乳液法制备球形化有机小分子单体或复合物的方法与流程
本发明属于材料球形化技术,具体为一种制备球形化有机小分子单体和复合物的方法。
背景技术:
随着现代战争形势的变化,对于炸药的要求也越来越高,在要求高爆速和能量大的同时,也要求炸药具有更好的安全性,以避免运输和使用过程中意外引发事故和爆炸。因此高能量和低感度这两种看似矛盾的特性,成为现在含能材料领域研究者的最高追求目标。目前降感的改性方法有复合、球形化和超细化等,由于含能材料特别是有机小分子含能材料分子间的相互作用和其危险特性等,使上述改性降感技术的实现具有较大的难度,并且单一的降感技术对高能炸药的降感不是很明显,因此迫切需要一种高效的降感技术。若将超细、球形化、复合这几种降感的方法结合在一起,可以大幅度降低炸药的感度,同时提高炸药的装填密度和流动性,对现有含能材料的研究有着重大意义,是一种有效的改性手段。王相元等以乙酸乙酯为溶剂,采用重结晶法通过控制反应温度和反溶剂滴加速率等条件制备出特性落高为15.5cm的低感度球形化太安(中北大学,2009,硕士学位论文)。高思静等以N-甲基吡咯烷酮为溶剂、丙酮为非溶剂采用溶剂非溶剂法将堆积密度较低的针状硝基胍重结晶制备成平均粒径为90μm、结构密实、晶体缺陷少、热分解温度提高的球形硝基胍(火炸药学报,2014,12,37(6))。赵雪等采用重结晶法得到了类球形的RDX晶体,该晶体形状规则、表面光滑、棱角少、流散性好,撞击感度、摩擦感度比普通RDX略有降低,以球形化RDX为基的PBX相比以普通RDX为基的PBX冲击波感度降低约25%,但该方法制备的晶体球形度较差,粒径也较大(北京理工大学学报,2011,1,31(1))。
另一方面,有机小分子广泛应用于药物、导电有机晶体、非线性光学晶体、染料、照相原料色素及农用化学品,其结构及形貌与性能密切相关。若将其制备成超细球形化单体或复合物,可以在较大程度上扩展其研究和应用领域。目前,药物复合研究大多仍停留在其结构的检测分析以及复合试剂的选择等基础理论方面,而对其应用研究尚待加强,比如改变药物的熔点,药物的溶解度,提高药物的稳定性和生物利用度及生物活性研究与药物学研究等。随着复合基础理论研究的深入发展,会有更多的药物以复合的形式进入应用研究阶段,进一步推动药物复合在药学领域的应用。
技术实现要素:
本发明的一个目的是解决至少上述问题和/或缺陷,并提供至少后面将说明的优点。
为了实现根据本发明的这些目的和其它优点,提供了一种乳液法制备球形化有机小分子单体或复合物的方法,包括以下步骤:
步骤一、将一种或多种有机小分子加入到溶剂-非溶剂中,在一定温度下搅拌并加入表面活性剂,待乳液分散均匀后,超声或机械乳化,得到均匀分散的乳液,采用晶体析出法让晶体析出,得到固液混合物;
步骤二、将固液混合物过滤、洗涤和干燥得到单体或复合物球形晶体。
优选的是,所述多种有机小分子采用两种有机小分子,其质量比为0.01:1~1:100。
