基于局部和全局的网络中心性分析的癌症驱动基因预测方法及系统
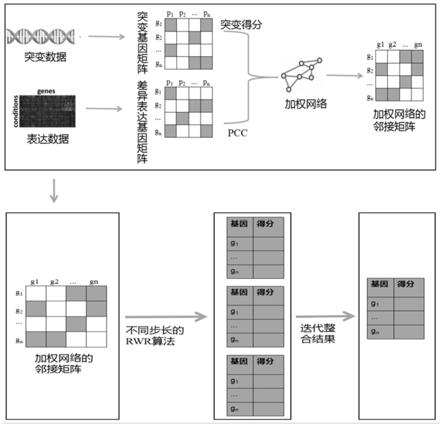
1.本发明涉及生物信息计算领域,特别是涉及一种基于局部和全局的网络中心性分析的癌症驱动基因预测方法及系统。
背景技术:
2.作为世界上最致命的疾病之一,癌症致病机理复杂,至今人类一直在不断的研究,目前人们普遍认为癌症是由一些体细胞突变引起的,这些突变中的一些赋予细胞生长和阳性选择优势,引起强烈的增殖和肿瘤。还有绝大多数的体细胞突变是中性的或引起细胞凋亡并不影响癌症的发生和发展也不会转化为癌细胞,所以区分哪些突变对肿瘤患者的发生和发展起作用是目前癌症治疗的主要目标之一。基于这一目标,很多识别驱动基因的算法应运而生,近年来基于网络识别驱动基因的方法越来越重要,也取得了较好的结果。然而,在网络中的无论是最短路径的长度还是节点的度,都不能反映整个网络的拓扑性质。为了改进这些策略,研究人员开始使用多种类型的中心性来提取局部和全局的拓扑性质。基于随机游动的方法表明,全局拓扑结构比局部信息对疾病基因预测更有价值。因此本发明设计新算法整合了局部和全局的网络特征来更好地识别驱动基因。另外,基于网络中心度识别网络中关键节点的方法忽略了基因节点的属性特征、邻居之间的拓扑影响、先验知识的影响以及网络本身的属性等。此外,除了基因组的数据外,还需要考虑其他组学的数据。
3.为了提高预测精度,需要对不同类型的数据进行适当的整合,以弥补它们的不足,可以使用多组学数据来增强预测。因此,通过设计一个可以整合多组学数据来增强网络的性能,并通过随机游走算法得到局部和全局拓扑结构信息的算法来解决上述问题。
技术实现要素:
4.本发明所要解决的技术问题是提供一种基于局部和全局的网络中心性分析的癌症驱动基因预测方法及系统,能够更好地对癌症数据进行驱动基因的预测与分类。
5.为解决上述技术问题,本发明采用的一个技术方案是:提供一种基于局部和全局的网络中心性分析的癌症驱动基因预测方法,包括以下步骤:
6.s1:对标准化的体细胞突变数据和基因表达数据进行预处理,表达成基因
‑
矩阵的形式;
7.s2:使用预处理后的数据对下载的ppi网络加权;
8.s3:模型构建,使用改进的重启随机游走算法通过控制跳转步长分析网络的全局和局部特征;
9.s4:使用构建好的模型对癌症数据集进行预测,以获取驱动基因的排名向量,实现对癌症驱动基因的预测。
10.在本发明一个较佳实施例中,所述体细胞突变数据包括单核苷酸变异数据、染色体畸变数据。
11.进一步的,对所述体细胞突变数据进行预处理的步骤包括:
12.s101:去除体细胞单核苷酸变异数据中“变异分类”项为“沉默”的基因,从染色体畸变数据的扩增片段和删除片段中选择基因,提取拷贝数变异信息;
13.s102:将体细胞突变数据汇总在一个二进制突变基因
‑
患者矩阵m中,在矩阵m中行表示基因,列表示癌症样本。
14.在本发明一个较佳实施例中,对所述基因表达数据进行预处理的步骤包括:
15.s111:采用k近邻对表达数中存在值为na的数据进行填充;
16.s112:根据每个基因在所有样本中的表达分布为高斯分布的假设,将表达数据转换成差异表达基因列表;
17.s113:收集每个患者的差异表达基因列表以获得全部患者集中的差异表达基因列表,将基因表达数据处理成差异表达基因
‑
患者的矩阵d,其中d
ij
表示差异表达基因i在患者j中的表达值。
18.在本发明一个较佳实施例中,步骤s2的具体步骤包括:
19.s201:输入预处理后的体细胞突变数据,根据得到的突变基因
‑
患者矩阵m,计算突变基因的突变得分,其中,每个基因i的突变分数m(i)表示突变对癌症的贡献,定义为:
[0020][0021]
k
i
为突变基因i的所有发生突变的患者集合,n
k
是样本k中突变基因的总数,n
max
为所有样本中突变基因的最大数目,如果基因i在所有样本中没有突变,即k
i
为空,则m(i)被赋予一个不大于任何突变基因的背景突变评分;
[0022]
s202:输入预处理完成的基因表达数据,根据差异表达基因
‑
患者的矩阵d,计算两个基因之间的皮尔森相关系数:
[0023][0024]
其中,x
i
和x
j
