一种单细胞ATAC-seq数据分析方法
本发明属于生物信息分析,具体涉及一种单细胞atac-seq数据分析方法。
背景技术:
1、单细胞染色质可达性靶向测序(single-cell assay for targetingaccessible-chromatin sequencing,scatac-seq)方法的兴起为刻画单细胞分辨率的染色质可达性图谱奠定了坚实的基础,其已经成为揭示基因转录过程中细胞特异性调控机制的重要手段。近年来,随着单细胞atac-seq数据规模的快速增长,即scatac-seq方法检测到的染色质可达性特征峰的规模的快速增长,生物信息学家们开始着手致力于利用单细胞atac-seq数据实现染色质可达性预测、细胞类型注释、染色质可达性图谱降噪、转录因子活性推断等下游分析任务。然而,单细胞atac-seq数据固有的高维性、二值性、稀疏性为计算分析带来了巨大挑战。因此,设计精准、鲁棒、高效的单细胞atac-seq数据分析方法已然成为生物信息学领域中亟待解决的关键问题。
2、截至目前为止,生物信息学家们已经提出了一系列单细胞atac-seq数据分析方法,它们的基本思想是利用数理统计、机器学习等手段从海量单细胞atac-seq数据中提取每个细胞的高阶特征,并将所提取的特征应用于下游分析任务中。例如,威库鲁文脑与疾病研究中心的斯坦因·艾茨教授领衔设计的cistopic算法、清华大学张强峰教授领衔设计的scale算法分别采用隐式迪利克雷分配模型、高斯变分自编码器模型等生成式模型将单个细胞映射到低维的、连续的、广义的特征空间中,有效地克服了单细胞atac-seq数据的高维性、二值性、稀疏性,成功地实现了细胞类型注释、染色质可达性图谱降噪等下游分析任务。然而,cistopic和scale算法仅通过染色体坐标表示染色质可达性特征峰,忽略了dna序列中潜藏的转录调控语法规则,导致了单细胞特征表示的不准性。斯坦福大学医学院的威廉·格林利夫教授领衔设计的chromvar算法、美国麻省理工学院-哈佛大学博德研究所的阿维夫·雷格夫教授领衔设计的brockman算法分别使用转录因子-dna结合基元的数量、染色质可达性特征峰中k-mer碱基字符串等生物学先验知识刻画不同细胞的特征,相比于cistopic和scale而言进一步提高了转录因子活性推断等下游分析任务的可用性、精准性、可解释性。“谷歌”公司旗下的计算生物学家大卫·凯利教授领衔设计的scbasset算法运用卷积神经网络从已知染色质可达性特征峰的一级序列中提取转录因子-dna结合基元的基元信息,开创性地实现了单细胞染色质可达性的预测任务。
3、但上述chromvar、brockman、scbasset等广泛使用的单细胞atac-seq数据分析方法依然存在一些不足之处。第一点,大量研究表明启动子和增强子等具有生物功能的dna短序列能够通过发生相互作用控制各类细胞的状态,然而现有的单细胞atac-seq数据分析方法难以有效地捕捉启动子和增强子等转录因子-dna结合基元的相对位置和长距离依赖关系,进而无法精准地刻画启动子和增强子之间的相互作用。第二点,现有的单细胞atac-seq数据分析方法呈现各司其职的状态,难以将染色质可达性预测、细胞类型注释、染色质可达性图谱降噪、转录因子活性推断等重要的下游分析任务集成到一个统一的框架中,导致了方法的通用性有一定局限。
