皮细胞吞噬不利于药物递送。纳米磷酸钙具有ph依赖性降解优势,肿瘤的弱酸微环境和内体、溶酶体酸性环境使得药物可以定位且缓慢释放,可以增强其抑瘤效果。但是这种优势仍不足以起到靶向递送的目的产物的作用,因而探寻具有颗粒稳定性、靶向性递送的纳米磷酸钙载药技术具有重要的意义。
目前,有许多不同的方法来生产具有不同形态和尺寸的磷酸钙颗粒。对于药物递送应用,多孔的磷酸钙颗粒由于增加的负载能力而特别重要。多孔磷酸钙纳米级材料的传统合成方法涉及将钙和磷酸盐的水溶性盐混合。利用这种技术,可以通过改变沉淀反应的条件来控制颗粒的尺寸,形状和结晶度。例如,为了获得多孔磷酸钙球形纳米颗粒,经常使用微波辅助水热法。通过将脱水氯化钙与atp混合并进一步进行微波处理,合成了平均直径为260nm的多孔磷酸钙颗粒,所得的纳米颗粒在不同ph的水溶液中显示了超过150小时的良好稳定性,这比以前报道的多孔磷酸钙颗粒要长得多。或者,可以通过改变前体的比例来制备具有不同形态和结晶度的多孔磷酸钙纳米颗粒。结果表明,可以通过在盐酸的存在下老化氢氧化钙和三磷酸钠的混合物来合成磷酸钙颗粒。此外,可以通过模板法获得空心磷酸钙纳米颗粒。例如,以壳聚糖-聚丙烯酸(cs-paa)为模板制备空心磷酸钙微球,形成机理是基于壳聚糖(cs)和聚丙烯酸(paa)之间的静电相互作用。
技术实现要素:
本发明的目的在于克服现有技术的不足之处而提供一种磷酸钙纳米颗粒及其制备方法和应用,本发明制备的磷酸钙纳米颗粒具有空心结构,粒径较小,稳定时间较长,且表面带有大量氨基,使磷酸钙纳米颗粒表面带有正电荷,可用于纳米颗粒表面荧光素,靶向基团的修饰。
为实现上述目的,本发明采取的技术方案如下:
一种磷酸钙纳米颗粒,所述磷酸钙纳米颗粒为空心磷酸钙纳米颗粒,药物负载量大;空心磷酸钙纳米颗粒的表面修饰有氨基,氨基可与多种基团反应,用于在磷酸钙表面进一步修饰荧光素、各种肿瘤或正常组织靶向的基团;且该空心磷酸钙纳米颗粒的粒径≤500nm,粒径较小,稳定时间较长。
进一步地,所述空心磷酸钙纳米颗粒的表面通过氨基结合有荧光素、靶向基团中的至少一种。
进一步地,所述靶向基团包括rgd多肽、叶酸、转铁蛋白中的至少一种,使得该磷酸钙纳米颗粒作为药物载体,起到靶向递送药物的作用,能够更加精准、有效治疗肿瘤并且减少目前临床药物治疗的副作用。
本发明还提供了上述的磷酸钙纳米颗粒作为抗肿瘤药物载体的应用。
本发明还提供了上述磷酸钙纳米颗粒的制备方法,包括以下步骤:
(1)聚阳离子高分子与含有磷酸根的生物分子在水中自组装形成纳米胶束,加入氯化钙,通过微波加热,得到表面修饰有氨基的空心磷酸钙纳米颗粒。
本发明采用模板法制备氨基修饰的磷酸钙纳米颗粒,将聚阳离子高分子与含有磷酸根的生物分子在水中混合,通过亲疏水作用在水中自组装形成纳米颗粒,作为形成磷酸钙的模板,在溶液中加入氯化钙,微波加热使含有磷酸根的生物分子水解释放磷酸根并于钙离子反应形成磷酸钙纳米颗粒。本发明制备得到粒径≤500nm的磷酸钙纳米颗粒,粒径较小,稳定时间较长,且磷酸钙表面含有大量氨基可与多种基团反应,用于在磷酸钙表面进一步修饰各种肿瘤或正常组织靶向的基团;本发明的磷酸钙纳米颗粒具有空心结构,对药物有比较高的装载量。