优选的是,所述的有机小分子为六硝基六氮杂异伍兹烷、黑索今、奥克托今、高氯酸铵、二硝酰胺铵、硝酸铵、5,5’-联四唑-1,1’-二氧羟铵盐、3,3′-二氨基-4,4′-偶氮呋咱、3,3′-二氨基-4,4′-氧化偶氮呋咱、1,1-二氨基-2,2-二硝基乙烯、2,4,6-三硝基甲苯、苦味酸、1,3-二硝基苯、1,2-二硝基苯、对硝基氯苯、对硝基苯胺、对硝基苯酚、3,5-二硝基苯胺、3,5-二硝基甲苯、2,4-二硝基甲苯、2,4-二硝基苯酚、3,5-二硝基苯甲酸、硝化纤维素、依托咪酯、三聚氰胺、立痛定、乙嘧啶、茶碱、环丙沙星、诺氟沙星、阿司匹林、昔多芬、布洛芬、呋喃苯胺酸、己酮可可碱、扑热息痛中的一种或多种。
优选的是,所述溶剂-非溶剂的质量比为0.01:1~1:100;所述搅拌的转速为0~2000rpm;所述一定温度为-10~90℃;所述表面活性剂的用量为0.0001~1倍的有机小分子总质量;所述超声乳化的时间为0.01~600min,超声乳化的功率为0~100000W。
优选的是,所述溶剂-非溶剂为水、甲醇、乙醇、乙酸、乙酸乙酯、乙酸丁酯、乙酸异戊酯、丙酮、正丁酮、甲基异丁基酮、环己烷、正丁烷、环己酮、甲苯环己酮、甲基丁酮、氯苯、二氯苯、二氯甲烷、氯仿、四氯化碳、苯、甲苯、二甲苯、二甲基亚砜、N,N二甲基甲酰胺、乙醚、石油醚、环氧丙烷、乙二醇醚、乙腈中的一种或多种。
优选的是,所述表面活性剂为阿拉伯胶、虫胶、聚乙烯吡咯烷酮、十二烷基苯磺酸钠、十六烷基吡啶、十二烷基硫酸钠、十六烷基硫酸钠、十八烷基硫酸钠、二辛烷琥珀酸磺酸钠、明胶、聚乙烯醇、聚乙二醇、二氯甲烷、司班20~80、吐温20~80中的一种或多种。
优选的是,所述晶体析出法为自然挥发、升温、减压蒸馏法、冷冻干燥、超临界萃取、电泳、加入非溶剂的萃取法中的一种或多种;所述萃取法中非溶剂的用量为0.01~100倍的溶剂质量;升温的温度范围为0~90℃;减压蒸馏的压力范围在0~0.9atm;所述冷冻干燥包括以下步骤:
步骤Ⅰ、预冷冻:冷冻温度-50~-70℃,冷冻时间1~2小时;
步骤Ⅱ、预冷冻后升温至20~25℃,保持1~2小时;
步骤Ⅲ、将步骤Ⅱ得到的晶体加入真空冷冻干燥机中,设置冷阱温度为-60~-95℃,真空度为20~60pa,冷冻干燥时间8-36h。
优选的是,所述有机小分子、溶剂和非溶剂的加入顺序可任意调换;所述表面活性剂的加入顺序为非溶剂加入之后。
优选的是,所述步骤一的过程替换为:将一种或多种有机小分子加入到装有超声波装置的超临界反应装置中,然后加入溶剂-非溶剂和表面活性剂,在体系密封后通入二氧化碳至15~45MPa,并设定体系温度在40~90℃;在施加超声的超临界二氧化碳体系中搅拌混合1~3小时,然后卸去二氧化碳压力,温度为-10~90℃,搅拌0.5~1小时,然后再次注入二氧化碳至压力为50~80MPa,在施加超声的超临界二氧化碳体系中搅拌1~2小时,卸压,得到固液混合物。
优选的是,所述搅拌的速度为800~1500rpm;所述超声的频率为20~60kHz,超声波功率密度为1000~2000W/L,超声波连续辐照或者间歇辐照,间歇辐照时的间歇时间为6~12s/6~12s。
