利用基于原子轨道的特征确定分子性质的系统和方法与流程
1.本发明大体上涉及基于分子系统性质设计和合成分子的系统和方法;并且更具体地,涉及利用基于原子轨道的特征和深度学习量子化学计算来确定合成化学品的性质的系统和方法。
背景技术:
2.分子模拟有助于包括固态材料、聚合物、精细化学品和药物的科学工业的发现努力。当前的方法采用基于物理学的方法,这些方法求解量子力学方程来描述原子和分子的行为。当前的方法虽然功能强大,但极具计算成本(消耗了世界超级计算资源的相当大一部分)和人力时间成本(必要的计算需要数月或更长的挂钟时间)。分子模拟的进展将扩大其在工业创新和开发过程中的应用。
技术实现要素:
3.根据本发明的各种实施例的系统和方法使得能够基于分子系统性质来设计和/或合成分子。在许多实施例中,具有特定分子系统性质的分子可以被合成,以用于广泛的产品开发过程,诸如用于制药工业的药物发现,以及用于化学、石油、电池和电子工业的材料设计。根据本发明的各种实施例合成的材料的示例包括(但不限于):催化剂、酶、药物、蛋白质和抗体、有机电子器件、表面涂层、纳米材料和有机材料。
4.许多实施例使用基于原子轨道的深度学习(orbnet)过程,根据基于原子轨道的特征来预测分子系统性质。在若干实施例中,基于原子轨道的特征包括(但不限于):基于原子轨道(ao)的特征、基于对称性适应原子轨道(saao)的特征、基于ao的特征的导数以及saao特征的导数。根据本发明的各种实施方式的分子系统性质的示例包括(但不限于):溶解度、对分子的结合亲和力、对蛋白质的结合亲和力、氧化还原势能、pka、电导率、离子电导率、热导率、光吸收频率、光吸收强度和光吸收效率。
5.在许多实施例中,orbnet过程可以允许在计算和挂钟时间上比现有的基于物理学的量子力学方法加速至少1000倍。在若干实施例中,这些过程允许人类效率提高至少100倍。通过使用云资源大规模部署orbnet,周转时间可以从几天缩短到几秒。根据本发明的若干实施例的orbnet能够实现至少10倍的预测准确度改进。一些其他实施例实施软件包,消除了计算预测的风险,降低了下游实验和生产成本,并且加快了上市时间。
6.本发明的一个实施例包括一种合成分子的方法,包括:使用计算机系统获得分子系统的原子轨道集;使用计算机系统,基于分子系统的原子轨道集来生成基于原子轨道的特征集;使用在计算机系统上实施的基于原子轨道的机器学习(orbnet)模型,基于特征集来确定至少一个分子系统性质;以及当所确定的至少一个分子系统性质满足计算机系统的至少一个标准时,合成分子系统。
7.在另一实施例中,基于原子轨道的特征集包括基于原子轨道的特征的属性图表示。
8.在进一步的实施例中,属性图表示的节点特征对应于对角线原子轨道块,并且属性图表示的边特征对应于非对角线原子轨道块。
9.在又一实施例中,原子轨道集包括对称性适应原子轨道(saao),并且基于原子轨道的特征集包括基于原子轨道的特征集、基于saao的特征集、基于原子轨道的特征集的导数或基于saao的特征集的导数。
10.在再一进一步的实施例中,分子系统是多个候选分子系统中的一个。此外,确定所确定的至少一个分子系统性质何时满足至少一个标准还包括:基于候选分子系统中的每个候选分子系统的原子轨道集来生成基于原子轨道的特征集;使用orbnet模型,基于候选分子系统中的每个候选分子系统的基于原子轨道的特征集,来确定候选分子系统中的每个候选分子系统的至少一个分子系统性质;基于为多个候选分子系统中的每个候选分子系统确定的至少一个分子系统性质来筛选多个候选分子系统;以及基于筛选标识分子系统。
11.又一进一步的实施例还包括使用描述多个分子系统及其分子系统性质的训练数据集来训练orbnet模型,以学习基于原子轨道的特征集与分子系统性质集之间的关系。
12.在再一实施例中,训练orbnet模型以学习基于原子轨道的特征集与分子系统性质集之间的关系还包括:获得分子系统的训练数据集中的每个分子系统的原子轨道集;以及基于原子轨道集获得基于原子轨道的特征集。
13.再次在进一步的实施例中,通过构建旋转不变的对称性适应原子轨道基集,获得分子系统的训练数据集中的每个分子系统的对称性适应原子轨道集;以及至少基于对称性适应原子轨道来获得基于对称性适应原子轨道的特征集。
14.在进一步的附加实施例中,获得原子轨道集包括计算选自由hartree-fock理论、密度泛函理论和半经验方法组成的组的一种平均场电子结构,并且获得基于原子轨道的特征集包括计算选自由hartree-fock理论、密度泛函理论和半经验方法组成的组的一种平均场电子结构。
15.在又一进一步的实施例中,获得原子轨道集包括通过神经网络参数化出现在选自由hartree-fock理论、密度泛函理论和半经验方法组成的组的电子结构方法的公式中的至少一个量子力学算符,并且获得基于原子轨道的特征集包括通过神经网络参数化出现在选自由hartree-fock理论、密度泛函理论和半经验方法组成的组的电子结构方法的公式中的至少一个量子力学算符。
16.在另一附加实施例中,神经网络包括图形神经网络,其中,图形神经网络的至少一个节点对应于至少一个原子,并且图形神经网络的至少一条边对应于至少一个原子间相互作用。
17.再次在另一实施例中,训练orbnet模型和神经网络同时发生。
18.在又一进一步的实施例中,确定对称性适应原子轨道包括对角化至少一个对角密度矩阵块。
19.在又一实施例中,训练orbnet模型包括图形神经网络。
20.在另一附加实施例中,图形神经网络包括至少一个消息传递层和至少一个解码层。
21.再次在进一步的实施例中,分子系统包括原子、分子键以及由原子和分子键形成的分子中的至少一个。
22.在又一实施例中,特征集包括基于原子轨道的特征,基于原子轨道的特征包括物理算符。
23.再次在又一进一步的实施例中,基于原子轨道的特征还包括选自由以下各项组成的组的至少一个特征:fock矩阵中的元素,coulomb矩阵中的元素,hartree-fock矩阵中的元素,密度矩阵中的元素;核心hamiltonian矩阵中的元素;以及重叠矩阵中的元素。
24.再次在又一实施例中,至少一个分子系统性质包括选自由量子相关能、构象能、平均场能、单点能、学习能、分子轨道能、势能面、力、原子间力、振动频率、偶极矩、电子密度、响应性质、热性质、激发态能、激发态力、线性响应激发态能、线性响应激发态力和光谱组成的组的至少一个性质。
25.在又一进一步的附加实施例中,合成分子系统包括选自由催化剂、酶、药物、蛋白质、抗体、表面涂层、纳米材料、半导体和有机材料组成的组的至少一个分子。
26.又一附加实施例包括筛选候选分子系统集的方法,包括:使用计算机系统获得多个候选分子系统的原子轨道集;使用计算机系统,基于候选分子系统中的每个候选分子系统的原子轨道集,为每个候选分子系统生成基于原子轨道集的特征集;使用在计算机系统上实施的基于原子轨道的机器学习(orbnet)模型,基于候选分子系统中的每个候选分子系统的基于原子轨道的特征集,来确定候选分子系统中的每个候选分子系统的至少一个分子系统性质;使用计算机系统基于为候选分子系统中的每个候选分子系统确定的至少一个分子系统性质来筛选候选分子系统,以标识具有满足至少一个标准的至少一个分子系统性质的至少一个分子系统;以及使用计算机系统生成描述在候选分子系统的筛选期间标识的至少一个分子系统的报告。
27.又一进一步的实施例还包括一种使用反向分子设计过程合成分子系统的方法,包括:使用计算机系统搜索具有由基于原子轨道的机器学习(orbnet)模型预测的满足至少一个标准的至少一个分子系统性质的基于原子轨道的特征集,其中,orbnet模型被训练,以接收分子系统的特征集并且输出至少一个分子系统性质的估计;使用计算机系统使用特征-结构图将定位的基于原子轨道的特征集映射到标识的分子系统,其中,特征-结构图被训练,以将基于原子轨道的特征集映射到对应的分子结构;使用计算机系统基于至少一个筛选标准筛选所标识的分子系统;以及当所标识的分子系统满足至少一个筛选标准时,合成所标识的分子系统。
28.再次在又一实施例中,搜索具有由orbnet模型预测的满足至少一个标准的至少一个分子系统性质的基于原子轨道的特征集还包括使用至少一个生成模型来生成候选特征集。
29.在又一进一步的实施例中,生成模型包括图形神经网络。
30.再次另一进一步的实施例包括一种训练基于原子轨道的机器学习(orbnet)模型以从分子系统的原子轨道集中预测至少一个分子系统性质的方法,包括:使用计算机系统获得多个分子系统及其分子系统性质的训练数据集;使用计算机系统,基于候选分子系统中的每个候选分子系统的原子轨道集,为训练数据集中的每个分子系统生成基于原子轨道的特征集;使用计算机系统训练ml模型,以学习训练数据集中的每个分子系统的基于原子轨道的特征集与训练数据集中的分子系统中的每个分子系统的分子系统性质之间的关系;以及利用orbnet模型,根据基于特定分子系统的原子轨道集为特定分子系统生成的基于原
子轨道的特征集,预测特定分子系统的至少一个分子系统性质。
31.在另一进一步的附加实施例中,获得多个分子系统及其分子系统性质的训练数据集还包括:使用计算机系统,基于特定分子系统的原子轨道集,生成特定分子系统的基于原子轨道的特征集;基于所检索的基于原子轨道的特征与特定分子系统的基于原子轨道的特征集的基于原子轨道的特征之间的接近度,从数据库中检索基于原子轨道的特征;以及使用所检索到的分子系统形成训练数据集。
32.在又一进一步的实施例中,训练orbnet模型以学习训练数据集中的每个分子系统的基于原子轨道的特征集与训练数据集中的分子系统中的每个分子系统的分子系统性质之间的关系还包括:利用迁移学习过程来训练先前训练的orbnet模型,以确定分子系统的基于原子轨道的特征与不同的分子系统性质集之间的关系。
33.再次在又一进一步的实施例中,训练orbnet模型以学习训练数据集中的每个分子系统的基于原子轨道的特征集与训练数据集中的分子系统中的每个分子系统的分子系统性质之间的关系还包括:利用在线学习过程来更新先前训练的orbnet模型。
34.附加的实施例和特征部分地在下面的描述中阐述,并且部分地对于本领域技术人员在检查说明书时将变得显而易见,或可以通过本公开的实践而获知。通过参考构成本公开一部分的说明书和附图的剩余部分,可以实现对本公开的本质和优点的进一步理解。
附图说明
35.参考以下附图,将更全面地理解该描述,这些附图是作为本发明的示例性实施例呈现的,并且不应被解释为对本发明的范围的完整叙述。需要说明的是,专利或申请文件包含至少一幅彩色附图。应要求并支付必要的费用,专利局将提供本专利或专利申请出版物的(一个或多个)彩色附图副本。
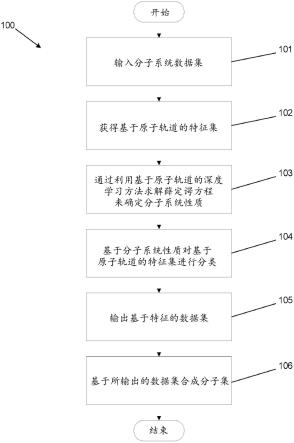
36.图1示出了根据本发明的实施例的基于原子轨道的机器学习过程。
37.图2示出了根据本发明的实施例的能够确定分子结构的软件的用户界面。
38.图3示出了根据本发明的实施例的用于ao特征的orbnet过程的架构。
39.图4示出了根据本发明的实施例的用于saao特征和saao特征的导数的orbnet过程的工作流程。
40.图5示出了根据本发明的实施例的用于saao特征和saao特征的导数的orbnet过程中的消息传递层的结构。
41.图6概念性地图示了根据本发明的实施例的原子轨道对的数据库。
42.图7示出了根据本发明的实施例的用于采集基于原子轨道的特征的orbnet过程。
43.图8示出了根据本发明的实施例的结合机器学习回归来确定分子系统性质的orbnet过程。
44.图9a示出了根据本发明的实施例的用于选择要合成的候选分子系统的过程,其中,该过程使用orbnet模型。
45.图9b示出了根据本发明的实施例的用于标识要合成的分子系统的过程,其中,该过程使用基于ml模型的反向分子设计过程。
46.图9c示出了根据本发明的实施例的orbnet过程,该过程用于生成与特定分子系统相关的训练数据,以便训练orbnet模型用于估计特定分子系统的至少一个化学性质。
fock算符的示例包括(但不限于):fock(f)矩阵的元素、coulomb(j)矩阵的元素、hartree-fock交换(k)矩阵的元素、密度(p)矩阵的元素、轨道质心距离(d)矩阵的元素、核心哈密顿(h)矩阵的元素和/或重叠(s)矩阵的元素。许多实施例规定,量子算符可以基于kohn-sham密度泛函理论,包括(但不限于):交换相关算符、交换相关算符的近似和交换相关算符的分量。许多实施例规定,量子算符可以是密度泛函紧束缚理论计算和/或其他经验电子结构理论方法,包括(但不限于):壳层解析电荷和coulomb、交换、fock和/或交换相关算符的近似。若干实施例包括可以是分子系统的性质的量子算符。这些性质的示例包括(但不限于):偶极矩、原子间距离矩阵、连续溶剂化能。许多实施例实施包括(但不限于)图形神经网络的神经网络,以参数化矩阵,包括(但不限于)fock(f)矩阵、coulomb(j)矩阵、hartree-fock交换(k)矩阵、密度(p)矩阵、轨道质心距离(d)矩阵、核心哈密顿(h)矩阵和重叠(s)矩阵,以生成基于ao的特征。可以容易地理解,根据本发明的各种实施例,用于描述分子系统的具体ao特征很大程度上仅受具体应用的要求限制。根据本发明若干实施例的orbnet过程利用对称的特征。如下面进一步讨论的,对称性不是必需的。根据本发明的许多实施例的orbnet过程适当地根据具体应用的要求利用对称或不对称的特征。
59.在许多实施例中,orbnet过程利用使用输入数据集训练的模型。许多实施例基于包括(但不限于)saao特征的输入ao特征与在训练orbnet模型期间学习的性质之间的关系,预测分子系统的某些性质作为输出。根据若干实施例,orbnet可以预测高质量的电子结构能量。在一些实施例中,输出性质可以包括(但不限于):(1)分子的可计算性质,诸如多体薛定谔方程的解,包括基态和/或激发态平均场能量、基态和/或激发态多体关联能量、势能面、总体/或相对构象能、电子能量、关联能量、saao对贡献、平均场能量、单点能量、分子轨道能量、热性质、力、原子间力、振动频率(hessian)、偶极矩、电子密度、激发态能量、线性响应激发态和力和/或光谱;和(2)分子的实验可测量的性质,诸如活性系数、溶解度、pka、ph、分配系数、蒸汽压、熔点、沸点、闪点、溶剂化自由能、氧化还原势能、电导率、离子电导率、热导率、光吸收频率、光吸收强度、光吸收效率、粘度、adme性质、毒性、药物毒性、结合亲和力和/或蛋白质结合亲和力。多个实施例实施saao特征的导数作为orbnet模型中的输入,并且能够预测响应性质,包括(但不限于):力、优化的几何结构、原子间力、偶极子和线性响应激发态。
60.在一些实施例中,力和/或hessian的预测可以用于将分子系统的几何结构优化到局部最小值或鞍点。若干实施例规定,力的预测可以用于运行分子动力学。在多个实施例中,能量和/或力的预测可以用于执行构型采样。在若干实施例中,基于由orbnet模型基于分子系统的包括(但不限于)saao特征的输入ao特征输出的分子系统的预测性质来选择分子系统。在多个实施例中,orbnet模型可以用于执行生成设计,其中,在特征空间内执行搜索,以标识提供期望的分子系统性质的包括(但不限于)saao特征的至少一个ao特征集。在若干实施例中,包括(但不限于)saao特征的ao特征可以使用特征-结构图被映射到分子结构,该特征-结构图可以使用深度学习过程从训练数据集推导。然后对应于包括(但不限于)saao特征的(一个或多个)所识别的ao特征集的(一个或多个)分子系统可以被进一步分析,以确定最适合特定应用的(一个或多个)分子系统。如可以容易地理解的,根据本发明的各种实施例的系统和方法可以适当地根据具体应用的要求利用分子系统的各种输入ao特征中的任何一种,来预测对应分子系统的各种不同性质中的任何一种。
61.许多实施例规定,orbnet过程可以基于一个特定和/或极小基集输入来预测对应于较大和/或不同的原子轨道基集的性质。若干实施例规定,orbnet过程可以预测对应于更昂贵和/或不同层面的电子结构理论的性质,该电子结构理论包括(但不限于)利用混合交换相关泛函的密度函数理论(dft),其基于包括(但不限于)利用局部密度近似或半经验电子结构方法的dft的一级电子结构理论的输入。
62.在若干实施例中,通过输出性质预测的分子系统可以与输入分子系统处于相同的分子家族中。在许多实施例中,通过输出性质预测的分子系统可以与输入分子系统处于不同的分子家族中。不同的分子家族的示例可以包括(但不限于):分子组成、分子几何结构和/或成键环境。在许多实施例中,包括(但不限于)saao特征的输入ao特征集不明确依赖于原子类型,因此orbnet过程可以增强训练结果的化学可迁移性。
63.在多个实施例中,orbnet过程被实施为软件应用。若干实施例在量子化学软件包中实施orbnet过程,这自动降低了分子模拟的计算和人工时间成本,同时保持用户界面不变。许多实施例规定,将orbnet集成到现有工业工作流程中可以提高计算速度,而不会降低准确度,并且不需要对用户进行再训练。