分别代表基因i和基因j的表达量,n代表样本数,利用r语言的cor()函数计算pcc值,将基因i和j之间的pcc值作为节点i和j在网络中的权值;
[0025]
s203:整合突变得分m(i)和pcc,以此计算基因之间的相互作用分数来为ppi网络加权,加强随机游走算法中的初始概率p
0t
;
[0026][0027]
m(i)和m(j)分别表示基因i和j的突变得分,w
ij
表示网络的边缘权重,所述边缘权重使用基因之间的皮尔森相关系数。
[0028]
在本发明一个较佳实施例中,步骤s3的具体步骤包括:
[0029]
s301:在加权的ppi网络上使用随机游走算法遍历网络节点,并将ppi网络的权值作为随机游走的初始概率p
0t
;
[0030]
s302:采用反馈中心性代替随机游走算法的跳转概率,通过控制随机游走的步长,得到在不同的步长下网络分析的结果;
[0031]
s303:整合不同步长所得到的结果,通过递归的方法得到在已知的驱动基因验证下精度趋近于1的结果,并返回不同步长所占的权值,整合公式如下:
[0032]
score=w1p(1)+w2p(2)+w3p(3)
[0033]
其中,p(1)代表步长为2的重启随机游走算法计算的节点向量,p(2)表示的是步长为3,p(3)表示的是迭代趋于平稳并且步长小于1000的结果;
[0034]
通过整合不同步长的rwr算法的结果,能够识别网络中的重要的基因节点。
[0035]
为解决上述技术问题,本发明采用的另一个技术方案是:提供一种基于局部和全局的网络中心性分析的癌症驱动基因预测系统,包括:
[0036]
数据预处理模块,用于对标准化的体细胞突变数据和基因表达数据进行预处理,表达成基因
‑
矩阵的形式;
[0037]
网络加权模块,用于使用预处理后的数据对下载的ppi网络加权;
[0038]
模型构建与分析模块,用于模型构建,使用改进的重启随机游走算法通过控制跳转步长分析网络的全局和局部特征;
[0039]
模型预测模块,用于使用构建好的模型对癌症数据集进行预测,以获取驱动基因的排名向量,实现对癌症驱动基因的预测。
[0040]
本发明的有益效果是:
[0041]
(1)基于网络的分析方法在识别驱动基因方面存在网络背景知识不完整,本发明使用基因突变和表达数据来为网络加权,提高识别驱动基因的精度;
[0042]
(2)随机游走算法是在游走的过程中是等概率的跳转到相邻节点的,这样可能会使得结果陷入局部最优的情况,本发明使用反馈中心性来代替跳转的概率;
[0043]
(3)网络中心性分析方法容易忽略那些节点度不高但作用很关键的节点,我们通过改变随机游走算法的步长,得到网络局部和全局分析,更好的表征网络中的关键节点;
[0044]
(4)本发明在识别驱动基因方面达到较好的性能,不仅能够识别那些显著突变的驱动基因,还可以识别罕见突变的驱动基因,大大提高对癌症驱动基因预测的精度,为癌症的诊断和精准医疗的发展做出了贡献。
附图说明
[0045]
图1是本发明基于局部和全局的网络中心性分析的癌症驱动基因预测方法的流程图;
[0046]
图2是本发明与基于网络的方法(mecorank、dnmax和dnsum)的实验结果对比图;
[0047]
图3是本发明与其它方法(scs,frequency和oncoimpact)的实验结果对比图;
[0048]
图4是所述基于局部和全局的网络中心性分析的癌症驱动基因预测系统的结构框图。
具体实施方式
[0049]
下面结合附图对本发明的较佳实施例进行详细阐述,以使本发明的优点和特征能更易于被本领域技术人员理解,从而对本发明的保护范围做出更为清楚明确的界定。
[0050]
请参阅图1,本发明实施例包括:
[0051]
一种基于局部和全局的网络中心性分析的癌症驱动基因预测方法,包括以下步骤:
[0052]
s1:对标准化的体细胞突变数据和基因表达数据进行预处理,表达成基因
‑
矩阵的形式;
[0053]
优选的,从癌症基因组图谱(tcga)数据库中下载标准化的体细胞突变数据和基因表达数据。进一步的,所述体细胞突变数据包括单核苷酸变异(snvs)和染色体畸变(cnvs)。从研究充分的癌症基因普查(cgc)数据库中获得已经验证的癌症基因作为正样本(即驱动基因),默认为不在已知的癌症基因阳性集中的所有其他的基因为负样本(即乘客基因)。