进一步地,所述聚阳离子高分子包括树状大分子、聚乙烯亚胺、聚氨基酯中的至少一种;所述含有磷酸根的生物分子包括腺苷-5'-二磷酸钠盐水合物、磷酸肌酸、磷酸果糖、磷酸核黄素、磷酸吡哆醛中的至少一种,作为磷酸根来源,制备磷酸钙,其中,相较于其它种类的含有磷酸根的生物分子,腺苷-5'-二磷酸钠盐水合物效果更好,更有利于制备得到具有空心球结构的磷酸钙纳米颗粒。
进一步地,所述聚阳离子高分子与含有磷酸根的生物分子的质量比为0.1-1:100,在此范围内,制备的磷酸钙具有较好的空心球结构,对药物有比较高的装载量。
微波加热的温度越高,加热时间越长,磷酸钙纳米颗粒空心球的尺寸越大,容易导致空心结构消失,进一步地,所述步骤(1)中微波加热的温度为70-100℃,在此温度范围内,制备得到的磷酸钙具有较好的空心球结构。
本发明通过优选聚阳离子高分子与含有磷酸根的生物分子的种类及其配比,以及微波加热条件,有利于制备得到表面带有大量氨基且粒径≤500nm的空心磷酸钙纳米颗粒。
进一步地,所述的磷酸钙纳米颗粒的制备方法还包括以下步骤:
(2)表面修饰有氨基的空心磷酸钙纳米颗粒与nhs-peg-靶向基团反应,形成靶向基团-peg-磷酸钙;
或表面修饰有氨基的空心磷酸钙纳米颗粒与荧光素反应,得到带有荧光成像功能的磷酸钙;
或表面修饰有氨基的空心磷酸钙纳米颗粒与nhs-peg-靶向基团反应,形成靶向基团-peg-磷酸钙,再将靶向基团-peg-磷酸钙与荧光素反应,获得带有荧光成像功能的靶向基团-peg-磷酸钙;
本发明中所述peg的分子量可以为500-15000,peg保护层可以避免磷酸钙纳米颗粒过早的被内皮质网系统吞噬,延长体内循环的时间,可用于体内药物递送。
所述步骤(2)中,表面修饰有氨基的空心磷酸钙纳米颗粒与nhs-peg-靶向基团的质量比为10:1。
进一步地,所述的磷酸钙纳米颗粒的制备方法还包括以下步骤:
(3)将抗肿瘤药物装载于靶向基团-peg-磷酸钙或带有荧光成像功能的靶向基团-peg-磷酸钙,
所述抗肿瘤药物包括烷化剂、抗代谢药、抗癌抗生素、中药类、激素类、抗体、蛋白质、多肽中的至少一种。
与现有技术相比,本发明的有益效果为:
(1)与其他方法制备的纳米磷酸钙纳米颗粒相比,本发明制备的纳米磷酸钙颗粒粒径得到有效的控制,将磷酸钙纳米颗粒的粒径控制在500nm以内,颗粒粒径较小,稳定时间较长,可以用于体内药物递送。
(2)与其他方法制备的磷酸钙相比,本发明制备的磷酸钙纳米颗粒表面带有大量氨基,可以与多种基团反应,以此在磷酸钙表面修饰其他具有靶向作用的官能团。
(3)本发明制备的磷酸钙具有空心结构,对药物有比较高的装载量。
附图说明
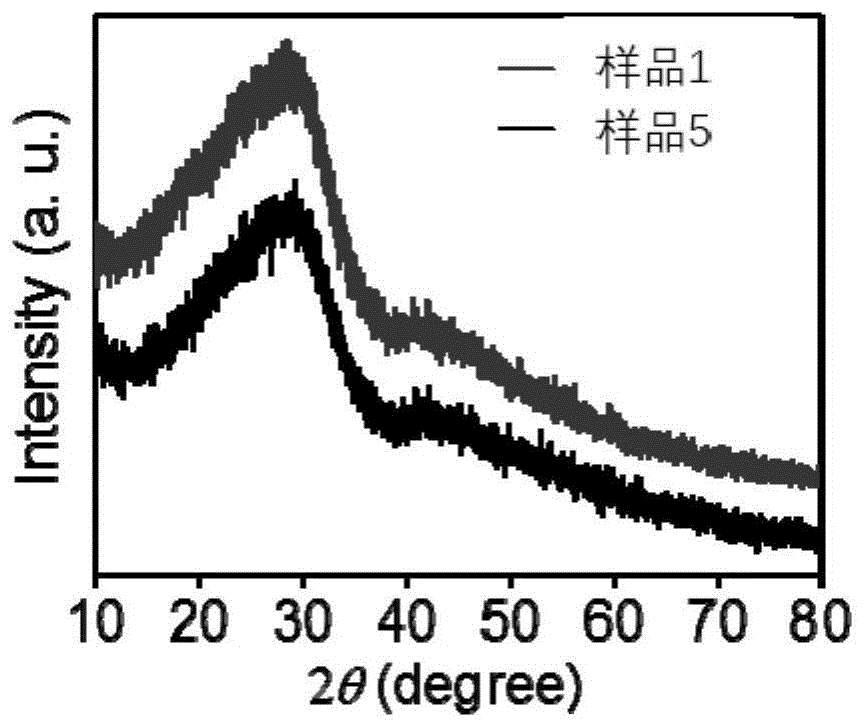
图1为样品1与样品5的x射线衍射图谱。
图2为样品1与样品5的热重图谱分析。
图3为样品1与样品5的zeta电位数值。