64.在许多实施例中,可以使用更复杂的分子系统模型,包括(但不限于)分子系统的属性图表示,作为矩阵组织表示的替代。在一些实施例中,图形表示的拓扑和连通性可以从ao特征和/或saao特征张量的集合和/或子集推导。在一些实施例中,量子化学信息可以表示为属性图g(v,e,x,xe)。在若干实施例中,属性图的节点特征对应于包括至少一个ao集的对角ao块,并且边特征对应于包括至少一个ao集的非对角ao块。在某些实施例中,属性图的节点特征对应于对角saao特征(xu=[f
uu
,j
uu
,k
uu
,p
uu
,h
uu
]),并且边特征对应于非对角saao特征(x
euv
=[f
uv
,j
uv
,k
uv
,d
uv
,p
uv
,s
uv
,h
uv
])。分子系统的基于图形的表示能够实现多任务学习。如可以容易地理解的,适当构建的图形表示可以提供置换不变性和大小扩展性的好处。在许多实施例中,可以利用包括消息传递层的图形神经网络(gnn)机器学习架构来执行从基于图形的表示到化学性质的不同集合的机器学习任务。根据一些实施例的gnn架构可以包括至少两个消息传递层。若干实施例可以包括gnn架构中的三个消息传递层。在多个实施例中,orbnet过程可以利用分子系统的图形表示来形成一般的化学性质分类。
[0065]
在若干实施例中,在机器学习回归过程中利用orbnet模型的可迁移性,机器学习回归过程利用被迁移到一般分子性质的预先训练的基于能量的模型。若干实施例利用图形神经网络(gnn)的回归训练。根据一些实施例的gnn可以包括消息传递层和解码功能。在某些实施例中,消息传递层可以通过对隐藏节点特征和边特征使用聚合函数来实现。在多个实施例中,解码功能可以通过对变换的节点属性使用求和函数来实现。许多实施例规定,解码功能可以使用图形读出函数来实现,包括(但不限于):对变换的边属性的求和、全局图形池函数和递归神经网络。根据本发明的许多实施例的orbnet过程可以支持基于几何运算的多类读出函数。若干实施例在orbnet过程中实施多任务学习以提高学习效率。在多任务学习期间,根据一些实施例,可以用分子能量和根据一些实施例的量子力学波函数的其他计算性质两者来训练根据一些实施例的orbnet过程。在若干实施例中,orbnet过程可以用包括(但不限于)溶剂化能的实验测量的量来训练。此外,随着生成的量子模拟数据量的增加,根据本发明的许多实施例的orbnet过程可以基于新数据主动更新底层orbnet模型,而不需要使用原始训练数据语料库进行再训练。
[0066]
许多实施例在orbnet过程中实施深度学习架构,以用于学习化学性质。根据若干实施例的orbnet过程实施量子力学分子表示和规范对称性。若干实施例基于紧束缚近似波函数和原子轨道(ao)构建分子表示。一些实施例规定,基于ao的分子表示更好地编码了物理先验,并且是无限可微分的。在许多实施例中,通过将orbnet公式化为作用于紧束缚量子算符的等变映射,具有基于ao的特征的orbnet过程整合了量子相互作用中的规范对称性。在多个实施例中,具有基于ao的特征的orbnet过程实施o(3)-协变嵌入和相互作用块,以参数化等变映射,从而基于ao进行学习,并且避免手动固定参考系统。根据一些实施例的具有基于ao的特征的orbnet过程从量子算符而不是中的向量获取输入,这不同于基于点云的等变网络。某些实施例规定,具有基于ao的特征的orbnet过程相对于通过跟踪球面张量的奇偶性的非方向保持变换是等变的,这在se(3)等变神经网络中可能没有被正确处理。许多等变神经网络中存在的表达能力限制可以通过根据许多实施例的归一化方案repnorm来减轻。若干实施例利用repnorm归一化方案来在orbnet设置和/或其他等变网络中获得更鲁棒的学习。
[0067]
根据本发明若干实施例的orbnet过程可以提高量子模拟的效率和准确度。在多个实施例中,由orbnet过程生成的输出性质是可迁移的,并且因此可以用于确定不同分子系统的分子。在一些实施例中,orbnet过程具有跨分子几何结构的可迁移性。若干实施例实施在分子家族内具有可迁移性的orbnet过程。一些实施例实施提供跨成键环境的可迁移性的orbnet过程。某些实施例实施提供跨化学元素的可迁移性的orbnet过程。在若干实施例中,orbnet在相同数据量的情况下提供了大约33%的预测准确度改进。在许多实施例中,orbnet过程提供了类似于dft的预测准确度,但是计算成本相对于dft方法降低了至少三个数量级。
[0068]
许多实施例实施orbnet过程跨分子系统的化学可迁移性,并且因此能够标识具有广泛性质的分子。具有特定分子系统性质的分子可以使用根据本发明各种实施例的过程合成,以用于广泛的产品开发过程,诸如药物发现和材料设计。这样的实施例的示例包括(但不限于)可以用于以下的过程:有机发光二极管材料设计、催化剂设计、酶反应和药物设计、蛋白质和抗体设计、有机材料设计、纳米材料设计和/或电池、化学和石油工业的材料设计。
[0069]
下面进一步详细讨论根据本发明的各种实施例的用于实施orbnet过程的系统和方法。
[0070]
量子化学中的机器学习
[0071]
用于分子的机器学习大多将分子系统编码为图形或点云,而缺乏关于其量子相互作用的基本信息。在基础层面上,化学可以用玻恩-奥本海默多体薛定谔方程来描述:
[0072][0073]
其中,ψ(re;r)是电子位置re和原子核位置r处的波函数,并且e(r)是分子系统的能量。概念上,eq.1可以用来模拟化学反应,但量子关联使其成为一个难以解决的问题。诸如密度泛函理论(dft)的近似数值方法会遭受惩罚性缩放和速度-准确度权衡,这对于诸如药物发现的大规模应用可能是不切实际的。势能面是分子和材料建模中的一个感兴趣的中心量。在化学、生物和材料系统中足够准确地计算这些能量可以在dft层面上充分地描述。然而,由于其成本相对较高,至少与力场和半经验量子力学理论相比,dft的应用限于相对较小的分子或适度的构象采样。量子化学的机器学习(ml)的主要焦点是提高预测分子
和材料系统势能的效率,同时保持准确度。尽管这样的方法在预测各种基准的能量方面取得了成功,但是深度神经网络模型在化学空间和不平衡几何结构中的可推广性被较少调研。从eq.1的稳态解推导的量,例如e(r),可以被学习来解决这一挑战。
[0074]
经验近似e(r)的问题被认为是确定分子的力场。虽然构建力场需要在设计其功能形式方面的广泛领域专业知识,但已经提出了机器学习方法,以使用手工制作的特征或基于距离信息的图形神经网络,以及最近的广义几何信息,以更高的灵活性从数据中进行近似e(r)。然而,这样的经验方法将分子视为原子核坐标(ψ(re;r)中的r)的(经典)点云,因此不知道电子(ψ(re;r)中的re)所携带的量子力学相互作用。另一方面,以前专注于构建具有量子力学签名的分子表示的工作在某些任务上显示了可观的准确度,但它们大多需要类似于dft的数值计算成本来获得表示,并且一些可能需要计算密集型特征处理来加强对称性。
[0075]
量子化学中的先前工作集中在基于原子特定特征或几何特定特征以及基于核的或神经网络机器学习架构来预测电子能量或密度。(参见,例如,j.s.smith等人的chem.sci,2017,8,3192-3203;l.zhang等人的phys.rev.lett.,2018,120,143001;m.rupp等人的phys.rev.lett.,2012,108,58301;k.hansen等人的j.chem.theory comput.,2013,9,3404;其公开内容通过引用整体并入本文中。)最近的研究集中在抽象表示中分子的特征化,诸如从低成本电子结构计算中获得的量子力学性质,以及利用基于图形的神经网络技术来提高可迁移性和学习效率。(参见,例如,m.welborn等人的j.chem.theory comput.,2018,14,4772-4779;l.cheng等人的j.chem.phys.,2019,150,131103;k.yang等人的j.chem.inf.model,2019,59,3370-3388;j.klicpera等人的international conference onlearning representations,2020;其公开内容通过引用整体并入本文中。)
[0076]
基于来自平均场层面(即hf理论或dft)电子结构计算的量子力学特征,已经开发了若干用于预测高级(即耦合簇)相关能量的ml方法。miller等人的美国专利申请第2020/0294630号描述了一种基于分子轨道的机器学习(mob-ml)方法,该方法使用定域分子轨道以生成输入特征来预测分子性质,其应用包括基于来自平均场参考理论的信息来预测相关波函数性质。
[0077]
在mob-ml中,定域分子轨道是经由轨道定域程序(诸如boys、ibo等)获得的,其中轨道是通过平均场电子结构计算获得的。然后根据分子轨道的矩阵元素相对于基内的各种算符(即fock、coulomb和交换算符)并使用特征排序方案,计算对角和非对角分子轨道对的特征向量。为与mob特征向量相关联的成对相关能量标签训练高斯过程或基于聚类的回归器。相比之下,根据许多实施例的orbnet过程使用ao来评估用于特征生成的算符的矩阵元素,并且采用gnn方案来对以下性质执行回归:包括(但不限于)saao解析性质(诸如saao对对相关能量的贡献)的ao解析性质,包括(但不限于)药物毒性、结合亲和力、pka、相关能量、平均场能量的全分子性质,包括(但不限于)部分电荷、fukui反应性、质子亲和力的原子解析性质,和/或包括(但不限于)键离解能、键级的键解析性质。
[0078]
在mob-ml中,lmo是使用迭代轨道定域程序生成的,该迭代轨道定域程序可以包括一系列o(n3)运算,因此妨碍了使用半经验方法和在大分子系统上的特征生成效率。在许多实施例中,orbnet过程允许使用比dft快1000倍的近似量子力学模型来构建表示并且将物理对称性公式化到神经网络架构设计中。根据若干实施例的orbnet过程使用saao进行特征
化,这可以在单次o(n)块对角化运算中获得,解决了当采用廉价的电子结构方法进行特征生成时的计算瓶颈。相比之下,许多实施例规定,通过消除单次o(n)块对角化运算,使用ao进行特征化的orbnet过程可以执行得更快。
[0079]
neuralxc(参见,例如,s.dick等人的machine learning accurate exchange and correlation functionals of the electronic densi,2019;其公开内容通过引用整体并入本文中)和deephf(参见,例如,ychen等人的groundstate energy functional with hartree-fock efficiency and chemical accuracy,2020;其公开内容通过引用整体并入本文中)是采用从电子结构计算中获得的基于ao的特征来执行分子能量的回归和预测的机器学习技术。neuralxc和deephf都依赖于使用cc-pvdz或更大的原子轨道基集从hartree-fock(hf)(在deephf中)或低级密度泛函理论(dft)(在neuralxc中)计算中获得的电子密度和轨道。两个模型都学习低级计算与高级(诸如,ccsd(t))参考能量之间的残差项。两个模型可能需要与高级(诸如,ccsd(t))预测相关联的ao基集相同(或比其更大)的ao基集用于平均场计算。neuralxc和deephf都不允许预测直接从最小ao基平均场计算获得的特征的大ao基集结果。相反,根据许多实施例的orbnet过程允许使用最小ao基计算(计算成本大大降低)用于特征生成。在若干实施例中,在具有和不具有到其他基集或轨道子空间的投影的情况下,orbnet过程包括使用除最小基集之外的ao基集,这在构建特征的方式方面与deephf不同。
[0080]
在neuralxc和deephf中,ao集或准ao集被用于生成机器学习的特征。neuralxc没有对不同原子或原子内不同量子数(主量子数或角量子数)壳层之间的相互作用进行特征化。例如,neuralxc在构建特征中使用来自平均场(dft)计算的密度矩阵的对角元素。deephf在构建特征中也使用来自平均场(hf)计算的密度矩阵的对角元素,并且在一些情况下包括不同原子上的量之间的相互作用。deephf不包括同一原子上不同壳层之间的相互作用,并且其引入了对基于原子间距离的预定加权函数的需求。
[0081]
相比之下,与现有方案相比,orbnet过程通过构建可以具有更丰富的信息。与neuralxc不同,壳层平均不需要在orbnet过程中执行。此外,与neuralxc和deephf两者相反,一些实施例规定,orbnet过程包括特征内的所有非对角算符矩阵元素(包括原子内和原子间元素,以及壳层内和壳层间元素),从而保留了信息内容并且使得能够描述远距离贡献。与deephf相比,根据本发明某些实施例的orbnet过程可以包括同一原子上不同壳层之间的相互作用,并且避免了对基于原子间距离的预定加权函数的需求。在多个实施例中,orbnet过程包括量子化学矩阵,包括fock(f)矩阵、coulomb(j)矩阵、交换(k)矩阵、密度(p)矩阵、核心hamiltonian(h)矩阵和/或重叠(s)矩阵,它们可以是能量预测任务的重要组成部分。neuralxc和deephf方法两者都没有被应用于基于低级半经验方法(诸如gfn-xtb)的dft质量结果的预测。
[0082]
在特征内如何实施旋转不变性的方式上出现了其他差异。在neuralxc中,可以通过对ao投影密度的所有子壳层分量(诸如,局部密度矩阵的轨迹)求和来保证特征的旋转不变性,从而不保留信息内容。在deephf中,可以通过使用局部密度矩阵的特征值而不是轨迹来为每个壳层构建特征向量来强迫执行特征的旋转不变性。相比之下,许多实施例规定,orbnet过程可以通过使用saao或通过使用ao结合旋转等变神经网络架构来实现特征的旋转不变性,这不涉及信息内容的损失。
[0083]
许多实施例规定,orbnet过程实施与neuralxc和deephf不同的机器学习方法。对于neuralxc,使用behler-parrinello型神经网络进行机器学习回归,其中标签与壳层上的单体求和相关联,以产生用于特征的理论层面与用于预测的理论层面之间的总能量差,即e
ccsd(t)-e
pbe
,其中,pbe指的是perdew-burke-ernzerhof密度泛函。对于deephf,使用密集神经网络进行ml回归,其中标签与壳层上的单体求和相关联,以产生总相关能量。
[0084]
相反,根据许多实施例的orbnet过程将gnn用于机器学习回归。某些实施例使用多头图形注意力机制和/或执行者注意力机制以及残差块来提供结果,这极大地提高了模型的表示能力,以学习复杂的化学环境。与deephf中预先调整的聚合系数不同,orbnet过程还为学习轨道相互作用提供了一个灵活的框架,并且可以自然地迁移到下游任务。
[0085]
若干实施例规定,与neuralxc和deephf相比,orbnet过程具有更好的推理和训练效率。在neuralxc和deephf中,可能需要大基集scf计算来获得高保真特征值。根据一些实施例的orbnet过程可能只需要scf的最小基以达到预测的化学准确度,这可以使特征生成加速大约100倍到大约1000倍。
[0086]
在许多实施例中,orbnet过程可以使用来自最小基hf计算的输入特征来提供相关能量的准确预测。一些实施例规定,对于给定相同量的训练数据的ccsd(t)相关能量的预测,orbnet方法可以比deephf准确大约10倍。
[0087]
在若干实施例中,orbnet过程可以比deephf和neuralxc提供更好的可迁移性。对于deephf,与根据实施例的orbnet过程相比,跨不同有机分子(qm7b-t数据集)的可迁移性显示出低得多的预测准确度。当在7重原子有机分子(qm7b-t数据集)上训练和在更大的13重原子有机分子(gdb13-t数据集)上测试时,orbnet过程表现出比deephf和neuralxc更好的预测准确度,并且提供了很好的可迁移性。
[0088]
下面进一步讨论根据本发明的各种实施例的用于合成具有特定分子系统性质的分子的系统和方法以及可以在分子的设计和/或合成中使用的基于原子轨道的机器学习(orbnet)过程。
[0089]
基于原子轨道的机器学习过程
[0090]
许多实施例使用包括(但不限于)自洽场计算的计算,基于输入特征,利用准确且可迁移的orbnet过程来预测包括(但不限于)相关波函数能量的性质。图1示出了根据本发明的实施例的使用orbnet过程合成分子的方法。过程100可以从获得分子系统数据集(101)开始。一些实施例包括输入数据集,该输入数据集包括具有相同元素的分子。在多个实施例中,输入数据集可以包括具有不同类型分子键的分子。在若干实施例中,输入数据集可以包括具有不同几何结构的分子。一些实施例包括输入数据集,这些输入数据集包括相同元素的不同组成。在许多实施例中,数据集可以包括不同的分子和元素。如可以容易地理解的,根据本发明的各种实施例,可以适当地根据具体应用的要求利用各种输入数据集中的任何一种。
[0091]