[0054]
具体的,对所述体细胞突变数据进行预处理的步骤包括:
[0055]
s101:去除体细胞单核苷酸变异数据中“变异分类”项为“沉默”的基因,从染色体畸变数据的扩增片段和删除片段中选择基因,提取拷贝数变异(cnv)信息;
[0056]
s102:将体细胞突变数据汇总在一个二进制突变基因
‑
患者矩阵m中,在矩阵m中行表示基因,列表示癌症样本。对于基因i,如果至少一个病人身上发生突变,则m(i,j)=1,否则m(i,j)=0。
[0057]
具体的,对所述基因表达数据进行预处理的步骤包括:
[0058]
s111:采用k近邻对表达数中存在值为na(not available)的数据进行填充;
[0059]
s112:根据每个基因在所有样本中的表达分布为高斯分布的假设,将表达数据转换成差异表达基因(degs)列表;
[0060]
为了表明每个患者的基因差异,计算配对肿瘤与正常样本之间基因表达的log2倍变化,绝对值大于1的基因定义为degs;
[0061]
s113:收集每个患者的差异表达基因列表以获得全部患者集中的差异表达基因列表,将基因表达数据处理成差异表达基因
‑
患者的矩阵d,其中d
ij
表示差异表达基因i在患者j中的表达值。
[0062]
s2:使用预处理后的数据对下载的ppi网络加权;
[0063]
首先下载ppi网络(蛋白质
‑
蛋白质相互作用,protein
‑
protein interaction,ppi)作为突变基因与degs的交互图,只要在ppi网络中存在基因i和j的边缘,则基因i和j就是存在交互网络中。本步骤中构建了一个以突变的基因作为源节点,degs作为一个目标节点的交互网络,通过计算为交互网络赋予权值,加强网络的性能。具体步骤包括:
[0064]
s201:输入预处理后的体细胞突变数据,根据得到的突变基因
‑
患者矩阵m,计算突变基因的突变得分,其中,每个基因i的突变分数m(i)表示突变对癌症的贡献,定义为:
[0065]
[0066]
k
i
为突变基因i的所有发生突变的患者集合,n
k
是样本k中突变基因的总数,n
max
为所有样本中突变基因的最大数目。如果基因i在所有样本中没有突变,即k
i
为空,则m(i)被赋予一个不大于任何突变基因的背景突变评分(bms);
[0067]
s202:输入预处理完成的基因表达数据,根据差异表达基因
‑
患者的矩阵d,计算两个基因之间的皮尔森相关系数:
[0068][0069]
其中,x
i
和x
j
分别代表基因i和基因j的表达量,n代表样本数,利用r函数中的cor()函数计算pcc值,将基因i和j之间的pcc值作为节点i和j在网络中的权值;然后计算突变基因的突变得分,整合突变得分m(i)和pcc以此计算基因之间的相互作用分数来加强随机游走算法中的初始概率p
0t
。
[0070]
s203:整合突变得分m(i)和pcc,以此计算基因之间的相互作用分数来为ppi网络加权,加强随机游走算法中的初始概率p
0t
;
[0071][0072]
m(i)和m(j)分别表示基因i和j的突变得分,w
ij
表示网络的边缘权重,所述边缘权重使用基因之间的皮尔森相关系数。这里整合了基因组的突变数据和转录组的表达数据来增强随机游走过程中的初始概率p
0t
。
[0073]
s3:模型构建,使用改进的重启随机游走算法分析网络的全局和局部特征;原理如下:
[0074]
基于中心性的方法在一定程度上提高了疾病基因的预测,在疾病基因预测中最成功的中心性是反馈中心性,如katz中心性。一个节点的中心性取决于其相邻节点的中心性,相邻节点的中心性进一步取决于其相邻节点的中心性。因此,使用反馈中心性的方法也与使用基于随机游走的方法相似的策略来预测疾病基因。本示例中使用重启的随机游走算法通过控制游走的步长得到网络的局部和全局的特征。
[0075]
使用随机漫步来预测疾病基因的算法,对于给定的基因g,用p0表示先验信息,其中p0(g)=1表示基因g是已知的驱动基因,p0(g)=0表示g是乘客基因。随机游走的公式如下:
[0076]
p
t+1
=wp
t
=w
t
p0[0077]