本发明至少包括以下有益效果:将有机小分子的复合、超细化、球形化三者结合起来,对于含能材料来说可以大幅度降低感度、提高流散性和装填密度,同时降低其在含能材料的3D打印或PBX炸药浆料的粘度;对于药物来说可以改变其熔点、溶解度,提高药物的稳定性和生物利用度,同时有利于将其压成药片;对于导电有机晶体、非线性光学晶体、染料、照相原料色素及农用化学品来说,可以扩展其研究及应用领域。其显著优点:(1)操作简单、效率高且成本地,可以批量化生产;(2)实验条件温和,产品品质较高;(3)溶剂或非溶剂可以采用水,环保安全。
本发明的其它优点、目标和特征将部分通过下面的说明体现,部分还将通过对本发明的研究和实践而为本领域的技术人员所理解。
附图说明:
图1为本发明实施例制备的球形化有机小分子单体或复合物的扫描电镜图;
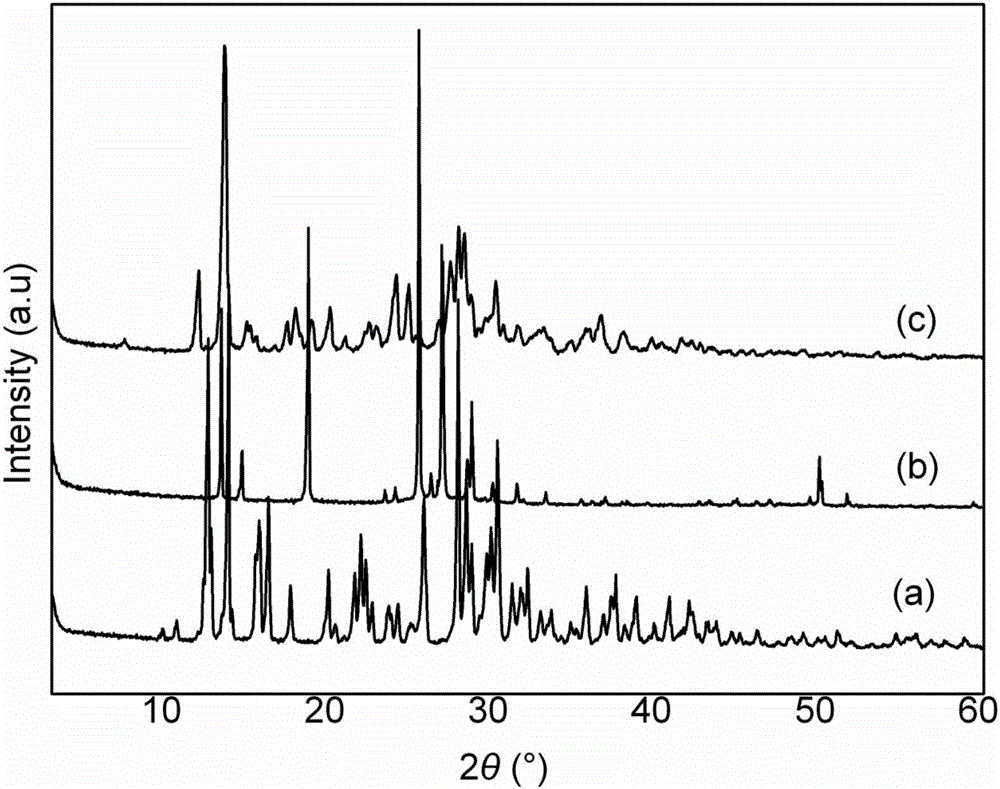
图2为本发明实施例制备的球形化六硝基六氮杂异伍兹烷(CL-20)/对硝基氯苯(PNCB)的XRD图。
具体实施方式:
下面结合附图对本发明做进一步的详细说明,以令本领域技术人员参照说明书文字能够据以实施。
应当理解,本文所使用的诸如“具有”、“包含”以及“包括”术语并不配出一个或多个其它元件或其组合的存在或添加。
实施例1:
步骤一、以六硝基六氮杂异伍兹烷(CL-20)/对硝基氯苯(PNCB)=1:3的摩尔比投料,分别称量0.438g的CL-20和0.479g的PNCB加入到盛有5mL乙酸乙酯的反应瓶中,再加入16mL蒸馏水,然后放在搅拌器中在40℃下以1000rpm进行搅拌,在搅拌过程中加入2×10-4g表面活性剂明胶,待乳液分散均匀后,以1200W的功率进行超声乳化60min,得到均匀分散的乳液;采用加入非溶剂乙醇萃取的方法让晶体析出,得到固液混合物;所述非溶剂乙醇的用量为10倍的溶质质量;
步骤二、将固液混合物过滤、洗涤和干燥得到复合物球形晶体;图1示出了c)CL-20/PNCB复合物共晶的扫描电镜示意图;从图1中可以看出,CL-20/PNCB复合物共晶的粒径分布均匀,颗粒表面光滑致密,该结构极大地降低了炸药的感度,同时增加了流散性和装填密度,为高能炸药的应用展现出了良好的应用前景;图2示出了CL-20/PNCB球形复合物的粉末XRD结构,其中a)CL-20,b)PNCB,c)CL-20/PNCB复合物,同两种原料相比,复合物在2θ为11.97°,13.57°,24.80°,and 28.21°的位置出现了新峰,在25.37°,27.14°,and 49.97°位置出现了偏移,分析认为,CL-20和PNCB分子间的氢键作用导致了其在重结晶的过程中出现了一种新的结构,该结构使得两者单质能更加均匀和稳定的结合,为其性能的提升提供了条件。
实施例2:
步骤一、以CL-20/2,4,6-三硝基甲苯(TNT)/PNCB=1:2:3的摩尔比投料,分别称量0.438g CL-20、0.2g TNT和0.479g PNCB加入到盛有7mL乙酸乙酯的反应瓶中,再加入28mL的水,然后放在搅拌器中在40℃下以800rpm进行搅拌,在搅拌过程中加入1.5×10-4g表面活性剂明胶,待乳液分散均匀后,以1200W的功率进行超声乳化60min,得到均匀分散的乳液;采用加入非溶剂乙醇萃取的方法让晶体析出;得到固液混合物;所述非溶剂乙醇的用量为15倍的溶质质量;