可以基于原子轨道来获得输入数据集的基于原子轨道(基于ao)的特征集(102)。在若干实施例中,基于ao的特征包括(但不限于)基于ao的特征集、基于(但不限于)对称性适应原子轨道(saao)的特征集、ao集的导数和/或saao集的导数。在一些实施例中,ao特征可以包括(但不限于)分子系统的量子算符。在若干实施例中,输入的基于ao的特征可以包括(但不限于):fock(f)矩阵的元素、coulomb(j)矩阵的元素、hartree-fock交换(k)矩阵的
元素、密度(p)矩阵的元素、轨道质心距离(d)矩阵的元素、核心hamiltonian(h)矩阵的元素和/或重叠(s)矩阵的元素。许多实施例规定,可以用kohn-sham密度泛函理论计算量子算符,包括(但不限于):交换相关算符、交换相关算符的近似和交换相关算符的分量。若干实施例规定,可以用密度泛函紧束缚理论计算和/或其他半经验电子结构理论方法(例如gfn1-xtb)来计算量子算符,包括(但不限于):壳层解析电荷和j、k、f、p、d、h、s的近似和/或交换相关算符。若干实施例包括量子算符可以是分子系统的性质。这些性质的示例包括(但不限于):偶极矩、原子间距离矩阵和/或连续溶剂化能。许多实施例实施包括(但不限于)图形神经网络的神经网络,以参数化矩阵,包括(但不限于)fock(f)矩阵、coulomb(j)矩阵、hartree-fock交换(k)矩阵、密度(p)矩阵、轨道质心距离(d)矩阵、核心hamiltonian(h)矩阵和重叠(s)矩阵,以生成基于ao的特征。如可以容易地理解的,可以适当地根据具体应用的要求利用各种输入的基于ao的特征中的任何一种。
[0092]
在某些实施例中,使用orbnet过程来执行量子化学计算(103)。在多个实施例中,可以在本地计算设备上执行计算。在若干实施例中,在远程服务器系统上执行计算。orbnet过程可以使用输入数据集的基于ao的特征进行训练。
[0093]
在训练过程(未示出)期间,orbnet过程可以使用训练数据集来学习基于ao的特征和分子系统的性质之间的关系。在一些实施例中,训练数据集可以是从输入数据集中随机选择的子集。在这样的实施例中,分子数据集的示例可以包括(但不限于):qm7b、qm7b-t、qm9、gdb-13、gdb-13-t、drugbank、drugbank-t、chembl27、jsch-2005、生物片段数据库的侧链-侧链相互作用子集、md17和bfdb-ssi。在若干实施例中,训练数据集可以是来自相同或不同分子系统的分子集。如可以容易地理解的,根据本发明的各种实施例,可以适当地根据具体应用的要求利用各种训练数据集中的任何一种。
[0094]
orbnet过程可以利用描述基于ao的特征与分子系统的性质之间的关系的训练模型来至少对输入数据集中的分子进行排序和/或分类(104)。在许多实施例中,orbnet过程还可以基于包含模型预测将具有期望性质的分子的特征空间的区域来标识新分子和/或不在输入数据集中的分子。下面进一步讨论根据包括具体示例的本发明的各种实施例的orbnet过程可以用于标识具有期望性质的分子系统的各种方式。
[0095]
在许多实施例中,训练后的orbnet过程生成分子系统性质的输出数据集(105)。分子系统性质可以包括(但不限于):(1)分子的可计算性质,诸如多体薛定谔方程的解,包括基态和/或激发态平均场能量、基态和/或激发态多体关联能量、势能面、总和/或相对构象能、电子能量、关联能量、ao对和/或saao对贡献、平均场能量、单点能量、分子轨道能量、热性质、力、原子间力、振动频率(hessian)、偶极矩、电子密度、激发态能量、线性响应激发态和力、电子光谱、旋转光谱、核共振光谱和/或振动光谱;和(2)分子的实验可测量的性质,诸如活性系数、溶解度、pka、ph、分配系数、蒸汽压、熔点、沸点、闪点、溶剂化自由能、氧化还原势能、电导率、离子电导率、热导率、光吸收频率、光吸收强度、光吸收效率、粘度、adme性质、毒性、药物毒性、结合亲和力和/或蛋白质结合亲和力。多个实施例实施ao和/或saao特征的导数作为输入,并且能够预测响应性质,包括(但不限于):力、优化的几何结构、原子间力、偶极子和/或线性响应激发态。如可以容易地理解的,用作分子系统性质的具体特征在很大程度上仅局限于具体应用的要求。基于输出数据集,可以标识和合成具有期望分子系统性质集的分子(106)。
[0096]
虽然上文参考图1描述了使用orbnet过程合成化学品的各种过程,但是根据本发明的各种实施例,利用机器学习来估计分子系统的性质的各种过程中的任何一种都可以根据具体应用的要求用于化学品的设计和/或合成。例如,分子系统可以在利用生成orbnet过程的过程中合成,以使用类似于下面讨论的那些技术来标识具有满足某些标准的分子性质的分子系统。下面进一步讨论根据本发明的各种实施例的设计具有所需性质的分子的过程。
[0097]
确定分子结构
[0098]
在许多实施例中,orbnet过程实现实时化学建模和设计,并且提供可以用于以协作方式执行这些活动的平台。在若干实施例中,orbnet过程在可以在本地计算机或远程服务器上执行的软件包中实施。附加地,根据一些实施例的软件包可以对许多可能的化学改进进行计算,并且返回最有可能的化学修改的排序推荐。通过并行计算,所有结果都可以在几秒钟内返回。以这种方式,可以执行类似于以上所描述的用于设计分子系统的各种过程的过程,并且结果用于生成直观的和交互式的图形用户界面,该图形用户界面使得各种实验化学家能够在化学物质的设计和/或合成中利用orbnet。
[0099]
图2概念性地示出了可以由软件使用根据本发明的实施例实施的ml过程生成的用户界面。在许多实施例中,该软件可以使任何实验化学家,而不仅仅是专家计算化学家,能够标识具有期望化学性质的分子系统。例如,可以为软件实施用户界面,其能够使各种实验化学家中的任何一种进行分子系统的设计和合成,这些实验化学家包括(但不限于):药物化学家、合成化学家、材料科学家和/或生物化学家。
[0100]
虽然上文参考图2描述了使用orbnet过程设计分子的各种过程,但是根据本发明的各种实施例,利用机器学习来估计分子系统的性质的各种过程中的任何一种都可以根据具体应用的要求用于化学品的设计和合成。下面进一步讨论根据本发明的各种实施例的用于执行基于ao的特征生成的过程。
[0101]
分子的原子轨道表示
[0102]
薛定谔方程(eq.1)可以用来模拟化学反应,但量子关联可以使其成为一个难以解决的问题。诸如dft的近似数值方法会遭受惩罚性缩放和速度-准确度权衡,这对于大规模应用可能是不切实际的。从eq.1的稳态解推导的量,例如e(r),可以被学习来解决这一挑战。经验近似e(r)的问题被认为是确定分子的力场。虽然构建力场可能需要在设计其功能形式方面的广泛领域专业知识,但已经提出了机器学习方法,以使用手工制作的特征或基于距离信息的图形神经网络以及最近的广义几何信息,以更高的灵活性从数据中近似e(r)。经验方法将分子视为原子核坐标(ψ(re;r)中的r)的(经典)点云,并且不知道电子(ψ(re;r)中的re)所携带的量子力学相互作用。另一方面,专注于构建具有量子力学签名的分子表示的工作可能需要类似于dft的数值计算成本来获得表示,并且一些可能需要计算密集型特征处理来加强对称性。
[0103]
在存在对称先验的情况下,等变性已经被提出作为深度学习的统一概念。利用
″
baked-in
″
对称性,等变神经网络已经被引入到均匀网格和欧几里得数据中,并且可以在几何或基于网格的观察和高能物理问题的背景下被推广到流形的规范对称性。一些方法可以应用于分子建模,其专注于3d旋转对称;而架构是为经典的基于点云的分子表示而设计的。
[0104]
许多实施例在orbnet过程中实施深度学习架构,以用于学习具有量子力学分子表
示和规范对称性的化学性质。若干实施例基于紧束缚近似波函数和原子轨道构建分子表示,其更好地编码了物理先验并且是无限可微的。许多实施例实施原子轨道中表示的量子算符的规范不变性。具有基于ao的特征的orbnet过程实施o(3)-协变嵌入和相互作用块,以参数化等变映射,从而基于原子轨道进行学习,并且避免手动固定参考系统。根据本发明的一些实施例的具有基于ao的特征的orbnet过程从量子算符而不是中的向量获取输入,这不同于基于点云的等变网络。某些实施例规定,具有基于ao的特征的orbnet过程相对于通过跟踪球面张量的奇偶性的非方向保持变换是等变的,这在se(3)等变神经网络中可能没有被正确处理。许多等变神经网络中存在的表达能力限制可以通过归一化方案来减轻,诸如(但不限于)根据本发明的许多实施例在orbnet过程中使用的repnorm。若干实施例规定,repnorm归一化方案可以在orbnet设置中产生更鲁棒的学习,并且可以应用于其他等变网络。
[0105]
代替仅依赖于核位置信息r直接学习e(r),若干实施例对从可以以低计算成本获得的近似波函数ψ0(re;r)学习目标性质y的泛函进行参数化:
[0106][0107]
对于分子系统,ψ0(re;r)可以用原子轨道和量子算符来表示。提供了与量子力学中使用的符号惯例相交的形式符号。
[0108]
狄拉克的bra-kets定义:设v是上的一个希尔伯特空间,其中,u,v∈v,它们的hermitian内积表示为《u|v》。|v》是ket,并且《u|是bra。当v是有限维向量空间时,bra《u|可以是行向量,并且ket|v》可以是列向量。在物理学中,|v》可以称为量子态。单电子量子态可以在实空间中使用,其中,希尔伯特空间是平方可积函数:的函数空间。内积由给出,其中,u
*
(r)表示u(r)的复共轭。
[0109]
原子轨道采用泛函形式其中,ra是原子a的原子核位置,za表示原子a的原子序数,称为不依赖于r-ra方向的径向函数,并且y
lm
是秩l和度m的球谐函数。在量子力学中,索引n,l,m是主量子数、角量子数和磁量子数。对于类氢原子,具有的某些形式的被称为eq.1的精确波函数解,并且对于分子系统,它们可以用作以数值表示多电子波函数的基函数。在大多数情况下,原子轨道的集合既不是相互正交的,也不是v的完整基,而是作为ψ(re;r)的计算上易处理的表示基。
[0110]
量子算符的定义:(单电子约化)量子算符是在上定义的自伴线性算子。表示作用于ket向量的量子算符。给定ket集{|φi》},是的矩阵表示。对于由分子的原子轨道给出的ket集,用速记符号表示中的的矩阵表示。
[0111]
该分子可以由通过基于给定分子的原子轨道的近似波函数ψ0(re;r)建立的量子算符的矩阵表示来表示。可以被构建为从而将在原子轨道中表示的映射到目标y,并且θ可以从数据中确定。是ao分子表示,用作的输入信号。近似量子算符可以经由紧束缚hamiltonian高效地计算。生成
所需的计算时间比获得基础真值(例如dft计算)至少低1000倍,并且是无限可微的。
[0112]
提供了ao分子表示中的等变性。给定变换g,如果则映射f被称为等变的。构建以正确地描述分子系统中的物理对称性可能需要映躬在某些规范变换下是等变的。ao分子表示上的规范变换由应用于原子坐标r的平移旋转反演应用于ψ0,的轨道相位变换和应用于|φa》的局部规范变换ga组成。可以看出,ao分子表示在构建上对和ga是不变的,并且没有信息内容的损失,但是由(即o(3))生成的全局规范变换需要在的公式中明确处理。和对的作用可以基于群表示理论获得,以解决后者的o(3)对称性。
[0113]
提供了作用于ao分子表示的引理1o(3)。对于全局旋转和全局反演对的作用由下式给出
[0114][0115][0116]
其中,a、b两者都是原子索引,并且称为wigner-d矩阵,已知用于变换给定旋转的球谐函数y
lm
。
[0117]
学习原子轨道相互作用
[0118]
若干实施例提供了规范等变映躬的构建。一些实施例实施作用于ao分子表示的o(3)-协变神经网络层。在一些实施例中,在公式中,
″
局部
″
块可以是并且
″
非局部
″
块可以
[0119]
某些实施例对球形原子嵌入实施wigner-eckart。因为原子a仅局部
″
看到
″oaa
而没有来自周围原子的几何约束,所以一些实施例提取不依赖于|φa》的取向的特征而没有信息损失。利用wigner-eckart定理的对应关系:
[0120][0121]
其中,是秩l和度m的不可约球面张量算符,定义为是秩l和度m的不可约球面张量算符,定义为其中,中,表示不依赖于m1,m2和m的旋转不变标量值。是clebsch-gordan(cg)系数,已知其用于将so(3)中表示的张量积与其不可约表示联系起来。形成任意球面张量算符的正交基,产生展开式在没有索引a和n的情况下,给定γ血可以被恢复为所需的独立于基的嵌入。保持这种动机,对于根据某些实施例的原子嵌入可以通过利用辅助基集来获得:
[0122][0123]
其中,被构建为高斯函数和球谐函数的乘积,并且基重叠系数
可以被分解为标量常数和cg系数;使用eq.5和cg系数的恒等式关系,可以表明在每一个(n,l)下的ha都是在旋转下被协变变换为的球面张量。p∈{0,1}是用于跟踪球面张量的奇偶性的索引;在反演下,具有偶数(p+l)的张量是不变的,但是具有奇数(p+l)的张量翻转了其符号。
[0124]
一些实施例提供了共变的原子轨道线性组合。非局部块o
ab
编码了以原子核位置ra和rb为中心的原子轨道之间的相互作用。由于原子轨道|φa》和|φb》在空间上是分离的,所以o
ab
不能像为o
aa
所做的那样分解成更简单的成分。某些实施例提供了基于张量收缩在o
ab
上学习的物理激励方案。为了对原子中心的属性进行更新可以为每对原子(a,b)学习根据一些实施例的规范张量集
[0125][0126]
其中,是原子中,心a与b之间的笛卡尔方向向量,||
·
||表示取球面张量的规范不变内容,若干实施例规定,和是可学习的线性函数。eq.7是球面张量和上的线性映射,则其遵循在o(3)的作用下也是球面张量协变的。由于相同秩的两个球面张量的内积是o(3)-不变标量,通过由组合索引(na,la,ma)形成的oab的bra-维收缩产生了在其ket-空间中定义的新的球面张量,即根据若干实施例的消息张量
[0127][0128]
将上述可学习的运算应用于eq.7和eq.8相当于在由原子轨道跨越的希尔伯特空间中的线性投影:
[0129][0130]
其中,是原子轨道的线性组合(lcao),是一个投影算符,去除了已经通过eq.6捕获的自相互作用的贡献。因此是原子轨道(bra侧)和lcao(ket侧)的混合基中的量子算符的期望值。eq.7被称为lcao层。
[0131]
多个实施例为ao-lcao相互作用提供消息传递。可以被聚合用于更新原子中心a上的表示,类似于图形神经网络实现中的节点与边之间的消息传递。一些实施例通过球谐函数结合了原子位置r的经典几何信息,并将其与耦合,通过以下提出的o(3)-协变消息传递方案给出:
[0132][0133]
其中,是可学习的线性函数,是用于提高网络容量的标量值权重,其被参数化为多头注意力:
[0134][0135]
其中,
[0136]
并且是在所有更新步骤t中共享的标量权重,其中,ξk是morlet小波径向基函数。mlp表示2层多层感知器,w
κ
是可学习的线性函数,并且na表示注意力头的数量(即的长度)。注意力机制eq.11与显式扩展o
ab
相反,在不增加存储器成本的情况下提升了通道宽度限制,并且这与se(3)-transformer中的注意力相一致。许多实施例规定,聚合的等变消息将通过等变相互作用块与进行相互作用以完成更新
[0137]
广义等变非线性
[0138]
许多实施例提供了归一化方案来缓解等变神经网络中的表达能力问题。等变神经网络可能在处理对称先验方面是成功的,但是许多实现显示出对非线性的限制。直接在球面张量(例如向量的xyz分量)上应用激活函数(诸如relu)可能会违反等变性。这个问题也存在于基于点云的等变分子神经网络中,并且可以在一些架构中通过对特征l>0应用由标量特征参数化的门控运算来缓解。然而,这样的方法可能无法与已知可改进学习的技术(诸如批归一化)相结合,并且可能对实际中建立神经网络训练提出挑战,诸如对权重初始化的敏感性。
[0139]
若干实施例实施归一化方案,包括(但不限于)球面张量上的repnorm,以缓解表达能力问题。给定球面张量x,repnorm可以被定义为:其中,和由下式给出:
[0140]
和
[0141]
其中,和是不变内容||x||的均值和方差估计值,其可以从批统计或层统计中获得;β
n,l,p
是正的、可学习的标量,其控制x中的张量尺度信息的部分在中保留,并且∈是在根据某些实施例的实施方式中被设置为10-3
的数值稳定性因子。eq.12中的repnorm运算将球面张量x分解为允许由标量nn变换的归一化标量值张量以及可以在以后重新组合以完成更新x的
″
纯规范
″
张量在eq.12中,0始终是映躬的固定点,并且没有明确触及x中的方向信息;因此,repnorm可以保持等变性,并且不会引入诸如非物理对称性破缺的伪影。根据一些实施例的repnorm提高了训练稳定性,并且消除了对跨不同任务手动调整权重初始化和学习率的需要。