图4为胰腺癌细胞株psn1在不同时间(1h、2h、4h、12h)对样品8(fam-capo)的摄取定性、定量结果。a.confocal显微镜观察、流式细胞仪检测psn1细胞株在1h、2h、4h、12h对样品8(fam-capo)的摄取情况;b.流式细胞仪cytoflex检测psn1细胞株在1h、2h、4h、12h对样品8(fam-capo)摄取的荧光信号强度;c.流式细胞仪cytoflex检测psn1细胞株在1h、2h、4h、12h对样品8(fam-capo)摄取的平均荧光强度(meanfluorescenceintensity,mfi)。
图5为样品6(rgd-peg-capo)、样品9(rgd-peg-capo/5fu)与商业化试剂5fu在不同浓度(100、50、25、12.5、6.25、3.125、1.5625mg/l)处理条件下,psn1细胞株的存活率。
图6为样品6(rgd-peg-capo)、样品9(rgd-peg-capo/5fu)与商业化试剂5fu浓度为50mg/l的药物浓度处理细胞株psn124h后的凋亡蛋白表达wb检测结果。
图7为不同浓度的样品5(100、50、25、12.5、6.25mg/l)处理条件下,hk2细胞株的存活率。
图8为样品5(capo)的溶血试验结果。a.溶血试验的图片。样品1至7加入样品5(capo)的浓度从高到低依次为100.00、50.00、25.00、12.50、6.25、3.12、1.56mg/l,试管8为阴性对照组(生理盐水),试管9为阳性对照组(去离子水);b.不同浓度磷酸钙的样品对应的溶血率。
具体实施方式
为更好地说明本发明的目的、技术方案和优点,下面将结合具体实施例对本发明进一步说明。本领域技术人员应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。
实施例中,所使用的实验方法如无特殊说明,均为常规方法,所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
实施例1:
本实施例的磷酸钙纳米颗粒表面没有氨基修饰,其制备方法为:
将100mg腺苷-5'-二磷酸钠盐水合物溶解在20ml超纯水中,室温搅拌10min,标记为溶液1;将0.147g二水合氯化钙溶解于20ml超纯水中,并加入溶液1中,搅拌10min,标记为溶液2;将溶液2微波加热至70℃,加热10min后离心,用超纯水洗涤1次,酒精洗涤3次,70℃烘干,即得到样品1,即没有氨基修饰的磷酸钙纳米颗粒。
实施例2:
本实施例的磷酸钙纳米颗粒为粒径≤500nm的空心磷酸钙纳米颗粒,其表面修饰有氨基,其制备方法为:
将100mg腺苷-5'-二磷酸钠盐水合物溶解在20ml超纯水中,加入100μl1mg/ml树状大分子(g5pamam),室温搅拌10min,标记为溶液1;将0.147g二水合氯化钙溶解于20ml超纯水中,并加入溶液1中,搅拌10min,标记为溶液2;将溶液2微波加热至70℃,加热10min后离心,用超纯水洗涤1次,酒精洗涤3次,70℃烘干,即得到样品2。
实施例3:
本实施例的磷酸钙纳米颗粒为粒径≤500nm的空心磷酸钙纳米颗粒,其表面修饰有氨基,其制备方法为:
将100mg腺苷-5'-二磷酸钠盐水合物溶解在20ml超纯水中,加入200μl1mg/mlg5pamam,室温搅拌10min,标记为溶液1;将0.147g二水合氯化钙溶解于20ml超纯水中,并加入溶液1中,搅拌10min,标记为溶液2;将溶液2微波加热至70℃,加热10min后离心,用超纯水洗涤1次,酒精洗涤3次,70℃烘干,即得到样品3。