其中w为ppi网络的列归一化邻接矩阵。如果w是一个随机矩阵,这个过程等价于网络上的随机漫步。如果允许每一步以r的概率重新开始随机漫步,可以得到rwr算法如下:
[0078]
p
t+1
=(1
‑
r)wp
t
+rp0[0079]
其中p
t
在稳定状态下包含了每个基因与疾病相关的概率。如果为不同的路径长度设置不同的权值,随机行走可能会更可控,从而产生更好的预测。控制步长为2和3得到局部预测的结果,然后设置最大游走步长为1000或结果趋于平稳,作为全局的拓扑结果。另一个问题是,本示例中改变了随机游走的概率矩阵,使节点在随机跳转的时候不是等概率的选
择邻居节点之一,而是用邻居节点的ketz中心性作为跳转概率。
[0080]
通过控制随机游走的步长,得到在不同的步长下网络分析的结果。设置迭代次数为2次,这样可以计算基因邻居和邻居的邻居的信息。设置迭代步长为3,使用三级邻居迭代的属性代替节点在网络中的地位。将迭代次数设置为1000次,并控制损失函数阈值为0.00001,这样得到趋于稳定的全局网络分析的结果。整合不同步长所得到的结果,通过递归的方法得到在已知的驱动基因验证下精度(precision)最好的结果,即精度趋近于1的结果,并返回不同步长所占的权值(w1、w2、w3)。整合公式如下:
[0081]
score=w1p(1)+w2p(2)+w3p(3)
[0082]
在上式中,p(1)代表步长为2的重启随机游走算法计算的节点向量,p(2)表示的是步长为3,p(3)表示的是迭代趋于平稳并且步长小于1000的结果。然后将各个步长的结果向量迭代加和,找到使得实验结果是最好的一组参数搭配,根据最好的得分排序驱动基因。通过整合不同步长的rwr的结果,可以更好的地识别网络中的重要的基因节点。
[0083]
随机游走算法是在游走的过程中是等概率的跳转到相邻节点的,这样可能会使得结果陷入局部最优的情况,本发明使用反馈中心性来代替跳转的概率;另外基于网络的分析方法在识别驱动基因方面存在网络背景知识不完整,本发明使用基因突变和表达数据来为网络加权。
[0084]
s4:使用构建好的模型对癌症数据集进行预测,以获取驱动基因的排名向量,实现对癌症驱动基因的预测。
[0085]
当有外部数据要进行预测时,经过数据预处理和网络加权计算后,可以直接输入到模型中进行预测,获得癌症数据的候选驱动基因。
[0086]
本实施例中,使用了两种类型的数据:编码区体细胞突变数据和基因表达数据。特别是编码区体细胞突变数据包括cnvs和snvs。这些数据来自328个胶质母细胞瘤(gbm)样本、379个膀胱癌(blca)样本、252个前列腺癌(prad)样本和316个卵巢癌(ovaria)样本,并从tcga数据门户网站(https://tcga
‑
data.nci.nih.gov/tcga/)下载。本示例中只使用了包含这两种情况的样本。癌症是一种多组基因相互作用的疾病,这些基因不仅与单个基因有关,而且在某些分子网络中相互作用,在示例中,所采用的基因相互作用网络都可能是从一个关于人类蛋白的参考资料库(human protein reference database,本文记为hprd)下载的,其中包含9617个基因和74078个相互作用边。构建无向无权图g(v,e)作为实验中的参考网络,其中v表示网络中的基因节点,e表示基因之间的边,当基因i与基因j存在相关性时,g
ij
=1,否则g
ij
=0。例如,卵巢癌数据集包括来自316个卵巢患者,他们都有体细胞突变数据和基因表达数据,其中有5309个差异表达基因和5705个突变基因映射在基因相互作用网络上。
[0087]
在缺乏基本事实的情况下,使用标准灵敏度/特定基准技术进行定量测量是不切实际的。为了帮助评估本发明结果的质量,本实施例从研究充分的癌症基因普查(cgc)数据库中获得已知的驱动基因,cgc是一个将与癌症因果相关的突变基因进行编目的数据库,在许多癌症研究中被广泛作为基准。在本实施例中,将其作为标准参考列表,cgc中共包含了616个已知驱动基因的列表,版本为(09/26/2016)。
[0088]
对于每次比较,本发明均使用三种测量方法(精度precision、召回率recall和flscore),这三种测量方法以排名前n个基因为基础。
[0089][0090][0091][0092]