[0142]
基于原子轨道的特征生成
[0143]
许多实施例通过基于ao的低成本电子结构计算实施特征。一些实施例包括可以用于生成基于ao的特征的各种过程。根据若干实施例的orbnet过程中的基于ao的特征可以通过平均场方法来确定。在某些实施例中,可以使用(但不限于)hartree-fock理论、密度泛函理论或半经验理论来计算orbnet过程中的基于ao的特征。这些方法的多个中心对象包括(但不限于)fock(f)矩阵、密度(p)矩阵和重叠(s)矩阵。根据某些实施例,这些矩阵可以通过执行平均场计算从分子几何结构中确定。若干实施例实施矩阵来确定orbnet过程的基于ao的输入特征。
[0144]
许多实施例提供了端到端框架来为orbnet过程生成基于ao的特征。在一些实施例
中,fock矩阵可以由包括(但不限于)图形神经网络(gnn)的神经网络来参数化。这些实施例避免使用平均场计算。某些实施例规定,fock矩阵参数化:
[0145]
f=dec[gnn(r,z)]
ꢀꢀꢀꢀ
(13)
[0146]
其中,r是分子中原子的核坐标,z是分子中原子的原子序数,dec是解码模块。若干实施例规定,gnn的节点对应于原子,并且边对应于原子间的相互作用。fock矩阵的元素是:
[0147][0148]
其中,μ和v索引ao基函数,并且l(μ)是对应于基函数l的总角动量,并且h(μ)[gnn(...)]是对应于基函数μ以其为中心的原子的节点表示。
[0149]
根据一些实施例,解码器的形式是多层感知器(mlp)。它由一对ao角动量来索引。在若干实施例中,它可以被实施为mlp集,其中每个角动量对具有一个mlp。在某些实施例中,它可以被实施为单个多任务mlp,其每个头对应于一角动量。若干实施例代表sto-6g基集中的量子力学矩阵。许多实施例规定,可以独立于orbnet模型或结合orbnet模型来训练gnn。
[0150]
许多实施例规定,可以从fock矩阵确定orbnet特征。在一些实施例中,密度矩阵可以通过对角化fock矩阵来确定:
[0151]
fc=sc
∈
ꢀꢀꢀꢀꢀ
(15)
[0152][0153]
其中,n
elec
/2是分子中的电子数,并且*表示复共轭。
[0154]
如可以容易地理解的,可以为ao评估各种运算中的任何一种,其可以用作输入的基于ao的特征,并且可以根据具体应用的要求选择各种输入的基于ao的特征中的任何一种。
[0155]
基于原子轨道特征的orbnet
[0156]
许多实施例提供等变相互作用块作为模块化组件以构建从而在给定另一球面张量ga(例如eq.10中的或本身)与相互作用的情况下执行更新
[0157]
其中,
[0158][0159]
其中,
[0160]
其中,是kronecker增量函数,并且mlp1和mlp2表示多层感知器。为了计算效率,在eq.18中的奇偶感知球面张量耦合中,根据一些实施例的角动量索引(l1,l2)被限制在该范围{(l1,l2);l1+l2<l
max
)内,
[0161]
其中,l
max
是实施方式中考虑的最大角动量。
[0162]
一旦表示被更新到最后一步根据若干实施例的池化操作根据若干实施例的池化操作可以被用来读出目标预测由于orbnet模型公式中的规范等变性,学习任务的物理先验可以通
过设计池化方案来灵活地解决,而无需修改模型架构。可以基于的代数结构以及y是广泛的还是密集的性质,为量子化学性质的代表性类别定义池化运算。这种设置能够学习具有挑战性的张量性质,包括(但不限于)偶极矩和电子密度。
[0163]
一些实施例通过堆叠nn构建块,组装orbnet模型,即图3示出了根据本发明的实施例的用于基于ao的特征的orbnet架构的顶层视图。
[0164]
基于对称性适应原子轨道的特征生成
[0165]
许多实施例通过基于saao的低成本电子结构计算实施特征。许多实施例包括可以用于生成saao特征的各种过程。在若干实施例中,saao可以从分子系统和/或其他外部势的变换的原子轨道基的集合和/或子集推导。某些实施例规定,saao可以经由原子轨道表示中的分子系统的约化密度矩阵获得。在多个实施例中,saao可以经由基于原子轨道表示中的fock矩阵的特征值和/或wigner旋转的方案来获得。若干实施例规定,saao特征可以是从量子算符的期望值和/或量子算符的期望值相对于saao的导数推导的标量和/或张量。量子算符的示例包括(但不限于):fock(f)矩阵的元素、coulomb(j)矩阵的元素、hartree-fock交换(k)矩阵的元素、密度(p)矩阵的元素、轨道质心距离(d)矩阵的元素、核心hamiltonian(h)矩阵的元素和/或重叠(s)矩阵的元素。一些实施例基于(紧束缚)密度泛函理论计算中的量子算符和/或其他半经验电子结构理论方法来实施saao特征,包括(但不限于):壳层解析电荷和j、k、f、p、d、h、s的近似和/或交换相关算符。许多实施例规定,算符可以是kohn-sham密度泛函理论,包括(但不限于):交换相关算符、交换相关算符的近似和交换相关算符的分量。若干实施例包括量子算符可以是分子系统的性质。这些性质的示例包括(但不限于):偶极矩、原子间距离矩阵、连续溶剂化能。如可以容易地理解的,可以为ao评估各种运算中的任何一种,其可以用作输入的saao的特征,并且可以根据具体应用的要求选择各种输入的saao的特征中的任何一种。
[0166]
在许多实施例中,saao特征集不明确依赖于原子类型,因此orbnet过程可以增强训练结果的化学可迁移性。在若干实施例中,作为不同分子几何结构和不同分子的saao特征的函数的配对相关能量的平滑变化和局部线性可以有利于orbnet过程的可迁移性。
[0167]
许多实施例实施从输入特征值{f}到作为量子力学能的回归标签的可迁移映射,
[0168]
e≈e
ml
[{f}]
ꢀꢀꢀꢀꢀ
(20)
[0169]
若干实施例提供了saao特征的生成。设是ao基函数的集合,具有原子索引a和标准主量子数和角动量量子数n、l和m。设c是从平均场电子结构计算(诸如hf理论、dft或半经验方法)获得的对应分子轨道系数矩阵。ao基中的分子系统的单电子密度矩阵为
[0170][0171]
(对于封闭壳层系统)。通过对角化与索引a、n和l相关联的对角密度矩阵块,可以构建旋转不变的对称性适应原子轨道(saao)基使得
[0172][0173]
其中,对于s轨道(l=0),这种对称化过程可以忽略不计,并且可以跳过。通过构建,saao相对于分子的几何扰动是定域的且与其一致的,并且与通过最小化定域目标函数(pipek-mezey、boys等)获得的定域分子轨道(lmo)相反,saao可以通过一
系列非常小的对角化来获得,而不需要迭代过程。saao特征向量被聚合,以形成指定从ao到saao的完整变换的块对角变换矩阵y:
[0174][0175]
其中,μ和p分别对ao和saao进行索引。
[0176]
若干实施例采用由通过在saao基中评估量子化学算符而获得的张量组成的ml特征{f}。此后,所有的量子力学矩阵都可以用saao基表示,包括fock矩阵(f)、coulomb矩阵(j)和hartree-fock交换矩阵(k)、密度矩阵(p)、轨道质心距离矩阵(d)、核心hamiltonian矩阵(h)和重叠矩阵(s)。
[0177]
许多实施例提供了近似的coulomb和交换saao特征生成。当采用半经验量子化学理论时,由于需要计算四索引电子排斥积分,所以saao特征生成的计算瓶颈变成了j和k项。如在stda-xtb方法中,一些实施例实施mataga-nishimoto-ohno-klopman公式的一般化形式,
[0178][0179]
这里,a和b是原子索引,p,q,r,s是saao索引,并且
[0180][0181]
其中,r
ab
是原子a与b之间的距离,η是原子a和b的平均化学硬度,并且y
{j,k}
是指定阻尼相互作用核的衰变行为的经验参数。在某些实施例中,使用y
{j}
=4和y
{k}
=10。跃迁密度是根据群体分析计算的,
[0182][0183]
其中,y
′
=ys
1/2
的pth列包含对称正交化ao基中的第p saao的展开系数。这产生了用于特征化的近似j矩阵和k矩阵,
[0184][0185][0186]
eq.27和eq.28的简单实施方式是即主要渐近成本。然而,通过紧束缚近似,这种缩放可以减少到准确度损失可以忽略不计。j
mnok
和k
mnok
的计算不是特征生成的主要成本,并且因此不采用这样的紧束缚近似。
[0187]
虽然上面描述了用于为orbnet过程生成saao特征的各种过程,但是根据本发明的各种实施例,能够生成saao特征的各种过程中的任何一种可以根据具体应用的要求用于orbnet过程中。下面进一步讨论根据本发明的各种实施例的用于为具有saao特征的orbnet过程设计图形神经网络模型的过程。
[0188]
基于对称性适应原子轨道特征的orbnet
[0189]
在许多实施例中,orbnet过程提供了对saao基中的特征的有效评估。本发明的多个实施例利用包括(但不限于)图形神经网络(gnn)模型的机器学习模型,这些机器学习模型接收saao特征作为直接输入,并且输出所接收的saao特征的分子性质的估计作为输出。若干实施例规定,orbnet利用具有边和节点注意力和消息传递层的gnn架构,以及预测阶段,以确保所得能量的广泛性。许多实施例利用orbnet过程提供了从半经验质量特征到dft质量标签的特征映射。某些实施例规定,orbnet过程可以在用于特征的平均场方法(即,允许hartree-fock、dft等)中并且在用于生成标签的理论层面(即,允许耦合簇和其他相关波函数方法参考数据)实施。下面进一步讨论根据本发明的不同实施例的orbnet过程可以根据描述分子系统的特征集来估计分子性质的各种方式。
[0190]
许多实施例实施用于saao特征的orbnet,以将分子系统编码为图形结构数据,并利用图形神经网络(gnn)机器学习架构。gnn将数据表示为属性图g(v,e,x,xe),具有节点v、边e、节点属性和边属性和边属性其中,n=|v|,ne=|e|,并且d和e分别是每个节点和边的属性数。图4示出了描述根据本发明的实施例的orbnet过程的工作流程的图表。可以对分子系统进行低成本的平均场电子结构计算(401)。可以构建所得saao和相关联的量子算符(402)。属性图表示(403)可以用对应于saao张量的对角元素和非对角元素的节点和边属性来构建。属性图可以由嵌入层和消息传递层(404)处理,以产生变换的节点和边属性。可以提取编码层和每个消息传递层的变换后的节点属性(405),并且将其传递给mpl特定解码网络(406)。可以通过对解码网络输出逐节点求和来获得节点解析的能量贡献∈u(407),并且可以从节点上的单体求和来获得最终的扩展能量预测(408)。
[0191]
在若干实施例中,orbnet采用分子系统的图形表示,其中,节点属性对应于对角线saao特征xu=[f
uu
,j
uu
,k
uu
,p
uu
,h
uu
],并且边属性对应于非对角线saao特征通过为要包括的边引入边属性截止值,将相距无限远的非相互作用的分子系统编码为不连通图,从而满足大小一致性。
[0192]
可以通过经由径向基函数将非线性输入特征变换引入图形表示来增强模型容量,
[0193][0194][0195]
其中,和是具有预规范化属性的n
×
d和m
×
e矩阵。正弦基函数用于节点嵌入。一些实施例采用0阶球形贝塞尔函数进行边嵌入,
[0196][0197]
其中,c
x
(x∈{f,j,k,d,p,s,h})是的算符特定的截止值。为了确保当节点进入截止时特征平滑变化,一些实施例实施软化子(mollifier)i
x
(r):
[0198][0199]
需要说明的是,当边接近截止时,衰减到零,以确保大小一致性,并且软化子在边界处是无限阶可微的,这消除了可能由分子的几何扰动引起的表示噪声。当添加任意
数量的零边特征时,为了强制输出在机器精度下是恒定的,这对于提取分析梯度和训练势能面是至关重要的,一些实施例实施与消息传递机制相集成的
″
辅助边
″
方案,
[0200][0201]
其中,w
aux
是可训练参数矩阵。径向基函数嵌入由神经网络模块变换以产生0阶节点和边属性,
[0202][0203]
其中,ench和ence是包括3个密集神经网络层的残差块。与根据一些实施例的基于原子的消息传递神经网络相比,这种附加的嵌入变换捕获了物理算符之间的相互作用。
[0204]
节点和边属性经由变换器驱动的消息传递机制进行更新。对于给定的消息传递层(mpl)t+1,每条边携带的信息可以编码成消息函数和相关联的注意力权重并可以通过图卷积运算累积成节点特征。整个消息传递机制由下式给出:
[0205][0206]
其中,是在每条边上计算的消息函数:
[0207][0208]
并且卷积核权重被评估为(多头)注意力分数,以表征轨道对的相对重要性,
[0209][0210]
其中,求和被应用于被加数中的向量的元素。这里,索引j指定单个注意力头,并且ne是隐藏边特征的维度,表示向量级联操作,表示hadamard乘积,并且
·
表示矩阵向量乘积。边属性可以根据下式更新
[0211][0212]
是mpl特定可训练参数矩阵,是mpl和注意力头特定可训练参数矩阵,σ(
·
)是具有归一化层的激活函数,并且σa(
·
)是用于生成注意力分数的激活函数。
[0213]
图5示出了根据本发明的实施例的用于saao的orbnet消息传递层(mpl)的图。对于t+1mpl,由于与最近邻节点(502和503)的相互作用,可以更新给定节点(501)的属性,这取决于最近邻节点属性和最近邻边属性两者。节点和边特征(即和)组合,以产生消息(eq.32)和多头注意力分数(eq.33),它们经历注意力混合。来自每个最近邻节点和边的注意力加权消息被组合并且传递到密集层,其结果被添加到原始节点属性以执行更新(eq.31)。
[0214]
根据若干实施例的orbnet的解码阶段可以被设计成确保能量预测的大小扩张性。所采用的机制输出嵌入层(t=0)和所有mpl(t=1,2,...,t)的节点解析能量贡献,以预测与所有节点和mpl相关联的能量分量。最终的能量预测e
ml
可以通过首先对每个节点u在l上求和,并且然后对节点(即轨道)执行单体求和来获得,使得
[0215][0216]
其中,解码网络dec
t
是多层感知器。
[0217]
许多实施例在orbnet过程中结合了多任务学习策略,以提高学习效率。在若干实施例中,可以用分子能量和量子力学波函数的其他计算性质两者来训练orbnet过程。为了实现多任务学习并且提高orbnet模型的学习能力,若干实施例实施原子特定属性阳全局分子级属性q
t
,其中,t是消息传递层索引,并且a是原子索引。全分子和原子特定属性允许通过多任务学习来预测辅助目标,从而对分子的电子结构提供物理激励约束,物理激励约束可以用于在基于ao的特征层面上细化表示。
[0218]
若干实施例提供了orbnet的分析梯度理论。根据某些实施例的orbnet的分析梯度理论对于计算原子间力和包括(但不限于)偶极子和线性响应激发态的其他响应性质可能是必不可少的。
[0219]
在许多实施例中,对于电子能量和辅助目标两者的预测,仅采用最终的原子特定属性因为它们自洽地结合了全分子和节点特定属性以及边特定属性的影响。电子能量可以通过结合来自扩展紧束缚计算的近似能量e
tb
和模型输出e
nn
来获得,后者是原子贡献的单体和;原子特定的辅助目标da可以从相同的属性中预测。
[0220][0221][0222]
这里,能量解码器dec和辅助目标解码器dec
aux
是用全连接层和归一化层构建的残差神经网络,并且是针对孤立原子对总能量的贡献的元素特定的常数移位参数。
[0223]
许多实施例规定,通过采用输入特征,orbnet过程可以是端到端可微分的,这些输入特征包括(但不限于)基于ao的特征,其是原子坐标和外部场的平滑函数。若干实施例提供了总能量e
out
相对于原子坐标的分析梯度。一些实施例采用关于分子结构的局部能量最小化来证明所学习的势能面的质量。
[0224]
使用拉格朗日形式,预测能量相对于原子坐标x的分析梯度可以关于来自紧束缚模型、神经网络和附加约束项的贡献来表示:
[0225][0226]
这里,右手边的第三和第四项分别是来自轨道正交性约束和brillouin条件的梯度贡献,其中,f
ao
和s
ao
是原子轨道(ao)基中的fock矩阵和轨道重叠矩阵。在一些实施例中,orbnet的分析梯度可以基于紧束缚(gfn-xtb)模型。根据若干实施例的紧束缚梯度可以是紧束缚梯度。在某些实施例中,可以使用反向模式自动微分来获得关于输入特征的神经网络梯度
[0227]
若干实施例实施图形和原子级别的辅助任务,以提高分子的学习表示的可推广
性。一些实施例采用关于总分子能量和原子特定辅助目标的多任务学习。原子特定的目标可以类似于在deephf模型中引入的特征,通过将密度矩阵投影到不依赖于原子元素的标识的基集中而获得,
[0228][0229]
这里,投影的密度矩阵由给出,并且投影的价占据密度矩阵由给出,其中,|ψ
i,j