技术实现思路
1、针对现有技术中的上述不足,本发明提供的单细胞atac-seq数据分析方法解决了现有分析方法中,无法精准地刻画启动子和增强子之间的相互作用,以及方法通用性存在局限的问题,本发明提供一种精准、统一、高效的基于dna语言模型的单细胞atac-seq数据计算分析方法。
2、为了达到上述发明目的,本发明采用的技术方案为:一种单细胞atac-seq数据分析方法,包括以下步骤:
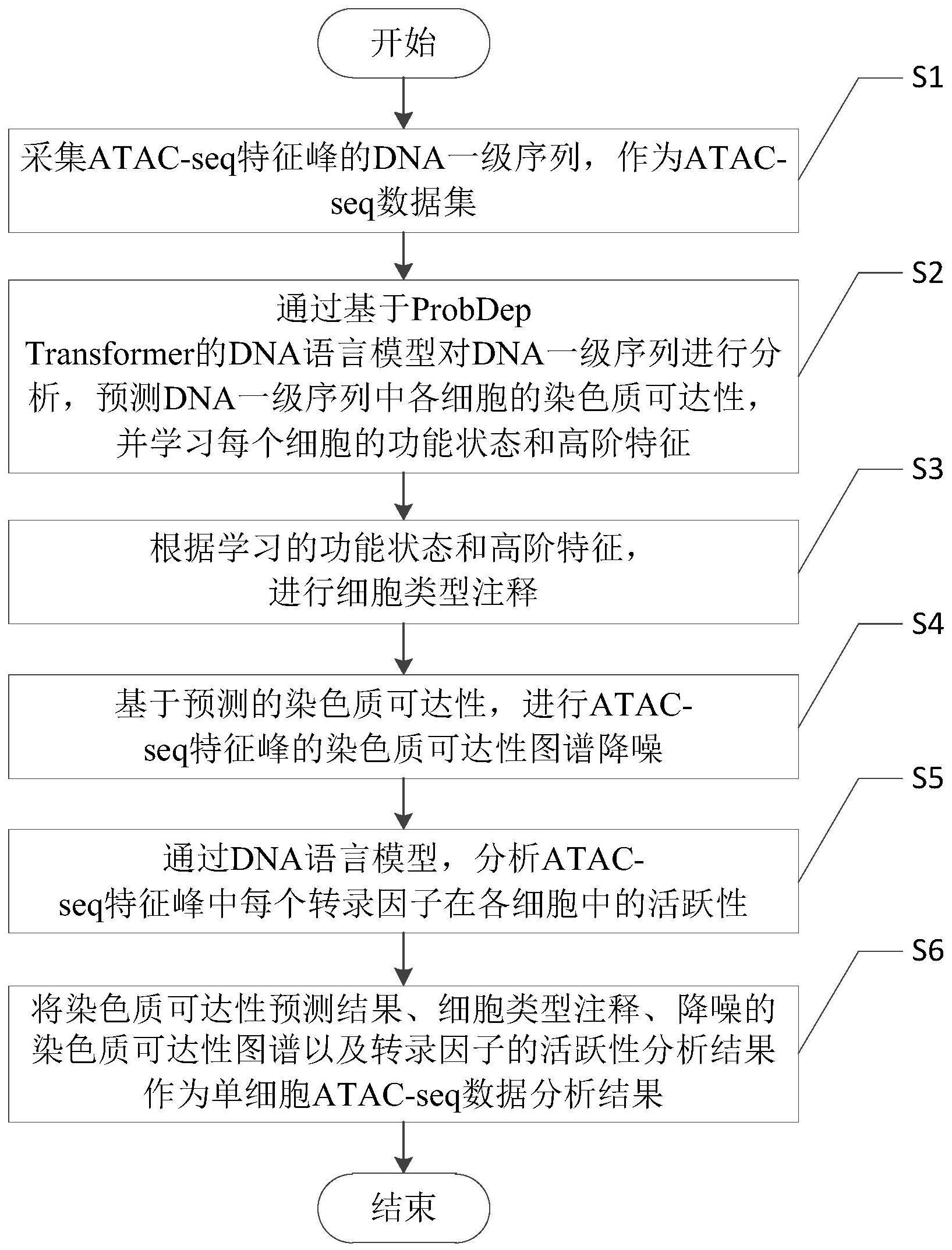
3、s1、采集atac-seq特征峰的dna一级序列,作为atac-seq数据集;
4、s2、通过基于probdep transformer的dna语言模型对dna一级序列进行分析,预测dna一级序列中各细胞的染色质可达性,并学习每个细胞的功能状态和高阶特征;
5、s3、根据学习的功能状态和高阶特征,进行细胞类型注释;
6、s4、基于预测的染色质可达性,进行atac-seq特征峰的染色质可达性图谱降噪;
7、s5、通过dna语言模型,分析atac-seq特征峰中每个转录因子在各细胞中的活跃性;
8、s6、将染色质可达性预测结果、细胞类型注释、降噪的染色质可达性图谱以及转录因子的活跃性分析结果作为单细胞atac-seq数据分析结果。
9、进一步地,所述步骤s2具体为:
10、s21、将长度为l的dna一级序列采用独热编码映射至维数为4×l的隐式特征空间中,并将其转换为基元编码矩阵;
11、对dna一级序列采用绝对位置编码生成维数为pos×2i的位置编码矩阵,将基元编码矩阵和位置编码矩阵相加作为dna语言模型的输入数据;其中,pos为当前转录因子-dna结合基元在dna一级序列中的位置下标,2i为当前转录因子-dna结合基元的位置编码向量的长度;
12、s22、在dna模型中,采用长距离依赖性测量评估查询与键之间的依赖性的方法对输入数据进行分析,获得每个键向量聚焦于排名最高的u个查询向量,进而得到dna模型的输出数据;
13、s23、将dna模型的输出数据作为dna一级序列的高维语义编码,通过序列高阶编码器将其映射到低维空间中,获得dna一级序列的高阶特征;
14、s24、根据获得的高阶特征,通过染色质可达性预测器预测当前dna一级序列在各细胞的染色质可达性大小,并学习得到每个细胞的功能状态和高阶特征。
15、进一步地,所述步骤s22中,长距离依赖性测量的表达式为:
16、
17、式中,为长距离依赖性测量操作,表示qi向量与全部键向量之间经过log-sum-exp操作之后的结果,表示log-sum-exp结果的算术平均值,qi为查询矩阵q的第i行,k为键矩阵,in为求对数操作,l为键矩阵k中当前行的下标,lk为键矩阵k中行的个数,为键矩阵k中的第k行的转置,d为键(key)矩阵k中列的个数;
18、dna模型的输出数据表示为:
19、
20、式中,为自注意力机制操作的输出,为自注意力机制操作,q,k,v分别为查询矩阵,键矩阵,值矩阵,softmax(·)为激活函数,为与矩阵q大小相同的稀疏矩阵,且只包含了了长距离依赖性测量中排名最高的u个查询向量。