其中,tp(true positive)表示真阳性数目,即cgc中的驱动基因被正确预测为癌症驱动基因的数目,tn(true negative)表示真阴性数目,即不在cgc中的其他基因被正确预测为乘客基因的数目,fp(false positive)是假阳性的数目,即原本是在cgc中的驱动基因而被预测为乘客基因的数目,fn(false negative)是假阴性数目,即原本是乘客基因而被预测为驱动基因的数目。
[0093]
这三种测量方法以排名前n个基因为基础。首先利用了tcga提供的4种癌症数据集,即blca、gbm、ov和prad,测试方法区分驱动基因的能力,将排名前100的候选基因结果与基于网络的三种竞争方法进行比较,分别为mecorank、dnmax和dnsum(muffin中的两种方法)的排序结果进行比较。如图2所示,精度曲线、召回率曲线和f1score的分数表明,本发明所述方法优于其它的方法。如图的实验结果部分只展示了blca和gbm两种癌症的结果。
[0094]
显然,一个驱动基因的算法排名越高,它的表现就越好。对于blca,在排名前100的候选驱动基因中,dnmax方法鉴定出31个基因在cgc中发现。dnsum鉴定出30个,mecorank方法鉴定出33个,本发明所述方法鉴定出36个。f1分数精度指标的值是对于检测精度与方法召回比率的可靠调和平均值,如图2所示,可以看出,对于f1的曲线得分来说,本发明所述方法增加更快。本发明所述方法在前100个识别的驱动基因的精度,召回率和f1分数都显著优于muffin和mecorank方法。
[0095]
对于gbm,在排名前100的候选驱动基因中,dnmax识别出36个基因在cgc中发现,dnsum方法识别出23个,mecorank方法识别出33个,本发明所述方法识别出39个。值得注意的是,本发明所述方法在其50个排名靠前的候选基因中检索到的已知驱动基因数量与mecorank在其100个排名靠前的候选基因中检索到的已知驱动基因数量大致相同,比dnsum在其前100个识别的候选基因中在cgc中发现的还多5个。本发明所述方法通过其20个、40个、60个、80个、100个排名靠前的候选基因,比其他方法识别出更多的已知癌症基因,在预测驱动基因方面达到了较好的精度。另外,f1的曲线得分本发明所述方法高于dnsum上升至30%。
[0096]
为了评估本发明所述方法的有效性,除了将它与基于网络的方法进行比较外,还将本发明所述方法与其它的基于各种原理的方法进行了比较,使用相同的数据和相同的参考癌症基因集,将本发明所述方法与scs,frequency和oncoimpact进行比较,其中scs是基于单个病人特异性的识别驱动基因的方法,对于它的结果,本实施例取在整体上的投票结果。前述已经证明了本发明所述方法优于基于网络的基因排序的方法,与其它的方法进行比较时,也得到了同样的预期结果。这些结果表明,本发明所述方法不仅为顶级候选基因提供了与当前最先进的基因中心算法一样高的精确度性,而且通过保持对排名候选基因的高灵敏度,为发现新癌症基因提供了实质上更多的机会。如图3所示,这里展示了在prad和ov
癌症数据上的效果。在cgc参考集上验证其它两个癌症数据集时也得到了类似的结果。
[0097]
本发明实施例还提供一种基于局部和全局的网络中心性分析的癌症驱动基因预测系统,包括:
[0098]
数据预处理模块,用于对标准化的体细胞突变数据和基因表达数据进行预处理,表达成基因
‑
矩阵的形式;
[0099]
网络加权模块,用于使用预处理后的数据对下载的ppi网络加权;
[0100]
模型构建与分析模块,用于模型构建,使用改进的重启随机游走算法通过控制跳转步长分析网络的全局和局部特征;
[0101]
模型预测模块,用于使用构建好的模型对癌症数据集进行预测,以获取驱动基因的排名向量,实现对癌症驱动基因的预测。
[0102]
以上所述仅为本发明的实施例,并非因此限制本发明的专利范围,凡是利用本发明说明书及附图内容所作的等效结构或等效流程变换,或直接或间接运用在其他相关的技术领域,均同理包括在本发明的专利保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1