实施例4:
本实施例的磷酸钙纳米颗粒为粒径≤500nm的空心磷酸钙纳米颗粒,其表面修饰有氨基,其制备方法为:
将100mg腺苷-5'-二磷酸钠盐水合物溶解在20ml超纯水中,加入400μl1mg/mlg5pamam,室温搅拌10min,标记为溶液1;将0.147g二水合氯化钙溶解于20ml超纯水中,并加入溶液1中,搅拌10min,标记为溶液2;将溶液2微波加热至70℃,加热10min后离心,用超纯水洗涤1次,酒精洗涤3次,70℃烘干,即得到样品4。
实施例5:
本实施例的磷酸钙纳米颗粒为粒径≤500nm的空心磷酸钙纳米颗粒,其表面修饰有氨基,其制备方法为:
将100mg腺苷-5'-二磷酸钠盐水合物溶解在20ml超纯水中,加入1ml1mg/mlg5pamam,室温搅拌10min,标记为溶液1;将0.147g二水合氯化钙溶解于20ml超纯水中,并加入溶液1中,搅拌10min,标记为溶液2;将溶液2微波加热至100℃,加热10min后离心,用超纯水洗涤1次,酒精洗涤3次,70℃烘干,即得到样品5。
相较于样品1,样品2-5呈粒径≤500nm的磷酸钙空心球结构。
样品1与样品5的x射线衍射图谱如图1所示。从图1来看,制备过程中无论有没有加入pamam高分子,得到的样品都是无定型磷酸钙。
样品1与样品5的热重图谱分析如图2所示。从图2可以看出,反应过程加入pamam,样品失重更多,这是因为pamam在高温下分解,导致样品失重增多。
样品1与样品5的zeta电位数值如图3所示。实施例5将聚阳离子高分子pamam与腺苷-5'-二磷酸钠盐水合物混合在水溶液中形成纳米胶束,作为模板制备得到的磷酸钙纳米颗粒表面带有氨基,使磷酸钙纳米颗粒表面带有正电荷,更有利于药物递送。
实施例6:
将10mgrgd多肽-聚乙二醇2000-n-羟基琥珀酰亚胺(rgd-peg2000-nhs)溶于10mldmso中,再加入100mg样品4分散在此溶液中,黑暗、常温下反应24小时,离心,透析,冻干,即得到rgd修饰的磷酸钙纳米材料,简称为rgd-peg-capo,标记为样品6。
实例7:
将1mg5(6)-羧基荧光素琥珀酰亚胺酯(fam)溶于1ml二甲基亚砜中,再加入10mg样品5,分散并于黑暗、常温下反应12小时,离心,透析,冻干即得到带有荧光成像功能的rgd修饰的磷酸钙纳米材料fam-rgd-peg-capo;标记为样品7。
实施例8:
将1mgfam溶于1ml二甲基亚砜中,再加入10mg样品5,分散并于黑暗、室温条件下反应12小时,离心,透析,冻干即得到带有荧光成像功能的磷酸钙材料,简称fam-capo,标记为样品8。
实施例9:
将100mg5-氟尿嘧啶溶解于10mldmso中,充分溶解,并将1mg样品6分散于此溶液中,黑暗、室温环境下于室温反应48小时,得到装载5-氟尿嘧啶的rgd-peg-capo纳米颗粒,简称为rgd-peg-capo/5fu,标记为样品9。
实施例10:测试人胰腺癌细胞psn1对样品8(fam-capo)的吞噬摄取情况。