21、进一步地,所述步骤s23中,dna一级序列的高阶特征为:
22、
23、式中,elu(·)为elu激活函数,wf和bf分别为序列高阶编码器的权重矩阵和截距向量,conv1(·)为1维卷积操作。
24、进一步地,当前dna一级序列在各细胞的染色质可达性大小y为:
25、
26、式中,σ(·)为sigmoid激活函数,wp和bp分为染色质可达性预测器的权重矩阵和截距向量,其中,权重矩阵作为全部细胞的功能状态和高阶特征矩阵。
27、进一步地,所述步骤s3具体为:
28、s31、根据学习的功能状态和高阶特征构建k-近邻图;
29、其中,在构建的k-近邻图中节点为细胞,边为细胞之间的相关性;
30、s32、在构建的k-近邻图中,划分每个细胞的类型,并进一步在二维空间中可视化细胞类型注释结果。
31、进一步地,所述步骤s4具体为:
32、s41、获取原始atac-seq数据,即染色质可达性特征峰矩阵;
33、s42、通过混合概率模型搜索染色质可达性特征峰矩阵中由实验误差导致的零计数,进而计算特征峰i在细胞类型k的细胞m中的丢失率;
34、s43、对于给定的细胞m,根据其每个特征峰的丢失率,将全部特征峰划分为待修正的特征峰集合am和无需修正的特征峰集合bm;
35、s44、将通过dna语言模型预测的特征峰i在每个细胞中的染色质可达性大小作为候选修正计数,对特征峰集合am中的特征峰进行修正,进而获得降噪后的atac-seq特征峰的染色质可达性图谱。
36、进一步地,所述步骤s42中的混合概率模型包括伽马分布模型和正态分布模型,其中伽马分布模型表示实验误差,正态分布模型表示scatac-seq特征峰的真实计数。
37、进一步地,所述步骤s42中,特征峰i在细胞类型k的细胞m中的丢失率dim为:
38、
39、式中,表示参数的估计值,为特征峰i在细胞类型k中的整体丢失率,和为伽马分布的形状参数和尺度参数,和为正态分布的均值和标准差。
40、进一步地,所述步骤s5具体为:
41、s51、随机选择s条scatac-seq特征峰序列进行二核苷酸打乱生成s条的背景序列;
42、s52、设定一个转录因子,将其与dna结合基元插入到s条背景序列中心位置,生成s条合成序列;
43、s53、通过dna模型分别预测s条背景序列和s条合成序列的染色质可达性;
44、s54、将单个细胞的背景序列和合成序列的平均染色质可达性预测差值作为当前转录因子在该单个细胞中的活跃性分析结果。
45、本发明的有益效果为:
46、1)本发明利用prob transformer有效捕捉了scatac-seq特征峰序列中各类转录因子-dna结合基元的所属种类、相对位置、长距离依赖关系,从而更精准、更全面地刻画了单个细胞的功能状态和高阶特征;
47、2)本发明方法能够将染色质可达性预测、细胞类型注释、染色质可达性图谱降噪、转录因子活性推断任务集成到了一个的框架中,显著地提高了单细胞atac-seq数据计算分析框架的通用性、统一性;
48、3)本发明方法支持在多gpu并行运算,可用于超大规模单细胞atac-seq数据的分析。
- 还没有人留言评论。精彩留言会获得点赞!