步骤二、将固液混合物过滤、洗涤和干燥得到复合物球形晶体。
实施例3:
步骤一、以CL-20/PNCB=1:3的摩尔比投料,分别称量0.438g的CL-20和0.479g的PNCB加入到盛有7mL乙酸乙酯的反应瓶中,再加入28mL的水,然后放在搅拌器中在40℃下以1000rpm进行搅拌,在搅拌过程中加入4×10-4g表面活性剂明胶,待乳液分散均匀后,以1200W的功率进行超声乳化60min,得到均匀分散的乳液;采用升温至90℃,让晶体析出;得到固液混合物;
步骤二、将固液混合物过滤、洗涤和干燥得到复合物球形晶体。
实施例4:
步骤一、以CL-20/PNCB=1:3的摩尔比投料,分别称量0.438g的CL-20和0.479g的PNCB加入到盛有3mL乙酸乙酯的反应瓶中,再加入28mL的水,然后放在搅拌器中在40℃下以1000rpm进行搅拌,在搅拌过程中加入5×10-4g表面活性剂二氯甲烷,待乳液分散均匀后,以1200W的功率进行超声乳化60min,得到均匀分散的乳液;采用减压蒸馏的方法让晶体析出,减压蒸馏的压力在0.5atm;得到固液混合物;
步骤二、将固液混合物过滤、静置、干燥,得到复合物晶体。
实施例5:
步骤一、以CL-20/PNCB=2:5的摩尔比投料,分别称量0.438g的CL-20和0.396g的PNCB加入到盛有10mL乙酸异戊酯的反应瓶中,再加入28mL的水,然后放在搅拌器中在40℃下以800rpm进行搅拌,在搅拌过程中加入5×10-4g表面活性剂二氯甲烷,待乳液分散均匀后,以1200W的功率进行超声乳化60min,得到均匀分散的乳液;采用减压蒸馏的方法让晶体析出,减压蒸馏的压力在0.5atm;得到固液混合物;
步骤二、将固液混合物过滤、静置、干燥,得到复合物晶体。
实施例6:
步骤一、以立痛定/对氨基苯甲酸=3:5的摩尔比投料,分别称量0.354g的立痛定和0.347g的对氨基苯甲酸加入到盛有10mL乙酸乙酯的反应瓶中,再加入8mL的水,然后放在搅拌器中在45℃下以1200rpm进行搅拌,在搅拌过程中加入2×10-4g表面活性剂聚乙烯醇,待乳液分散均匀后,以1200W的功率进行超声乳化60min,得到均匀分散的乳液;采用加入非溶剂乙醇萃取的方法让晶体析出;得到固液混合物;所述非溶剂乙醇的用量为15倍的溶质质量;
步骤二、将固液混合物过滤、洗涤和干燥得到复合物球形晶体。
实施例7:
以立痛定/对氨基苯甲酸=3:5的摩尔比投料,分别称量0.354g的立痛定和0.347g的对氨基苯甲酸加入到盛有8mL乙酸乙酯的反应瓶中,再加入28mL的水,然后放在搅拌器中在45℃下以1200rpm进行搅拌,在搅拌过程中加入1.5×10-4g表面活性剂吐温,待乳液分散均匀后,以1200W的功率进行超声乳化60min,得到均匀分散的乳液。采用加入非溶剂乙醇萃取的方法让晶体析出;得到固液混合物;所述非溶剂乙醇的用量为15倍的溶质质量;
步骤二、将固液混合物过滤、洗涤和干燥得到复合物球形晶体。
实施例8:
以立痛定/对氨基苯甲酸=3:5的摩尔比投料,分别称量0.354g的立痛定和0.347g的对氨基苯甲酸加入到盛有8mL乙酸乙酯的反应瓶中,再加入28mL的水,然后放在搅拌器中在60℃下以500rpm进行搅拌,在搅拌过程中加入4×10-4g表面活性剂司班-20,待乳液分散均匀后,以1200W的功率进行超声乳化60min,得到均匀分散的乳液;采用升温至90℃,让晶体析出;得到固液混合物;
步骤二、将固液混合物过滤、洗涤和干燥得到复合物球形晶体。
实施例9:
步骤一、以立痛定/对氨基苯甲酸=3:5的摩尔比投料,分别称量0.354g的立痛定和0.347g的对氨基苯甲酸加入到盛有5mL乙酸乙酯的反应瓶中,再加入28mL的水,然后放在搅拌器中在70℃下以2000rpm进行搅拌,在搅拌过程中加入5×10-4g表面活性剂司班-80,待乳液分散均匀后,以1800W的功率进行超声乳化60min,得到均匀分散的乳液;采用减压蒸馏的方法让晶体析出,减压蒸馏的压力在0.5atm;得到固液混合物;
步骤二、将固液混合物过滤、静置、干燥,得到复合物晶体。
实施例10:
步骤一、以立痛定/对氨基苯甲酸=2:7的摩尔比投料,分别称量0.236g的立痛定和0.503g的对氨基苯甲酸加入到盛有15mL乙酸乙酯的反应瓶中,再加入40mL的水,然后放在搅拌器中在50℃下以1800rpm进行搅拌,在搅拌过程中加入5×10-4g表面活性剂虫胶,待乳液分散均匀后,以2000W的功率进行超声乳化120min,得到均匀分散的乳液;采用减压蒸馏的方法让晶体析出,减压蒸馏的压力在0.5atm;得到固液混合物;
步骤二、将固液混合物过滤、静置、干燥,得到复合物晶体。
实施例11:
步骤一、以CL-20/TNT=1:2的摩尔比投料,分别称量0.438g CL-20、0.2g TNT加入到盛有7mL乙酸乙酯的反应瓶中,再加入8mL的水,然后放在搅拌器中在60℃下以1600rpm进行搅拌,在搅拌过程中加入2.5×10-4g表面活性剂明胶,待乳液分散均匀后,以1600W的功率进行超声乳化240min,得到均匀分散的乳液;采用加入非溶剂乙醇萃取的方法让晶体析出;得到固液混合物;所述非溶剂乙醇的用量为12倍的溶质质量;
步骤二、将固液混合物过滤、洗涤和干燥得到复合物球形晶体;图1示出了d)CL-20/TNT共晶的扫描电镜示意图;从图1中可以看出,CL-20/TNT共晶的粒径分布均匀,颗粒表面光滑致密,该结构极大地降低了炸药的感度,同时增加了流散性和装填密度,为高能炸药的应用展现出了良好的应用前景。
实施例12:
步骤一、取质量比为1:2:2的CL-20、TNT和3,3′-二氨基-4,4′-氧化偶氮呋咱(DAOAF)有机小分子加入到盛有乙酸乙酯的反应器中,再加入水,然后放在搅拌器中在60℃下以1600rpm进行搅拌,在搅拌过程中加入表面活性剂十八烷基硫酸钠,待乳液分散均匀后,以1600W的功率进行超声乳化240min,得到均匀分散的乳液;采用加入非溶剂乙醇萃取的方法让晶体析出;得到固液混合物;所述非溶剂乙醇的用量为12倍的溶质质量;所述乙酸乙酯与水的质量比为1:10;所述乙酸乙酯和水的总质量为25倍的有机小分子的总质量;所述表面活性剂的用量为0.01倍的有机小分子总质量;