(1)将人胰腺癌细胞psn1以2*105密度铺板接种在六孔板中,培养过夜。细胞附着后,除去原始培养基,并用预冷pbs小心洗涤细胞3次。然后每孔加入无血清培养基1ml,放37℃,5%的co2培养箱孵育30min,待培养基稳定30min后,加入浓度为10mg/l的样品8(fam-capo)分别孵育12h、4h、2h与1h,吸出测试溶液,并加入2ml4℃pbs溶液以停止细胞摄取并洗涤细胞。然后,加入500μl胰蛋白酶–edta,并将细胞消化3min。此后,添加0.5ml完全培养基以终止消化,收集细胞并以1500rpm离心3min。最后,弃去上清液后,加入500μlpbs将细胞分散在均匀的单细胞悬液中。通过流式细胞术(激发/发射=492nm/518nm)测量来自每个样品的10000个细胞中fam的荧光强度,并进行统计学分析,结果如图4(b)、4(c)所示。
(2)基于流式细胞仪的结果,confocal显微镜用于目测研究psn1细胞对样品8(fam-capo)的吸收。该过程如下进行:首先将psn1细胞以每孔1.0×105的密度接种在15毫米玻璃底细胞培养皿中,并在37℃下培养过夜。细胞充分贴合后,弃去旧培养基,并用1ml浓度为20mg/lfam-capo的代替。孵育1h、2h、4h、12h后移出测试溶液,然后用pbs洗涤3次。然后,加入1ml含dapi的pbs(10μg*ml-1)(激发/发射=364nm/454nm)作为核标记添加10min。抽吸弃去dapi溶液,并用冷pbs冲洗细胞。之后,将细胞再用4%多聚甲醛固定10min,然后用pbs漂洗1次,再用0.2%的triton通透5min,然后用pbs漂洗3次,然后用100nmdid染料作为细胞膜标记物染色(激发/发射=644nm/663nm)10min。最后,将细胞再用pbs洗涤两次,并用1mlpbs覆盖。随后通过confocal显微镜对荧光图像进行可视化和分析,结果如图4(a)所示。
实施例11:
评估不同浓度的样品6(rgd-peg-capo)、样品9(rgd-peg-capo/5fu)与商业化试剂5fu对胰腺癌细胞株psn1的毒性作用。
首先将以8*104/ml的细胞浓度,100μl/孔接种于96孔板,每个浓度设置4个复孔,然后放37℃,5%的co2培养箱孵育24h,待细胞贴壁后达到70%细胞密度开始给药处理,样品6(rgd-peg-capo)、5fu与rgd-peg-capo/5fu分别以无血清培养基1640配成1.5625、3.125、6.25、12.5、25、50和100mg/l七个药物浓度,除去原始培养基然后每孔加入100μl的不同药物浓度,每个药物浓度设置4个复孔。药物处理时间分别设为24h、48h与72h。在每个药物处理时间点结束时,每孔分别加入10μl的cck-8,然后放在37℃,5%的co2培养箱避光孵育2h,再使用酶标仪450nm波长进行测量。不同药物处理的细胞存活率百分比分别与对照组(未处理组)进行比较分析,结果如图5所示。相较于rgd-peg-capo组和5fu组,rgd-peg-capo/5fu提升了药物的抗肿瘤效果。