)是来自参考dft计算的分子轨道,是以原子a为中心的基函数,具有径向索引n和球谐度l和阶m。索引i和j分别在所有占据的轨道和价占据轨道索引上运行,并且||表示向量级联操作。分子中每个原子a的辅助目标向量da是通过级联所有n和l的获得的。
[0230]
虽然上面参考图4和图5描述了用于为saao特征的orbnet过程设计具有消息传递层的图形神经网络的各种过程,但是根据本发明的各种实施例,利用深度学习模型的各种过程中的任何一种都可以根据具体应用的要求用于实施saao特征的orbnet过程的设计。下面进一步讨论根据本发明的各种实施例的用于标识基于ao的特征距离度量的过程。
[0231]
化学空间结构发现
[0232]
根据本发明的各种实施例的过程可以依赖于距离度量的使用,该距离度量测量特征空间中不同分子系统的包括(但不限于)saao特征的基于ao的特征之间的距离。在许多实施例中,通过利用子空间嵌入技术来发现ao特征空间的局部和全局结构,进一步提升了化学空间结构发现。如下面进一步讨论的,根据本发明的各种实施例,可以根据具体应用的要求利用各种距离测量和/或结构发现技术中的任何一种。
[0233]
许多实施例实施ao特征,包括(但不限于)在ao特征空间中的多个ao(包括(但不限于):一对、三个、四个)之间的距离测量的集合。在这个空间中,可以定义基于它们的ao特征来区分这些对的距离。如可以容易地理解的,根据本发明的各种实施例,可以根据具体应用的要求利用各种距离度量实施方式中的任何一种。
[0234]
虽然上面描述了包括各种ao特征距离度量的系统和方法,但是根据本发明的各种实施例,用于测量不同分子系统的基于ao的特征之间的距离的各种过程中的任何一种都可以根据具体应用的要求用于orbnet过程中。下面进一步讨论根据本发明的各种实施例的用于生成基于ao的特征的数据库的过程。
[0235]
生成基于ao的特征的数据库
[0236]
根据本发明的各种实施例的过程能够生成基于ao的特征的数据库。如下面进一步讨论的,根据本发明的各种实施例,可以根据具体应用的要求利用各种基于ao的特征数据库中的任何一种。
[0237]
许多实施例实施orbnet过程,该过程存储、组织和分类包括(但不限于)原子轨道的数据库,这些原子轨道形成与ao基和/或saao相关联的特征值的基础。在一些实施例中,可以使用类似于上面参考图1描述的过程,从orbnet过程中输出ao基和/或saao关联的特征值。在一些实施例中,利用基于ao的特征数据库,该数据库是基于ao原始特征空间和/或ao特征空间的子空间和/或潜在空间中的多个(包括(但不限于):一对、三个和四个)原子轨道之间的距离测量的集合来组织的。图6示意性地示出了根据本发明的实施例的数据库结构。数据库610可以包含分子性质620。分子性质可以包括(但不限于)相关联的对能量630。可以
使用包括(但不限于)耦合簇理论和/或dft理论的过程来计算相关联的对能量。相关联的对能量可以用于确定输入的基于ao的特征,包括(但不限于)saao特征640。基于ao的特征可以通过(但不限于)应用各种层面的量子化学理论的特征生成协议来确定,量子化学理论诸如半经验紧束缚、来自hartree-fock(hf)的不同基集或来自密度函数理论(dft)的不同基集。如可以容易地理解的,在基于ao的特征数据库的生成中使用的具体特征很大程度上仅局限于具体应用的要求。此外,可以使用包括(但不限于)属性图的更复杂的量子化学信息表示来生成数据库。在若干实施例中,构建数据库,其中,使用属性图描述分子系统的量子化学信息,属性图使用基于原子轨道的特征g(v,e,x,xe)构建的,其具有对应于对角线ao块的节点特征和对应于非对角线ao块的边特征。一些实施例实施对应于saao特征(xu=[f
uu
,j
uu
,k
uu
,p
uu
,h
uu
])的节点特征和对应于非对角线saao特征(x
euv
=[f
uv
,j
uv
,k
uv
,d
uv
,p
uv
,s
uv
,h
uv
])的边特征。在多个实施例中,以这种方式表示为属性图的量子化学信息可以在各种orbnet过程中使用,包括(但不限于)执行多任务学习以从训练数据集中学习属性图结构与化学性质之间的关联的orbnet过程。图形表示的好处在于,它们可以提供排列不变性和大小扩张性,并且通过利用包括(但不限于)结合了通用消息传递机制的图形神经网络的技术,用于一般的化学性质分类或回归。如可以容易地理解的,量子化学信息可以使用数据库内的各种技术和/或结构中的任何一种来表示,并且所表示的信息可以在类似于本文所描述的各种机器学习和/或生成过程中使用,以便于根据具体应用的要求合成具有期望化学性质的分子系统。因此,本发明的实施例应理解为不限于量子化学信息的任何特定表示,而是理解为适用于量子化学信息的任何表示的通用技术。
[0238]
可以查询数据库610以生成对应于特定分子集、分子几何结构、理论层面或其任何组合的数据集。各种实施例采用分布在一台或多台计算机上的诸如mysql的sql数据库或诸如mongodb的非sql数据库。根据各种实施例,数据库可以被查询,以基于空间中的基于ao的特征集之间测量的距离度量,来找到给定的基于ao的特征集附近的基于ao的特征。若干实施例使得数据库能够被查询,以基于与那些分子系统相关联的原子轨道相关联的基于ao的特征值来找到分子系统。这样的实施例的示例可以包括(但不限于):在基于ao的特征的空间中采用k-d树。如可以容易地理解的,根据本发明的各种实施例,可以根据具体应用的要求利用数据库索引和/或便于搜索的各种实施方式中的任何一种。
[0239]
虽然上面描述了用于生成轨道对数据库的各种过程,但是根据本发明的各种实施例,不同分子系统的任何种类的轨道对数据库可以根据具体应用的要求用于orbnet过程中。下面进一步讨论根据本发明的各种实施例的采集ao特征的过程。
[0240]
基于原子轨道的特征采集器
[0241]
根据本发明的各种实施例的过程依赖于从量子化学计算中采集基于ao的特征,包括(但不限于)saao特征。如下面进一步讨论的,根据本发明的各种实施例,可以根据具体应用的要求利用各种基于ao的特征采集器中的任何一种。
[0242]
许多实施例实施orbnet过程来从量子化学计算的输出中收集和采集基于ao的特征值。从orbnet过程收集的包括(但不限于)saao特征值的基于ao的特征值的一些实施例可以包括基于一对/三个/四个分子轨道与存储在原子轨道数据库中的基于ao的特征值之间的距离的基于ao的特征值。从orbnet过程收集的基于ao的特征值的一些其他实施例基于一对原子轨道与存储在原子轨道数据库中的基于ao的特征值之间的距离来消除基于ao的特
征值。
[0243]
图7示出了根据实施例的使用orbnet过程来收集和采集基于ao的特征的方法。可以生成分子系统的数据集作为输入701。量子化学计算可以应用于输入数据集702。根据一些实施例的量子化学计算可以在包括(但不限于)互联网云的远程服务器上执行。该计算可以生成并且输出对应的基于ao的特征703。这些特征可以存储在基于ao的特征705的数据库中。来自计算结果的分子也可以用于合成这样的分子704。
[0244]
虽然上面描述了用于采集ao特征的各种过程,但是根据本发明的各种实施例,能够收集和采集不同分子系统的ao特征的任何种类的过程可以根据具体应用的要求用于orbnet过程中。下面进一步讨论根据本发明的各种实施例的机器学习回归方法的过程。
[0245]
机器学习回归
[0246]
根据本发明各种实施例的过程依赖于机器学习技术,包括(但不限于)机器学习回归。如下面进一步讨论的,根据本发明的各种实施例,可以根据具体应用的要求利用各种机器学习回归方法中的任何一种。
[0247]
许多实施例包括orbnet过程,其结合了基于ao的特征数据库以确定准确的分子系统性质。若干实施例使用任意分子系统的数据库及其相关性质以及分子性质之间的差异作为训练集来回归模型,该模型包括(但不限于)作为基于ao的特征和/或其他特征的函数的分子性质的orbnet模型。一些实施例基于(一个或多个)训练模型对候选分子进行分级和/或排序。某些实施例基于(一个或多个)训练模型对候选分子进行分类和/或排序。多个实施例提出候选分子,并且然后基于(一个或多个)训练模型优化它们。若干实施例反演(一个或多个)训练模型以预测基于ao的特征值,包括(但不限于)可以导致分子性质的期望值的saao特征值。许多实施例实施(一个或多个)反演模型来优化、分级、排序、分类和/或预测具有期望分子性质的分子。这样的性质的示例包括(但不限于)溶解度、结合亲和力、对蛋白质的结合亲和力、氧化还原势能、pka、电导率、离子电导率、热导率、光吸收频率、光吸收强度和光吸收效率。
[0248]
图8中示出了这样的实施例的示例。可以从ao数据库801中提取来自准确参考计算的基于ao的特征和标签。许多实施例使用ao来评估用于特征生成的算符的矩阵元素。可以基于包括(但不限于)saao特征的所选择的基于ao的特征来训练机器学习模型802。训练模型可以用于从这些特征预测标签803,和/或可以在生成过程中使用。该模型可以用于预测准确的分子系统性质,包括(但不限于)saao解析性质、全分子性质和量子力学能804。机器学习回归的这样实施例可以包括但不限于:图形神经网络(gnn)。一些实施例实施具有多头图形注意力机制和/或执行者注意力机制以及残差块的gnn,以提高学习复杂化学环境的表示能力。如可以容易地理解的,根据本发明的各种实施例,可以根据具体应用的要求利用各种机器学习回归过程中的任何一种。
[0249]
在许多实施例中,使用orbnet过程确定的分子系统性质包括但不限于ao对对相关能量、量子力学能、力、振动频率(hessian)、偶极矩、响应性质、激发态能量和力、原子间力、优化的几何结构和光谱的贡献。可以容易理解的是,根据本发明的各种实施例,可以根据具体应用的要求利用各种分子系统性质中的任何一种。一些实施例实施力和hessians的预测,其可以用于将分子系统的几何结构优化到局部最小值或鞍点。若干实施例包括力的预测可以用于运行分子动力学。还有一些实施例包括可以用于执行构型采样的能量和力的预
测。根据若干实施例,可以基于使用一级电子结构理论获得的基于ao的特征值针对高级理论进行预测。高级理论的示例可以包括(但不限于)具有混合交换相关泛函的dft。如可以容易地理解的,用作高级理论的具体特征在很大程度上仅局限于具体应用的要求。在一些实施例中,可以根据基于ao的特征值对大基集进行预测,该基于ao的特征值可以包括小基集中的数据。小基集的示例可以包括(但不限于)最小基集。可以容易地理解的是,用作小基集的具体特征很大程度上仅局限于具体应用的要求。大基集的示例可以包括(但不限于)与小基集相比不同且更大的基集。可以容易地理解的是,用作大基集的具体特征很大程度上仅局限于具体应用的要求。
[0250]
随着量子模拟数据量的增加,根据本发明的许多实施例的orbnet过程可以利用在线学习技术来连续更新orbnet模型,而无需使用全部原始训练数据集来重新训练模型。可以容易地理解的是,根据本发明的各种实施例,可以根据具体应用的要求利用各种在线ml技术中的任何一种,以使用附加的量子模拟数据来更新先前训练的orbnet模型。在若干实施例中,orbnet模型的软件实施方式可以提供用户界面,该用户界面使得用户能够使用由用户选择的附加的量子模拟数据源来高效地更新现有的orbnet模型,该附加的量子模拟数据源包括(但不限于)量子模拟数据流。
[0251]
虽然上面描述了用于机器学习回归的各种过程,但是根据本发明的各种实施例,任何种类的机器学习回归方法都可以根据具体应用的要求在ml过程中使用,包括(但不限于)使用量子化学信息的图形表示来训练的ml过程(参见上面的讨论)。下面进一步讨论根据本发明的各种实施例的分子合成过程。
[0252]
分子合成
[0253]
根据本发明的各种实施例的过程可以用于合成分子。在若干实施例中,基于与由orbnet模型预测的化学性质相关的一个或多个标准的集合,orbnet过程被用于进行候选分子系统集的虚拟筛选。在多个实施例中,使用反向设计或生成过程来标识分子系统,其中,基于与由orbnet预测的化学性质相关的一个或多个标准的集合来执行基于ao的特征空间的搜索(或其合适的嵌入)。由orbnet模型预测为具有期望化学性质的包括(但不限于)saao特征的基于ao的特征集然后可以被用于标识对应于可能具有期望化学性质的基于ao的特征的分子结构。如下面进一步讨论的,根据本发明的各种实施例,可以根据具体应用的要求利用各种化学性质标准中的任何一种来进行虚拟筛选和/或反向分子设计。
[0254]
许多实施例实施orbnet过程,该过程基于与一种或多种期望的化学性质相关的标准集合来筛选候选分子系统集,以标识要合成的分子结构。图9a中示出了根据本发明的实施例的使用orbnet过程作为合成具有期望特性集的分子系统的过程的一部分来筛选候选分子系统分子的方法。过程900包括获得(901)候选分子系统集,其作为虚拟筛选过程的输入而提供。在若干实施例中,获得候选分子系统的量子化学表示。在所示实施例中,候选分子系统由基于原子轨道的特征集描述(902)。
[0255]
在若干实施例中,基于分子系统的量子化学表示估计一种或多种化学性质的ml模型可以用于候选分子系统集的虚拟筛选。在所示实施例中,使用orbnet模型预测(903)候选分子系统的分子系统性质,该orbnet模型使用类似于以上所描述的各种过程中的任何一种的过程来训练。可以容易地理解的是,特定ml模型很大程度上取决于用于表示候选分子系统的量子化学表示、用于减少量子化学表示的特征空间的维度的任何过程、由ml模型预测
的特定化学性质和/或具体应用的要求。
[0256]
候选分子系统的预测化学性质可以用于根据与期望的分子系统化学性质集相关的一个或多个标准筛选候选分子系统。在许多实施例中,附加的标准也可用作筛选的一部分,包括特定分子系统的已知化学性质,诸如(但不限于)水溶性和/或毒性。在若干实施例中,合成过程还可以进一步优化所标识的分子系统的化学结构,以进一步增强一种或多种期望的化学性质。可以容易地理解的是,降低不期望的化学性质可以以等同于增加期望的化学性质的方式来处理。被确定为满足筛选过程的标准集的(一个或多个)候选分子系统可以作为报告信息输出,和/或被合成(905)。
[0257]
虽然许多量子化学ml过程利用候选分子系统作为起点,但是基于从量子化学信息推导的特征训练ml模型的过程可以固有地定义可以用于反向分子设计的特征空间。因此,根据本发明的许多实施例的系统和方法利用量子化学特征空间来标识可能导致具有期望化学性质的分子系统的量子化学特征集,并且然后标识对应于所标识的量子化学特征集的分子系统。
[0258]
图9b示出了根据本发明的实施例的使用反向分子设计过程来合成具有期望的化学性质集的分子系统的过程。过程920包括获得(921)描述特征集与化学性质集之间的关系的ml模型。可以容易地理解的是,可以利用orbnet模型,该orbnet模型是使用类似于以上所描述的用于训练orbnet模型的各种过程中的任何一种的过程获得的。在多个实施例中,也可以利用基于包括(但不限于)属性图表示的分子系统的替代量子化学表示训练的ml模型。可以容易地理解的是,所使用的特定ml模型很大程度上取决于具体应用的要求。
[0259]
然后可以在ml模型的特征空间内执行搜索(922),以标识ml模型预测将具有满足搜索标准集的化学性质集的特征集。
[0260]
可以容易地理解的是,特征空间对应于分子系统的量子化学表示。因此,反向分子设计过程涉及标识(923)具有对应于所标识的特征集的量子化学表示的分子系统。在多个实施例中,可以使用特征-结构图来实现ml模型的特征空间中的特征集到分子系统的映射。在若干实施例中,可以从训练数据集中学习特征-结构图,在该训练数据中,用特征空间中的特征集来注释具有成键信息和/或任何其他原子表示的分子结构。可以容易地理解的是,可以利用各种训练数据集和/或机器学习过程中的任何一种来学习从特征空间到特定分子结构的映射过程。
[0261]
在多个实施例中,反向分子设计过程产生具有预测化学性质的候选分子系统集。可以进行(924)附加的筛选,以基于各种标准过滤候选分子系统的列表,这些标准包括(但不限于):化学合成的复杂性、已知毒性、水溶性和/或各种替代化学性质中的任何一种。当标识了合适的候选分子系统时,可以生成报告和/或合成所选择的分子系统(925)。
[0262]
虽然上面描述了用于标识用于合成的分子结构的各种过程,但是根据本发明的各种实施例,使用ml模型来标识分子结构的各种过程中的任何一种都可以根据具体应用的要求用于执行化学合成。ml过程也可以在量子化学计算的环境中用于各种附加的目的。下面进一步讨论根据本发明的各种实施例在量子化学计算中使用ml的过程。
[0263]
分子
″
试衣间
″
[0264]
在多个实施例中,感兴趣的特定分子系统可以用于从化学性质已知的分子系统的数据库中标识相关的基于ao的特征训练数据集。分子系统的数据库可以被查询,以基于数