步骤二、将固液混合物过滤、洗涤和干燥得到复合物球形晶体。
实施例13:
步骤一、取质量比为3:1:2的黑索今(RDX)、硝酸铵(AN)和呋喃苯胺酸有机小分子加入到盛有乙酸异戊酯的反应器中,再加入水,然后放在搅拌器中在50℃下以1200rpm进行搅拌,在搅拌过程中加入表面活性剂十八烷基硫酸钠,待乳液分散均匀后,以1600W的功率进行超声乳化120min,得到均匀分散的乳液;采用超临界萃取的方法让晶体析出;得到固液混合物;所述乙酸异戊酯与水的质量比为1:12;所述乙酸异戊酯和水的总质量为30倍的有机小分子的总质量;所述表面活性剂的用量为0.01倍的有机小分子总质量;
步骤二、将固液混合物过滤、洗涤和干燥得到复合物球形晶体。
实施例14:
步骤一、取质量比为2:1:2的奥克托今(HMX)、1,1-二氨基-2,2-二硝基乙烯(FOX-7)和己酮可可碱有机小分子加入到盛有甲苯环己酮的反应器中,再加入水,然后放在搅拌器中在50℃下以1200rpm进行搅拌,在搅拌过程中加入表面活性剂十六烷基吡啶,待乳液分散均匀后,以1600W的功率进行超声乳化120min,得到均匀分散的乳液;采用减压蒸馏的方法让晶体析出,减压蒸馏的压力在0.5atm;得到固液混合物;所述甲苯环己酮与水的质量比为1:12;所述甲苯环己酮和水的总质量为30倍的有机小分子的总质量;所述表面活性剂的用量为0.01倍的有机小分子总质量;
步骤二、将固液混合物过滤、洗涤和干燥得到复合物球形晶体。
实施例15:
步骤一、取质量比为2:1:2:1的奥克托今(HMX)、CL-20、5,5’-联四唑-1,1’-二氧羟铵盐(TKX-50)和三聚氰胺有机小分子加入到盛有甲苯环己酮的反应器中,再加入水,然后放在搅拌器中在50℃下以1500rpm进行搅拌,在搅拌过程中加入表面活性剂虫胶,待乳液分散均匀后,以1600W的功率进行超声乳化120min,得到均匀分散的乳液;采用升温至90℃,让晶体析出;得到固液混合物;所述甲苯环己酮与水的质量比为1:18;所述甲苯环己酮和水的总质量为25倍的有机小分子的总质量;所述表面活性剂的用量为0.008倍的有机小分子总质量;
步骤二、将固液混合物过滤、洗涤和干燥得到复合物球形晶体。
实施例16:
步骤一、取质量比为2:3:2:1的黑索今(RDX)、CL-20、对硝基苯酚(PNP)和立痛定有机小分子加入到盛有二氯苯的反应器中,再加入水,然后放在搅拌器中在50℃下以1500rpm进行搅拌,在搅拌过程中加入表面活性剂聚乙烯吡咯烷酮,待乳液分散均匀后,以800W的功率进行超声乳化360min,得到均匀分散的乳液;采用冷冻干燥让晶体析出;得到固液混合物;所述二氯苯与水的质量比为1:15;所述二氯苯和水的总质量为50倍的有机小分子的总质量;所述表面活性剂的用量为0.01倍的有机小分子总质量;所述冷冻干燥包括以下步骤:
步骤Ⅰ、预冷冻:冷冻温度-50℃,冷冻时间1小时;
步骤Ⅱ、预冷冻后升温至20℃,保持1小时;
步骤Ⅲ、将步骤Ⅱ得到的晶体加入真空冷冻干燥机中,设置冷阱温度为-60℃,真空度为20pa,冷冻干燥时间8h;