细胞存活率=【(处理组-空白组)/(对照组-空白组)】*100%
实施例12:
比较样品6(rgd-peg-capo)、样品9(rgd-peg-capo/5fu)与商业化试剂5fu处理胰腺癌细胞株psn1的细胞凋亡相关蛋白(bcl-2,bax,cleavedcaspases-3)的表达水平。
首先将psn1细胞以每孔50×104密度接种于6孔板中,培养过夜,待细胞密度达70%时,除去原始培养基,加入含有浓度为50mg/l不同药物的新鲜无血清培养基1ml,放回培养箱继续孵育24h。药物处理时间结束后,除去原始培养基,采用4℃的预冷pbs清洗3次。每孔加入100μl的pmsf与ripa按照1:100配置的蛋白裂解液进行总蛋白提取,采用bca蛋白浓度测定试剂盒进行浓度测定并校准。每孔加入20μg蛋白进行十二烷基硫酸钠聚丙烯酰胺凝胶电泳,然后进行聚乙二烯二氟化物膜反应,进行孵育一抗过夜,二抗孵育2h,然后在分子成像仪进行曝光获取图像,结果如图6所示。凋亡蛋白抗体bcl-2,bax,cleavescaspases-3的配置浓度为1:1000,细胞膜骨架蛋白β-actin作为内参配置浓度为1:5000,二抗配置浓度为1:1000。
实施例13:
不同浓度的样品5(capo)对正常肾小管上皮细胞hk2的毒性作用。
首先将以80000/ml的细胞浓度,100μl/孔接种于96孔板,每个浓度设置4个复孔,然后放37℃,5%的co2培养箱孵育24h,待细胞贴壁后达到70%细胞密度开始给药处理,样品5(capo)以无血清培养基1640配成6.25、12.5、25、50和100mg/l五个药物浓度,除去原始培养基然后每孔加入100μl的不同药物浓度,每个药物浓度设置4个复孔。药物处理时间分别设为24h、48h与72h。在每个药物处理时间点结束时,每孔分别加入10μl的cck-8,然后放在37℃,5%的co2培养箱避光孵育2h,再使用酶标仪450nm波长进行测量,结果如图7所示。可见,本发明的capo对细胞明显无毒。
不同药物处理的细胞存活率百分比分别与对照组(未处理组)进行比较分析。细胞存活率=【(处理组-空白组)/(对照组-空白组)】*100%
实施例15:
不同浓度的样品5(capo)处理下的溶血实验用于测试样品5的生物相容性。
从大鼠的心脏获得血液然后进行分离、pbs清洗获得2%的红细胞。生理盐水作为阴性对照组,去离子水作为阳性对照组。将不同浓度的capo的生理盐水混合液与2%的红细胞溶液混合,室温条件下静止3h,之后1500r/min离心3min,取出100μl上清于96孔板上,用570nm的波长于酶标仪上检测,每个样品4个复孔,结果如图8所示。由图可知,样品5的溶血率低于3%,具有良好的生物相容性。
溶血率=【(样品组吸收光-阴性对照吸收光)/(阳性对照吸收光-阴性对照吸收光)】*100%。溶血率超过5%视为溶血。
最后所应当说明的是,以上实施例仅用以说明本发明的技术方案而非对本发明保护范围的限制,尽管参照较佳实施例对本发明作了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的实质和范围。
- 还没有人留言评论。精彩留言会获得点赞!