据库中表示的ao与感兴趣的分子系统的ao之间的特征空间中的距离来标识ao。可以利用距离度量来测量数据库中分子的基于ao的特征与感兴趣的分子系统的基于ao的特征之间的距离。以这种方式,可以生成分子系统特定的训练数据集,以用于训练orbnet模型来预测感兴趣的分子系统的化学性质(例如量子力学能)的目的。
[0265]
图9c示出了根据本发明实施例的用于训练orbnet模型以估计特定候选分子系统的化学性质的具体过程。orbnet过程接收(931)特定分子系统作为输入。生成特定分子系统的分子轨道的基于ao的特征集,包括(但不限于)saao特征。在所示实施例中,通过执行(932)平均场计算并且基于计算结果获得(933)基于ao的特征来生成基于ao的特征。然后可以利用基于ao的特征来查询(934)数据库,以标识数据库中描述的、在基于ao的特征空间中接近感兴趣的特定分子系统的ao的ao。然后可以利用邻近ao的基于ao的特征和它们的化学性质来训练(935)orbnet模型,然后可以利用该orbnet模型来准确预测(936)作为该过程的输入的特定分子系统的化学性质。可以容易地理解,在由特定分子系统占据的特征空间中的特定区域中训练orbnet模型可以极大地增加对该特定分子系统的化学性质进行估计的准确度。
[0266]
虽然上面参考图9c描述的过程的讨论主要集中在用于在基于ao的特征空间中标识训练数据的过程,但是可以使用分子系统的各种基于ao的表示(包括(但不限于)属性图表示)中的任何一种来执行类似的过程。下面进一步讨论利用类似于以上所描述的ml过程和ml模型的ml过程和ml模型来为特定分子系统提供量子化学计算的系统和方法。
[0267]
量子化学程序
[0268]
根据本发明的各种实施例的过程依赖于量子化学性质。如下面进一步讨论的,根据本发明的各种实施例,可以根据具体应用的要求利用不同分子系统的基于ao的特征的各种量子化学预测中的任何一种。
[0269]
许多实施例实施基于物理学的量子化学预测作为orbnet过程期间的输入的分子系统的基于ao的特征,包括(但不限于)saao特征。若干实施例根据基于ao的特征实施对分子系统的基于物理学的量子化学的预测。一些实施例包括输出结果可以包括分子系统性质。量子化学程序的各种实施例包括(但不限于)耦合簇理论和密度泛函理论。可以容易地理解,用作量子化学程序的具体特征在很大程度上仅局限于具体应用的要求。许多实施例被结合到软件包中。
[0270]
图10示出了根据本发明的实施例的用于将orbnet过程结合到软件包中的系统。用户可以向量子化学软件包1001提供输入。用户可以执行基于物理的计算1102。计算的结果可以用来自对应于用户输入1003的基于ao的特征的ml模型的预测来代替。一般化可以包括使用根据基于ao的特征的模型来加速而不是取代基于物理的计算,以预测中间量1004;以及使用这些策略生成机器学习模型。
[0271]
在一些实施例中,结合orbnet过程的软件包可以在用户友好的平台上运行,这样的实施例的示例包括(但不限于):智能手机、平板电脑和计算机。可以容易地理解,用作用户平台的具体特征很大程度上仅局限于具体应用的要求。根据一些实施例,软件包经由orbnet过程的基于云的后端部署在几秒钟内执行量子模拟。
[0272]
虽然上面描述了用于从基于ao的特征生成量子化学预测的各种过程,但是根据本发明的各种实施例,根据基于ao的特征预测分子系统性质的任何种类的过程可以根据具体
应用的要求用于orbnet过程。下面进一步讨论根据本发明的各种实施例实施orbnet过程的各种示例。
[0273]
示例性实施例
[0274]
以下部分提供了使用不同orbnet过程来确定用于合成的分子组成和结构的具体示例。示例1至示例9实施了具有saao特征的orbnet过程。示例10至示例13实施了具有ao特征的orbnet过程。可以容易地理解,orbnet过程可以以各种不同方式中的任何一种和/或使用各种不同软件包中的任何一种来实施。应理解,具体实施例是出于示例性目的而提供的,并不限制本公开的整体范围,本公开的整体范围必须根据整个说明书、附图和权利要求来考虑。
[0275]
示例1和示例2的计算细节
[0276]
示例1和示例2使用qm7b-t(具有最多七个c、o、n、s和cl重原子的7211个分子的qm7b集的热化版本)和gdb-13-t(具有十三个c、o、n、s和c1重原子的分子的gdb-13集的热化版本)。对于这些数据集,从使用b3lyp/6-31g*理论层面和350k下的langevin恒温器执行的自始分子动力学轨迹,以50fs的间隔对训练和测试几何结构进行采样。
[0277]
最小基hartree-fock(hf)计算使用sto-3g ao基进行。使用cc-pvtzao基进行大基hf计算。并且使用非自洽gfn0-xtb方法执行半经验xtb计算。这些计算和对应的saao生成是使用entos qcore包执行的。对于dft标签值,b97x-d泛函在def2-tzvp ao基集中使用;这些计算也是使用e
ntosqcore
进行的。
[0278]
对于示例1和示例2中的hartree-fock和dft结果,采用coulomb积分和交换积分两者的密度拟合。冻结核心近似被用于所有情况。
[0279]
示例1:具有saao特征的orbnet的最小基到大基hf能量
[0280]
许多实施例实施orbnet来从使用廉价的最小基(即,sto-3g)hf计算计算的特征预测分子系统的大基集(即,cc-pvtz)hartree-fock(hf)能量。回归标签是大基与小基hf原子化能之间的差值,即
[0281][0282]
其中,e
tz
和e
sz
表示从大基集和最小基集中获得的hf能量;和分别表示从大基集和最小基集获得的分子的基态自由原子能量的总和。
[0283]
表1报告了ml预测的准确度。表1包括用于学习sto-3g以预测cc-pvtz hf原子化目标的mae结果,在saao基下使用f,d和p进行图形特征化。该模型在6500个qm7b-t分子上进行训练,并且结果是从使用每个分子的1或7个热采样几何结构训练的模型中报告的。在qm7b-t和gdb-13-t两者上的归一化mae达到了化学准确度。
[0284]
表1.用于学习sto-3g以预测cc-pvtz hf原子化目标的mae结果
[0285][0286]
示例2:具有saao特征的orbnet的xtb到dft能量
[0287]
许多实施例实施orbnet来从使用低计算成本半经验方法(即,gfn0-xtb)计算的特
征预测分子系统的高级理论(即,具有ωb97x-d范围分离的杂化泛函和def2-tzvp ao基的dft)的能量。由于gfn0-xtb是一种非自洽的基于场的方法,所以在避免可能困扰大分子系统的收敛困难可能性的同时,以小的o(n3)运算的前置因子获得特征。回归标签是高级dft与gfn0-xtb原子化能之间的差值,即
[0288][0289]
其中,δe
fit
是从训练集到原子化能差的线性拟合获得的校正项,原子化能差与每个元素的分子中的原子数量有关。
[0290]
表2报告了ml预测的准确度。表2包括用于学习gfn0-x tb以预测ωb97x-d/def2-tzvp dft原子化目标的mae结果,在saao基下使用f,j,k,d和p进行图形特征化。该模型在6500个qm7b-t分子上进行训练,并且结果是从使用每个分子的1或7个热采样几何结构训练的模型中报告的。与流行的ωb97x-d/def2-tzvp理论层面的全部计算成本相比,成本降低是从gfn0-xtb计算来计算特征的大约1000倍或更多。
[0291]
表2.用于学习gfn0-x tb以预测ωb97x-d/def2-tzvp dft原子化目标的mae结果
[0292][0293]
示例3和示例4计算细节
[0294]
示例3至示例4实施了以下数据集:qm7b-t数据集(其对于具有多达七个c、o、n、s和cl型重原子的7211个分子中的每个分子具有七种构象),qm9数据集(其对于具有多达九个c、o、n和f型重原子的133885个分子具有局部优化的几何结构),gdb-13-t数据集(其对于具有多达十三个c、o、n、s和cl型重原子的gdb-13数据集中的1000个分子中的每个分子具有六种构象),drugbank-t(其对于drugbank数据库中具有十四至30个c、o、n、s和cl型重原子的的168个分子中的每个分子具有六种构象),以及hutchison构象数据集(其对于具有九至50个之间的c、o、n、f、p、s、cl、br和i型重原子的622个分子中的每个分子具有多达10种构象)。可以从使用b3lyp/6-31g理论层面和350k下的langevin恒温器执行的自始分子动力学轨迹,以50fs的间隔对来自drugbank数据集的热化几何结构进行采样。对于示例3中报告的结果,采用了ramakrishnan等人预先计算的dft标签。(参见,例如,r.ramakrishnan等人,sci.data,2014,1,1-7;其公开内容通过引用并入本文中。)对于示例4中报告的结果,可以使用ωb97x-d泛函和def2-tzvp ao基集,并且使用def2-universal-jkfit基集对coulomb积分和交换积分两者进行密度拟合,来计算所有dft标签;这些计算使用psi4进行。使用e
ntos q
core
包,使用gfn1-xtb方法进行半经验计算,该包也用于saao特征生成。
[0295]
对于示例3至示例4中的结果,可以使用数据集的以下训练测试分割来训练orbnet模型。对于qm9数据集上的结果,由于几何一致性检查失败,3054个分子被移除。然后随机采样110000个分子进行训练,并使用10831个分子进行测试。示例3中的25000和50000个分子的训练集是从110000个分子的数据集中下采样的。对于qm7b-t数据集,生成两组训练测试分割;对于仅在qm7b-t数据集上训练的模型(示例4中的模型1),从总共7211个分子中随机选择6500个不同的分子(每个分子有7种几何结构)用于训练,选出500个分子(每个分子有7
种几何结构)用于测试;对于示例4中的模型2至模型4,这500个分子集中的361个分子子集用于测试,并且qm7b-t的剩余6850个分子用于训练。对于gdb13-t数据集,随机采样948个不同的分子(每个分子有6种几何结构)用于训练,选出48个分子(每个分子有6种几何结构)用于测试。对于drugbank-t数据集,随机采样158个不同的分子(每个分子有6种几何结构)用于训练,选出10个分子(每个分子有6种几何结构)用于测试。没有在hutchison构象数据集上进行训练。由于orbnet的训练数据集都不包括具有p、br和i类型元素的分子,所以hutchison数据集中包括这些类型元素的分子被排除。由于缺少dlpno-lccsd(t)参考数据,所以排除了十六个分子;基于使用psi 4的至少一个构象的dft收敛问题,排除了附加的八个分子。
[0296]
表3总结了用于为示例3和4中的结果训练orbnet的超参数。从f,j,k,d,p,h,和s对输入特征执行预变换以获得和对于每个要获得的算符类型,所有对角saao张量值x
uu
被归一化到范围[0,1);对于非对角线saao张量值,取其中,x∈f,j,k,p,s,h,,并且在有限的搜索空间内选择模型超参数;通过检查qm7b-t和gdb13-t数据集之间的特征元素分布之间的重叠来获得截止超参数c
x
。在整个示例3和示例4中使用相同的超参数集。
[0297]
表3.在示例3和示例4的orbnet中采用的模型超参数。所有截止值都以原子为单位。
[0298][0299][0300]
为了从构型自由度为预测能量变化提供附加的正则化,对以下形式的损失函数进
行训练
[0301][0302]
对于小批量中的构象i,随机采样相同分子的另一构象t(i)来与i进行配对,以评估相对构象损失从而对构型能量变化的预测误差施加附加的惩罚。e表示小批量的基础真值能量值,表示小批量的模型预测值,并且表示l2损失函数对于示例3中的所有模型,使用α=0,因为只有优化的几何图形是可用的;对于示例4中的模型,α=0.9被用于所有训练设置。
[0303]
示例3和示例4中的所有模型都是使用adam优化器在单个nvidia tesla v100-sxm2-32gb gpu上训练的。对于所有的训练运行,小批量大小被设置为64,并且使用循环学习率调度,其对最初的100个纪元(epoch)执行从3
×
10-5
到3
×
10-3
的线性学习率增加,对接下来的100个纪元执行从3
×
10-3
到3
×
10-5
的线性衰减,以及对最后的100个纪元执行每个纪元因子为0.9的指数式衰减。除了在注意力头σa中使用的之外,在每个激活函数σ之前使用批归一化。
[0304]
示例3:具有saao特征的orbnet的qm9形成能
[0305]
许多实施例使用从gfn1-xtb方法获得的输入特征来提供准确dft能量的预测。gfn系列方法可以用于模拟大分子系统(1000个原子或更多),其能量和力的求解时间为秒级。然而,这种适用性会受到半经验方法的准确度的限制,从而为基于gfn1特征的gfn1与dft能量之间的差异的
″
增量学习
″
创造了自然的机会。在若干实施例中,回归标签可以与高级dft与gfn1-xtb总原子化能之间的差值相关联,
[0306][0307]
其中,最后一项是由线性模型确定的dft与gfn1之间的孤立原子能量的差值的总和。给定gfn1-xtb计算的结果,这种方法产生总dft能量的直接ml预测。
[0308]
许多实施例在从qm9数据集预测总能量任务u0时将orbnet过程与其他ml方法进行比较。qm9由具有多达9个重原子的处于局部优化几何结构的有机分子组成。该测试检查了相似化学环境中系统的ml模型的表达能力。orbnet的结果没有对独立训练的模型进行系综平均(即,仅在第一个训练的模型的基础上进行预测),而是对五个独立训练的模型的结果进行系综平均(orbnet-ens5)。根据一些实施例的orbnet的集合可以帮助将orbnet预测误差减少大约10%到大约20%。若干实施例实施具有多任务学习的orbnet。利用分子能量和量子力学波函数的其他计算性质来训练具有多任务学习的orbnet。通过多任务学习,结合电子结构上的物理激励约束,可以提高学习效率。具有多任务学习的orbnet在qm9数据集的能量预测任务上显示出改善的准确度,与提供类似准确度的传统量子化学计算(诸如密度泛函理论)相比,计算成本降低了一千倍或更多。提供了来自利用基于原子的特征的图形表示的方法的qm9数据集的预测结果,包括schnet、physnet、dimenet和deepmolenet。(参见,例如k.schutt等人的advances in neural information processing systems,2017,991-1001;o.t.unke等人的j.chem.theory comput.,2019,15,3678-3693;j.klcpera等人的international conference on learning representations,2019;其公开内容通过引用并入本文中。)dimenet采用定向消息传递机制,并且physnet和deepmolenet采用基于先验
物理信息的监督来提高模型的可迁移性。许多实施例规定,orbnet提供比所有先前的深度学习方法高的准确度和学习效率。
[0309]
表4列出了在b3lyp/6-31g(2df,p)理论层面上预测总能量的qm9数据集的mae(单位为mev)。列出了单个模型(orbnet)、5个模型的集合(orbnet-ens5)、具有多任务学习的orbnet(orbnet-multi)、schnet、physnet、dimenet和deepmolenet的结果。
[0310]
表4.用于预测不同ml模型的总能量的qm9数据集的mae
[0311][0312][0313]
示例4:具有saao特征的orbnet的可迁移性和构象能预测
[0314]
许多实施例提供了orbnet过程的可迁移性。在若干实施例中,orbnet在相对小分子的数据集上被训练(对于这些小分子,高准确度数据更容易获得),然后在更大和更多样的分子的数据集上被测试。一些实施例提供了orbnet在包含有机和类药物分子的一系列数据集上的性能。
[0315]
图11a和图11b分别示出了根据本发明的实施例使用orbnet模型对分子总能量和相对构象能的预测误差。在图11a和图11b中,orbnet模型用不断增加的数据量进行训练。平均绝对误差(mae)由条形高度表示,绝对误差的中值由黑点表示,绝对误差的第一分位数和第三分位数由下条形和上条形表示。使用示例3和示例4的计算细节中描述的训练-测试分割,模型1是使用qm7b-t数据集中的数据训练的;模型2是使用qm7b-t、gdb13-t和drugbank-t数据集中的数据来训练的;模型3是使用qm7b-t、qm9、gdb13-t和drugbank-t数据集中的数据来训练的;并且模型4是通过用与用于模型3相同的数据来集合五次独立的训练运行而获得的。对这些数据集以及hutchison构象数据集中的每一个数据集的保留分子的总能量(图11a)和相对构象能(图11b)进行了预测。训练和预测采用ωb97x-d/def2-tzvp理论层面的能量。所有能量单位为kcal/mol。
[0316]
orbnet预测用附加的数据和集成建模改进。除了drugbank-t mae中的非单调性之外,从模型1到模型4,绝对误差的中值和平均值持续下降,这可能是由于该数据集相对较小。图11b显示,仅包含qm7b-t中的数据的模型1在drugbank-t和hutchison数据集(包括多达50个重原子的分子)上产生相对构象能预测,其准确度可与更严格训练的模型相比。在所有四个测试数据集上,所有orbnet模型预测的相对构象能的mae和中值预测误差都较好地在1kcal/mol的化学准确度阈值范围内。不包括使用模型1和模型2对qm9的预测,因为qm9包括f原子,而那些模型中的训练数据不包括f原子;没有预测qm9的相对构象能,因为它们在
该数据集中不可用。尽管对于hutchison数据集上的每个重原子,orbnet的总能量预测误差略大于其他数据集,但hutchison数据集的相对构象能预测误差略小于gdb13-t和drugbank-t。这可能是由于hutchison数据集涉及局部最小化的构象,这些构象在每个重原子上的能量分布小于热化数据集的构象。
[0317]
图12示出了根据本发明的实施例的hutchison构象基准数据集的一系列势能方法的准确度与计算成本权衡的比较。图12呈现了orbnet相比于各种其他力场、半经验、机器学习、dft和波函数方法的准确度和计算成本的直接比较。对于大小范围从九到50个重原子的类药物分子的hutchison构象数据集,使用预测构象能的中值r2与dlpno-ccsd(t)参考数据进行比较,并使用在单个cpu内核上评估的计算时间,来评估各种方法的准确度。
[0318]
图12中的orbnet构象能预测是使用模型4报告的(即,使用来自qm7b-t、gdb13-t、drugbank-t和qm9的训练数据,以及五次独立训练运行的系综平均)。与其他方法一样,黑色实心圆圈表示orbnet预测相对于dlpno-ccsd(t)参考数据的中值r2值(0.81);这一点提供了与其他方法准确度的直接比较。黑色空心圆圈表示orbnet预测相对于ωb97x-d/def2-tzvp参考数据的中值r2值(0.90),模型就是根据这些参考数据进行训练的;这一点表明,如果orbnet的模型4实施方式采用了耦合簇训练数据而不是dft训练数据,则它将具有预期的准确度。误差条对应于95%的置信区间,由统计自举确定。我们在intel
tm core i5-1038ng7 cpu@2.00ghz的单核上对orbnet进行了计时,发现orbnet的计算成本主要由用于特征生成的gfn1-xtb计算决定。orbnet使用e
ntos q
core
进行gfn1-xtb计算。hutchison中gfn1-xtb的报告的计时较慢,具体是与gfn0-xtb计时相比。(参见,例如,g.hutchison等人int.j.quantum chem.,2020,e26381;其公开内容通过引用并入本文中。)对于gfn0-xtb,e
ntos q
core
的计时类似于hutchison中报告的计时,这是明智的,因为该方法不涉及自洽场(scf)迭代。然而,hutchison表明gfn1-xtb的计时比gfn0-xtb慢43倍,而orbnet显示使用e
ntos q
core
时这一比率大约为4.5。为了解决gfn1-xtb实施方式中的代码效率问题,并且控制计时与hutchison中使用的单个cpu内核的细节,orbnet计时在图13中相对于hutchison的gfn0-xtb计时进行了归一化。orbnet的cpu神经网络推理成本对这种计时的贡献可以忽略不计。
[0319]
许多实施例表明,orbnet使得能够预测类药物分子的相对构象能,其准确度可与dft相比,但是计算成本从dft到半经验方法领域降低了1000倍。若干实施例表明,orbnet在实际应用中提供了优于当前可用的ml和半经验方法的预测准确度的改进,而没有显著增加计算成本。
[0320]
示例5的计算细节
[0321]
许多实施例规定,示例5中orbnet的训练包括来自qm7b-t、qm9、gdb 13-t和drugbank-t数据集中的多达30个重原子的分子的优化和热化几何结构。模型训练使用示例4中模型3的数据集分割。使用ωb97x-d3泛函和def2-tzvp ao基集,并且使用def2-universal-jkfit基集对coulomb积分和交换积分两者进行密度拟合,来计算dft标签。
[0322]
对于示例5中的结果,通过使用具有平移-旋转坐标(tric)的bfgs算法来最小化势能,对dft、orbnet和gfn-xtb计算进行几何结构优化;gfn2-xtb的几何结构优化是使用xtb包中的默认算法执行的。所有局部几何结构优化都从ωb97x-d3/def2-tzvp理论层面的预优化结构初始化。对于b97-3c方法,采用mtzvp基。
[0323]
所有的dft和gfn-xtb计算都是使用e
ntos q
core
进行的;gfn2-xtb计算使用xtb包进
行。
[0324]
示例5:具有saao特征的orbnet的分子几何结构优化
[0325]
若干实施例实施具有多任务学习的orbnet。利用分子能量和量子力学波函数的其他计算性质来训练具有多任务学习的orbnet。通过多任务学习,结合电子结构上的物理激励约束,可以提高学习效率。具有多任务学习模型的orbnet在对构象数据集的分子几何结构优化上显示出改进的准确度,与提供类似准确度的传统量子化学计算(诸如密度泛函理论)相比,计算成本降低了一千倍或更多。
[0326]
能量梯度(即力)计算的实际应用是通过局部最小化能量来优化分子结构。与计算成本相当和更高的其他方法相比,许多实施例提供了orbnet势能面的准确度。对rot34和mconf数据集进行测试,其中初始结构在ωb97x-d3/def2-tzvp dft的高质量层面上进行局部优化,其具有严格的收敛参数。rot34包括具有多达13个重原子的12个有机小分子的构象;mconf包括具有17个重原子的褪黑素分子的52个构象。从这些初始结构,使用各种能量方法进行局部几何结构优化,包括orbnet、gfn族半经验方法和相对低成本的dft泛函b97-3c。所得结构相对于在ωb97x-d3/def2-tzvp层面优化的参考结构的误差,按照最佳分子排列被计算为均方根距离(rmsd)。该测试调查每种方法的势能面是否与高质量的dft描述局部一致。
[0327]
图13a和图13b示出了根据本发明的实施例的rot34和mconf数据集的分子几何结构优化准确度,与ωb97x-d3/def2-tzvp层面的参考dft几何结构相比,其被报告为最佳排列均方根偏差(rmsd)。误差的分布被绘制成直方图(具有重叠的核密度估计)。计时对应于在单个intel
tm xeon gold 6130@2.10ghz cpu内核上对mconf数据集进行单次力评估的平均成本。图13a和图13b示出了各种方法在每个数据集上的所得误差分布。表5报告了平均误差和对应于不正确几何结构(即rmsd>0.6埃)的优化结构的百分比。虽然gfn半经验方法提供了与orbnet相当的计算成本,但是所得的几何结构优化实质上不太准确,其中,很大一部分局部几何结构优化松弛成与优化的参考dft结构不一致的结构(即,rmsd超过0.6埃)。与使用b97-3c泛函的dft相比,orbnet提供了优化的结构,其具有与rot34相当的准确度,并且对于mconf更准确。然而,orbnet的计算开销要低100倍以上。
[0328]
表5.平均误差和对应于不正确几何结构的优化结构的百分比。
[0329][0330][0331]
示例6至示例9的计算细节
[0332]
根据若干实施例的orbnet denali过程在示例6至示例9中实施。与示例1至示例5
中的orbnet方法相比,orbnet denali过程具有以下修改:1).注意力机制被执行者注意力所取代。执行者注意力机制会导致内存使用的减少和可忽略的测试准确度下降。2).消息传递步骤的数量从2增加到3。3).批归一化层被替换为层归一化层。4).回归标签被修改以考虑带电分子。根据若干实施例使用orbnet denali模型的示例6至示例9实施了增加的模型和数据规模,这可以得到接近dft的性能。在一些实施例中,orbnet denali模型使用大约2100万个可训练参数和大约250万个训练数据。
[0333]
在示例6至示例9中,许多实施例提供了用于orbnet denali的大量训练数据集合。一些实施例在训练数据中实施chembl分子。chembl27数据库可以从chembl网络服务下载。简化分子线性输入规范(smiles)字符串包含50个或更少的元素c、o、n、f、s、cl、br、i、p、si、b、na、k、li、ca或mg的原子,并且没有保留同位素规格。不解析为封闭壳层lewis结构的smiles字符串被丢弃。从训练数据集中移除对应于hutchison构象基准集中的分子的所有smiles字符串。
[0334]
从这个子集,随机选择对应于中性分子的116,943个独特smiles字符串的最终幸存选择。每个smiles字符串的最多四个构象最初通过entosbreeze构象生成器生成,并在gfn1-xtb层面进行优化。对于这四个能量最小化的构象中的每个,使用entos breeze通过300k下的正常模式采样(nms)或500k下200fs的自始分子动力学(aimd)采样生成非平衡几何结构;在这两种情况下,在gfn1-xtb理论层面上。这些热化方法是随机选择的,其权重相等。这一过程产生总共1,771,191个平衡和非平衡几何结构。
[0335]
若干实施例在训练数据中实施质子化状态和互变异构体。从已过滤的chembl smiles字符串列表中随机选择26,186个smiles字符串的子集。对于这些中的每一个,使用dimorphite-dl版本1.2.4标识了多达独特的128个质子化状态,并且随机选择了这些质子化状态中的四个。相同的构象生成算法和非平衡几何采样算法被应用于这四个质子化状态,产生总共215,866个独特的几何结构。
[0336]
一些实施例在训练数据中实施盐络合物和非键合的相互作用。从已过滤的chembl smiles字符串列表中,选择多个smiles字符串,并与chembl结构管线中的常见盐列表中的一到三个盐分子随机配对。这一过程可以产生总共21,735种盐络合物。对于这些络合物中的每种,通过构象管线生成四个构象,并且nms采样用于为每个构象生成四个非平衡几何结构。这产生了271,084个独特的几何结构。附加地,jsch-2005中的结构和生物片段数据库的侧链-侧链相互作用(ssi)子集被添加到数据集中。
[0337]
某些实施例在训练数据中实施小分子。创建了有机分子中常见化学部分和键合模式的列表,以避免使数据集偏向于仅代表大的类药物分子,并且用于列举具有相对
″
奇异
″
成分的小分子的化学,从而产生大约15,000个smiles字符串。对于其中的每一种,smiles字符串都是通过随机用氢原子取代卤素,用碳原子取代硅原子而产生的。该过程可以生成总共40,565个smiles字符串,通过构象管线为其生成构象,从而产生总共94,588个独特的几何结构。
[0338]
示例6至示例9中的所有dft单点计算都是在e
ntos q
core
版本0.8.17中,在ωb97x-d3/def2-tzvp理论层面上,使用neese=4dft积分网格的堆芯密度拟合进行的。
[0339]
许多实施例在示例6至示例9中提供了orbnet denali过程的训练细节。pytorch v1.7.1和深度图形库(dgl)v0.6用于实施和训练模型。pytorch的分布式数据并行(ddp)策
略用于使用数据并行性在多个gpu上训练模型。orbnet denali模型在olcf summit超级计算机上使用96个nvidia v100-sxm2(32g)gpu进行训练,其中每个gpu的批量大小为4个,持续300个纪元,总共训练6912个gpu小时。学习率在前100个纪元线性预热,在剩余的200个纪元余弦退火至零。在这个过程中,最大学习率是3e-4。使用adam优化器。1.8tb的数据集被随机分成四个碎片。每个summit节点(包括6个gpu)被分配给这四个碎片中的一个,使得每个碎片在1/4的节点上使用。
[0340]
orbnet denali模型中的回归标签在eq.45中描述。在eq.45中,e
dft
是参考dft(即ωb97x-d3/def2-tzvp)能量,并且e
gfn1
是gfn1-xtb能量。在orbnet denali模型中,由下式给出
[0341][0342]
其中,i索引分子内的原子,zi是原子i的原子序数,并且q是分子的总电荷。和是参数,并且在orbnet训练之前用普通最小二乘法拟合到e
dft-e
gfn1
。
[0343]
根据一些实施例的orbnet denali 10%模型在随机采样的10%的训练数据上被训练。所有其他训练细节都是一样的。表6提供了示例6至示例9和示例1至示例5中给出的模型的比较。
[0344]
表6.示例1至示例9中orbnet模型的比较
[0345][0346][0347]
示例6:具有saao特征的orbnet的gmtkn55集合
[0348]
通用主族热化学、动力学和非共价相互作用55(gmtkn55)数据集是55个数据集的集合,旨在探索量子力学(qm)方法在从反应能量和电子性质到非共价相互作用能量和构象性质的各种化学问题上的准确度。该数据集由55个单独的子集组成,具有基于2462个单点计算的总共1505个相对能量。gmtkn55中分子的高层面参考能量可以是使用一系列外推方
案基于从若干不同来源收集的ccsd和ccsd(t)计算而计算的最佳估计。
[0349]
gmtkn55上qm方法的性能可以经由基于相对于参考的平均绝对偏差加权的聚合分数、wtmad-1或wtmad-2分数来表示,其中两者之间的差异是各个子集的相对加权。
[0350]
对于orbnet训练数据中的子集,与高层面参考能量相比,wtmad-1和wtmad-2分数为5.97和9.84。考虑到所有子集,其中,元素和自旋状态存在于训练集中,但化学空间不一定被orbnet训练数据覆盖(例如过渡态、无机系统等),wtmad-1和wtmad-2没有显著增加,相对于高层面参考能量为7.19和9.85。
[0351]
当相对于ωb97x-d3/def2-tzvp参考能量(用于生成orbnet训练数据的相同方法)计算加权分数时,wtmad-1和wtmad-2分数分别为3.67和6.37。对于在10%的数据上训练的orbnet denali版本,相对于ωb97x-d3/def2-tzvp参考,wtmad-1和wtmad-2分数分别为7.77和12.16,证明了增加数据集大小的积极影响。ωb97x-d3/def2-tzvp和高层面参考能量之间的wtmad-1和wtmad-2分别为3.67和6.37,这在某种意义上构成了相对于在ωb97x-d3/def2-tzvp数据上训练的orbnet模型的高层面参考能量的准确度的上限。
[0352]
相比之下,与高层面参考相比,流行的半低成本dft方法b97-3c对于gmtkn55的wtmad-1值和wtmad-2值分别为5.76和10.22,非常接近orbnet分数。对于这个数据集,orbnet大约比b97-3c快100倍。另一低成本的qm方法是gfnn-xtb(n∈{0,1,2})方法系列。对于这些方法,对于gfn0-xtb、gfn1-xtb和gfn2-xtb,wtmad-1值分别为45.9、20.9和15.4,相同系列的wtmad-2数为75.8、35.9和27.4。gfn1-xtb是用于为orbnetdenali生成输入的基准方法,尽管在gmtkn55上表现相对较差,orbnet仍能生成dft质量的能量预测。
[0353]
对于机器学习潜力ani-1ccx和ani-2x,可以计算仅包含具有由单独方法覆盖的元素的中性单线态分子的子集上的wtmad-n分数。对于根据ccsd(t)参考数据参数化的ani模型,即ani-1cc,wtmad-1值和wtmad-2值分别为15.5和24.2。对于ani-2x模型,与orbnet denali相似,在dft层面数据上进行参数化,wtmad-1和wtmad-2分别为14.2和23.9。
[0354]
在通用机器学习潜力可以应用的常见化学问题的覆盖范围方面,orbnetdenali可以提供gmtkn55的广泛覆盖。orbnet denali覆盖55个子集中的37个,因为orbnet训练集不覆盖元素he、be和al以及一些重金属,以及除单线态之外的自旋态,例如用于计算电离势和电子亲合势。当从训练分布外推至这些其他子集时,orbnet denali提供了合理但不太准确的结果,这是因为它基于gfn1-xtb。ani-1ccx和ani-2x的对应数字分别为14和20。ani-1ccx仅涵盖具有元素h、c、n和o的中性单线态分子,而ani-2x将这一涵盖范围扩展至元素f、cl和s。gfnn-xtb方法系列已在高达radon(z=87)的数据上进行了参数化,并且还处理具有奇数个电子的系统,并且因此能够涵盖gmtkn55。
[0355]
图14示出了根据本发明的实施例的每种方法的wtmad-n值和gmtkn55子集覆盖范围的图形概览。对于选择的方法示出了gmtkn55数据集的准确度和覆盖范围的统计数据,这些方法按照相对于参考高层面估计值的wtmad-2分数排序。在1401中示出了由每种方法覆盖的在子集上计算的任意单位的聚合wtmad-1和wtmad-2度量。在1402中示出了gmtkn55子集的百分比,该子集包括每个模型中允许的具有元素、带电状态和自旋状态的分子。def2-tzvp基集用于ωb97x-d3计算。
[0356]
图15示出了根据本发明的一个实施例的相对于ωb97x-d3/def2-tzvp的orbnet denali训练数据所覆盖的gmtkn55的子集的mae,单位为kcal/mol。对于ani-1ccx和ani-2x,
包含这些模型中不允许的元素或电荷状态的子集的值被忽略。
[0357]
示例7:具有saao特征的orbnet的构象评分
[0358]
准确确定热可及构象的集合是建模分子的关键。示例7包括构象能的基准的结果。该基准包含了~700个类药物分子中的每个分子的多达十个姿态。每个分子由集合c、h、n、o、s、cl、f、p、br、i中的元素组成,并且包含九至五十个重原子,其总电荷在-1至+2之间。
[0359]
给定方法在该基准中的准确度被报告为中值r2,并且确定如下。对于每个分子,计算该分子的构象能与参考(dlpno-ccsd(t))能之间的相关系数(r2)。然后,取对应于基准中所有分子的一组r2值的中值。
[0360]
图16示出了根据本发明的实施例的hutchison构象基准集的多种方法的计算成本和所得准确度之间的比较。示出了orbnet denali与计算化学方法的代表性样本的比较,计算化学方法包括力场、机器学习、半经验、密度泛函理论和波泛函理论。横轴表示单个构象能计算的平均时间,而纵轴表示数据集中分子的中值r2相关系数。误差条表示该数字的95%置信区间,并且通过自举获得。示出了所有方法的中值相关系数与dlpno-ccsd(t)的关系(实心圆圈、orbnet的白色空心圆圈)。附加地,示出了orbnet(黑色实心圆圈)的中值相关系数与ωb97x-d3/def2-tzvp参考能量的关系。该参考对应于用于训练模型的理论层面。
[0361]
对于orbnet以外的方法,可以观察到准确度与方法平均执行时间的对数之间有很强的相关性。相反,orbnet denali在每个分子大约一秒的平均执行时间下,提供了相对于参考dlpno-ccsd(t)的大约0.90
±
0.02的中值r2。不确定性是指95%的置信区间,并且通过自举数据集获得。gfn1-xtb是用于为orbnet生成输入的方法,它提供的0.62
±
0.04的中值r2与orbnet的执行时间相似。orbnet与ωb97x-d3/def2-tzvp(用于为orbnet生成训练数据的相同方法)之间的中值r2是0.973
±
0.004,突出了orbnet能够以高准确度学习其底层方法。与ωb97x-d3/def2-tzvp级理论(提供的准确度与dlplo-ccsd(t)相似,具有0.92
±
0.02的中值r2)相比,orbnet的速度提升了1000倍。这个数字也作为在ωb97x-d3/def2-tzvp数据上训练的模型的准确度的上限,并且表明与dlpno-ccsd(t)相比,为了增加orbnet的中值r2,可能有必要在超过dft准确度的数据上训练。
[0362]
示例8:具有saao特征的orbnet的非共价相互作用(s66x10)
[0363]
非共价相互作用准确度的标准基准可以是s66x10基准集。该数据集包括66种不同的分子二聚体及其平衡几何结构,以及沿质心轴的9个附加位移和对应的ccsd(t)/cbs外推束缚能。
[0364]
对于orbnet denali,对ccsd(t)/cbs的mae和rmse分别为0.75和1.01kcal/mol。这些数字接近用于生成训练数据的方法(ωb97x-d3/def2-tzvp)的mae和rmse,为0.70和0.91kcal/mol。将orbnetdenali与ωb97x-d3/def2-tzvp进行比较,可以发现较小的mae值和rmse值分别为0.46和0.65kcal/mol。对于在10%的数据上进行训练的orbnet denali,这些数字分别增加到0.67和0.85,这表明增加的训练数据大小可能是有益的,但也表明模型不可能大大超过训练数据的准确度。表7中总结了本部分提到的数字。对于标有星号(*)的行,将orbnet预测与在ωb97x-d3/def2-tzvp层面计算的束缚能进行比较。后一个参考对应于用于为orbnet denali生成训练数据的相同方法
[0365]
表7.s66x10基准的mae和rmse束缚能与ccsd(t)/cbs参考束缚能
[0366][0367]
示例9:具有saao特征的orbnet的类药物分子的扭转曲线
[0368]
经验电势的基准可以是扭转曲线可以再现的准确度。torsionnet500基准编译了500种化学性质不同的片段的扭转曲线,这些片段包含元素h、c、n、o、f、s和cl。对于这些扭转曲线,计算了ωb97x-d3/def2-tzvp层面的参考能量,对应于用于训练orbnet denali的理论层面。一些实施例通过比较若干不同的准确度测量来对orbnet denali的性能进行基准测试。概述见表8。在表8中,参考能量是在ωb97x-d3/def2-tzvp理论层面上计算的,标有星号(*)的行除外,这些行针对b3lyp/6-31g*参考进行基准测试。对于多种方法,示出了以下统计数据:pearson相关系数(r)大于0.9的500个扭转曲线的百分比,扭转曲线上的平均pearson r,扭转曲线的相对能量的mae和rmse,以及最后,扭转曲线的百分比,其中,参考曲线的局部最小角度对应于测试曲线中20
°
内的点,该点距离全局最小值也不超过1kcal/mol。
[0369]
第一个测量是参考能量与预测能量之间的pearson相关系数(r)大于0.9的扭转曲线的数量。对于orbnet denali,这对于大约99.4%的曲线是真实的,而对于orbnet denali(10%),对应的数字是大约98.8/%,平均pearson r值分别为0.995和0.988。第二,对于完整的orbnet denali模型,扭转曲线的平均mae和rmse为0.12和0.18kcal/mol,并且对于orbnet denali(10%)为0.23和0.34kcal/mol。最后,两个orbnet denali模型都正确地预测了所有500个曲线的全局最小值的位置,误差在20
°
内,能量误差在1kcal/mol内。实施例规定,当orbnet denali训练集不包含扭转曲线时实现这些结果。
[0370]
对于orbnet的基线方法(gfn1-xtb),相同的数字要低得多,只有65.6%的曲线具有r>0.9,并且平均r值为0.832,平均mae和rmse为0.94和1.3kcal/mol,同时捕获了89.4%的预测曲线的良好最小值。图18示出了根据实施例的通过orbnet denali的误差分层的orbnet denali对gfn1-xtb的25扭转能量曲线。图18示出了来自torionnet500数据库的25种类药物分子的扭转曲线,这些分子被分层以表示相对于在ωb97x-d3/def2-tzvp理论层面计算的相同扭转曲线的orbnet误差的五分位数,其被示出为参考(黑色)。orbnet的基线方法gfn1-xtb示出了相同的扭转曲线(红色)。在每种情况下,orbnet denali在1kcal/mol的化学准确度内再现了沿着每个扭转的每个点。除了5个最坏的情况,orbnet denali扭转曲线在质量上与参考方法相同。另一方面,gfn1-xtb示出了大多数扭转曲线的大误差。在一
些情况下,gfn 1-xtb曲线的形状在质量上是不正确的。总的来说,这表明orbnet的准确度不仅仅是由于使用了良好的基线方法,而是orbnet能够正确地捕获扭转曲线中的细微差异。
[0371]
将使用其他dft方法b97-3c计算的扭转曲线与参考曲线进行比较。对于b97-3c,与ωb97x-d3曲线相关的mae和rmse分别为0.29和0.43kcal/mol。这些数字可以突出orbnet denali比这两种dft方法之间的变化更接近dft参考值几乎三倍。因此,对于这种应用,orbnet可以被视为等同于dft方法。
[0372]
orbnet还与merck分子力学力场94(mmff94)和两种基于ml的方法(即ani-2x和torsionnet)进行了比较。发现mmff94力场捕获ωb97x-d3/def2-tzvp预测最小值的准确度最低,仅在大约75.2%的时间内找到公差范围内的正确最小值,并且在扭转曲线上具有较高的mae和rmse,分别为1.4kcal/mol和5.2kcal/mol。与ωb97x-d3/def2-tzvp参考扭转曲线相比,ani-2x在20。公差范围内捕获到低能量最小值,成功率为91.8%,优于mmff94、gfn0-xtb和gfn1-xtb。ani-2x在正确找到低能最小值方面可能具有更好的准确度,但它比gfn0-xtb和gfn1-xtb具有更大的mae和rmse,这可能是由于低估了旋转势垒。
[0373]
除了数字之外,一些实施例突出了在相同结构上比较ani-2x和torsionnet,但是相对于b3lyp/6-31g(d)单点能量的基准测试。ani-2x可以相对于ωb97x/6-31g(d)参考数据进行参数化,而torsionnet可以根据b3lyp/6-31g(d)参考数据进行参数化,因此参考数据可能为ani-2x提供更合理的参考。相对于b3lyp/6-31g(d)参考,torsionnet能够以大约83%的成功率定位低能量最小值,并且ani-2x为大约66%的成功率。torsionnet的mae和rmse相对于其在自身理论参考层面下计算的扭矩曲线分别为0.7和1.3kcal/mol,而ani-2x的mae和rmse分别为1.4和2.0kcal/mol,与ωb97x-d3/def2-tzvp参考值相比,在相同值的0.1kcal/mol之内。
[0374]
表8.torsionnet500基准集上的八种方法的性能。
[0375][0376]
示例10:利用基于ao的特征的orbnet在qm9数据集中预测能量和偶极矩
[0377]
许多实施例在学习量子化学性质中实施具有基于ao的特征的orbnet,这些性质包括(但不限于)在各种机器学习数据集上的单点能量、力、偶极矩、电子密度、分子轨道能量和热性质。若干实施例针对下游化学任务执行针对能量预训练的orbnet模型的零样本一般化测试,下游化学任务已经被开发用于基准量子化学模拟方法。在示例10至示例13中使用相同的模型超参数集。
[0378]
在许多实施例中,orbnet过程在qm9数据集上比其他方法高出至少150%,在md17数据集上比其他方法高出至少114%,在电子密度上比其他方法高出至少50-75%。除了其学习效率之外,经过能量训练的orbnet可以在各种实际的下游化学任务上实现鲁棒的性能,而无需任何模型微调。其准确度可与dft方法相媲美,但速度提高了3个数量级。
[0379]
若干实施例在qm9数据集上学习包括(但不限于)能量和偶极矩的量子化学性质时,实施具有基于ao的特征的orbnet。qm9数据集包含134k个有机小分子,在其平衡几何结构中有多达9个重(cnof)原子,其标量值化学性质由dft计算。由于其简单的化学成分和多重任务,qm9可以用于基准测试深度学习方法。使用110,000个随机样本作为训练集,并且另外10,831个样本作为测试集,对qm9目标进行训练。根据若干实施例的orbnet过程在所有12个目标上相对于其他模型提供了至少大约150%的平均mae减少。在一些实施例中,orbnet可以在偶极范数μ、电子空间范围《r2》、homo/lumo能量和间隙∈h
omo
,∈
lumo
,δ∈(它们在其公式中深深植根于电子结构中)方面实现质的改进。对能量u0和偶极子向量两个代表性的目标进行了实验。在不同大小的训练数据上,orbnet优于深度学习方法以及预先设计的方法。
[0380]
表9列出了在110k样本上训练的模型在qm9目标上的预测mae。每项任务的最佳/次佳结果用粗体/下划线标出。在所有12个目标上,orbnet比第二好的模型(spherenet)平均高出150%。
[0381]
图18a至图18b示出了根据本发明的实施例的基于ao的特征的orbnet的能量和偶极矩预测。图19a将orbnet与任务特定模型和深度学习方法在不同训练数据大小下在qm9数据集上的能量u0(单位mev)向量方面进行了比较。图19b将orbnet与任务特定模型和深度学习方法在不同训练数据大小下在qm9数据集上的mdebye向量中的偶极矩方面进行了比较。在不同大小的训练数据上,orbnet优于深度学习方法和预先设计的方法。
[0382]
表9.qm9数据集上的预测mae
[0383][0384][0385]
示例11:利用基于ao特征的orbnet在md17数据集中预测能量和力
[0386]
许多实施例在学习包括(但不限于)md17数据集上的能量和力的量子化学性质时,实施具有基于ao的特征的orbnet。md 17数据集包含来自八个小有机分子的分子动力学轨迹的能量和力标签,并且可以用于对建模势能面的单个实例的ml方法进行基准测试。orbnet使用报告的数据集分割和修订的标签,在每个分子的1000种几何结构的能量和力上进行训练,并且在另外1000个分子上进行测试。当与结合了核回归、核方法和图形神经网络的手工设计的特征相比时,orbnet可以在能量和力预测上实现超过110%的平均改进。不确定性被估计为3个独立训练模型的测试集的mae的标准偏差。
[0387]
表10列出了在1000个样本上训练的模型对md17能量(单位为kca1/mol)和力(单位为)的mae预测。平均而言,orbnet比其他能量模型(即fchl19/gpr)至少高出138%,并且比其他力模型(即nequip)至少高出114%。
[0388]
表10.对md17能量(单位为kcal/mol)和力(单位为)的预测
[0389][0390]
[0391]
示例12:利用基于ao特征的orbnet预测电子密度
[0392]
许多实施例在学习包括(但不限于)bfdb-ssi和qm9数据集上的电子密度的量子化学性质时,实施具有基于ao的特征的orbnet。若干实施例提供了对分子的电子密度的预测,这在dft的理论公式和实际构建中都起着重要作用。orbnet的o(3)等变性使得能够在紧凑的类原子轨道基上高效地学习与专门为学习开发的两个基线相比,orbnet2在平均l-1密度误差方面实现了大约50%至75%的降低,其中,表示模型预测的电子密度。与具有立方阶训练时间复杂度的sa-gpr相比,orbnet在训练方面更高效,并且与需要在每个网格点评估部分神经网络的deepdft相比,orbnet在推理方面更高效。
[0393]
表11列出了电子电荷密度学习统计。在∈
ρ
方面,orbnet在bfdb-ssi上比基线高出至少52%,在qm9上比基线高出至少75%,在训练和推理方面具有大的效率优势。
[0394]
表11.电子电荷密度学习统计
[0395][0396]
示例13:利用基于ao特征的orbnet的下游化学任务
[0397]
许多实施例提供了orbnet在化学家感兴趣的度量上的性能。在若干实施例中,orbnet2模型可以在具有宽化学空间覆盖和非平衡几何结构的237k样本的dft能量上训练,并且无需任何模型微调,直接将其应用于通常用于基准测试量子化学模拟方法的下游任务。在这种零样本设置中,预训练的orbnet模型实现了与dft泛函相似和/或更好的准确度,同时速度至少快200倍左右(如果在gpu上运行orbnet,速度将快1000倍以上),并且明显优于代表性的半经验量子力学或机器学习方法,后者提供了相当的速度。
[0398]
表12列出了针对下游任务的代表性的半经验量子力学(seqm)、机器学习(ml)和密度泛函理论(dft)方法的orbnet基准测试。
[0399]
表12.针对下游任务的代表性的半经验量子力学(seqm)、机器学习(ml)和密度泛函理论(dft)方法的orbnet基准测试
[0400][0401][0402]
等同原则
[0403]
从以上讨论中可以推断出,上述概念可以在根据本发明的实施例的各种布置中实施。因此,尽管已经在某些具体方面描述了本发明,但是许多附加的修改和变化对于本领域技术人员来说是显而易见的。因此,应理解,本发明可以以不同于具体描述的方式实施。因此,本发明的实施例在所有方面都应该被认为是说明性的而非限制性的。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1