步骤二、将固液混合物过滤、洗涤和干燥得到复合物球形晶体。
实施例17:
步骤一、取质量比为3:2:1的CL-20、奥克托今和依托咪酯有机小分子加入到盛有环氧丙烷的反应器中,再加入水,然后放在搅拌器中在50℃下以1500rpm进行搅拌,在搅拌过程中加入表面活性剂聚乙烯吡咯烷酮,待乳液分散均匀后,以800W的功率进行超声乳化360min,得到均匀分散的乳液;采用冷冻干燥让晶体析出;得到固液混合物;所述环氧丙烷与水的质量比为1:12;所述环氧丙烷和水的总质量为40倍的有机小分子的总质量;所述表面活性剂的用量为0.005倍的有机小分子总质量;所述冷冻干燥包括以下步骤:
步骤Ⅰ、预冷冻:冷冻温度-60℃,冷冻时间2小时;
步骤Ⅱ、预冷冻后升温至25℃,保持1.5小时;
步骤Ⅲ、将步骤Ⅱ得到的晶体加入真空冷冻干燥机中,设置冷阱温度为-80℃,真空度为50pa,冷冻干燥时间12h;
步骤二、将固液混合物过滤、洗涤和干燥得到复合物球形晶体。
实施例18:
步骤一、取质量比为3:2:1:1的CL-20、1,1-二氨基-2,2-二硝基乙烯、硝化纤维素和扑热息痛有机小分子加入到盛有环己烷的反应器中,再加入水,然后放在搅拌器中在60℃下以1000rpm进行搅拌,在搅拌过程中加入表面活性剂聚乙二醇,待乳液分散均匀后,以3000W的功率进行超声乳化360min,得到均匀分散的乳液;采用超临界让晶体析出;得到固液混合物;所述环己烷与水的质量比为1:20;所述环己烷和水的总质量为60倍的有机小分子的总质量;所述表面活性剂的用量为0.008倍的有机小分子总质量;
步骤二、将固液混合物过滤、洗涤和干燥得到复合物球形晶体。
实施例19:
步骤一、取质量比为3:2:1:1的3,3′-二氨基-4,4′-偶氮呋咱、3,5-二硝基苯胺、乙嘧啶和昔多芬有机小分子加入到盛有N,N二甲基甲酰胺的反应器中,再加入水,然后放在搅拌器中在60℃下以1000rpm进行搅拌,在搅拌过程中加入表面活性剂聚乙二醇,待乳液分散均匀后,以1800W的功率进行超声乳化120min,得到均匀分散的乳液;采用超临界让晶体析出;得到固液混合物;所述N,N二甲基甲酰胺与水的质量比为1:10;所述N,N二甲基甲酰胺和水的总质量为30倍的有机小分子的总质量;所述表面活性剂的用量为0.005倍的有机小分子总质量;
步骤二、将固液混合物过滤、洗涤和干燥得到复合物球形晶体。
实施例20:
步骤一、取质量比为3:2:1:1的CL-20、3,3′-二氨基-4,4′-偶氮呋咱、乙嘧啶和阿司匹林有机小分子加入到盛有N,N二甲基甲酰胺的反应器中,再加入水,然后放在搅拌器中在60℃下以1000rpm进行搅拌,在搅拌过程中加入表面活性剂环己酮,待乳液分散均匀后,以1800W的功率进行超声乳化120min,得到均匀分散的乳液;采用超临界让晶体析出;得到固液混合物;所述环己酮与水的质量比为1:20;所述环己酮和水的总质量为40倍的有机小分子的总质量;所述表面活性剂的用量为0.008倍的有机小分子总质量;
步骤二、将固液混合物过滤、洗涤和干燥得到复合物球形晶体。
实施例21:
步骤一、取质量比为2:2:1:1的CL-20、苦味酸、环丙沙星和布洛芬有机小分子加入到装有超声波装置的超临界反应装置中,然后加入二甲基亚砜、水和表面活性剂十二烷基苯磺酸钠,在体系密封后通入二氧化碳至15MPa,并设定体系温度在40℃;在施加超声的超临界二氧化碳体系中搅拌混合1小时,然后卸去二氧化碳压力,温度为20℃,搅拌0.5小时,然后再次注入二氧化碳至压力为50MPa,在施加超声的超临界二氧化碳体系中搅拌1小时,卸压,得到固液混合物;所述搅拌的速度为800rpm;所述超声的频率为20kHz,超声波功率密度为1000W/L,超声波采用间歇辐照,间歇辐照时的间歇时间为6s/6s(超声连续辐照时间/超声间歇时间);
步骤二、将固液混合物过滤、洗涤和干燥得到复合物球形晶体。
实施例22:
步骤一、取质量比为2:1的CL-20和3,5-二硝基甲苯有机小分子加入到装有超声波装置的超临界反应装置中,然后加入二甲基亚砜、水和表面活性剂十二烷基苯磺酸钠,在体系密封后通入二氧化碳至45MPa,并设定体系温度在90℃;在施加超声的超临界二氧化碳体系中搅拌混合3小时,然后卸去二氧化碳压力,温度为20℃,搅拌1小时,然后再次注入二氧化碳至压力为80MPa,在施加超声的超临界二氧化碳体系中搅拌2小时,卸压,采用电泳法让晶体析出,得到固液混合物;所述搅拌的速度为800rpm;所述超声的频率为60kHz,超声波功率密度为2000W/L,超声波采用间歇辐照,间歇辐照时的间歇时间为12s/12s(超声连续辐照时间/超声间歇时间);
步骤二、将固液混合物过滤、洗涤和干燥得到复合物球形晶体。
实施例23:
步骤一、取质量比为1:2的奥克托今和硝化纤维素有机小分子加入到装有超声波装置的超临界反应装置中,然后加入四氯化碳、水和表面活性剂阿拉伯胶,在体系密封后通入二氧化碳至30MPa,并设定体系温度在50℃;在施加超声的超临界二氧化碳体系中搅拌混合2小时,然后卸去二氧化碳压力,温度为25℃,搅拌0.6小时,然后再次注入二氧化碳至压力为60MPa,在施加超声的超临界二氧化碳体系中搅拌2小时,卸压,采用电泳法让晶体析出,得到固液混合物;所述搅拌的速度为1000rpm;所述超声的频率为40kHz,超声波功率密度为1500W/L,超声波采用间歇辐照,间歇辐照时的间歇时间为6s/8s(超声连续辐照时间/超声间歇时间);
步骤二、将固液混合物过滤、洗涤和干燥得到复合物球形晶体。
实施例24:
步骤一、取质量比为2:1:2:1的CL-20、黑索今、茶碱和昔多芬有机小分子加入到装有超声波装置的超临界反应装置中,然后加入乙醚、水和表面活性剂二辛烷琥珀酸磺酸钠,在体系密封后通入二氧化碳至30MPa,并设定体系温度在60℃;在施加超声的超临界二氧化碳体系中搅拌混合1小时,然后卸去二氧化碳压力,温度为5℃,搅拌0.5小时,然后再次注入二氧化碳至压力为70MPa,在施加超声的超临界二氧化碳体系中搅拌2小时,卸压,采用冷冻干燥法让晶体析出,得到固液混合物;所述搅拌的速度为800rpm;所述超声的频率为20kHz,超声波功率密度为1000W/L,超声波采用间歇辐照,间歇辐照时的间歇时间为6s/6s(超声连续辐照时间/超声间歇时间);所述冷冻干燥包括以下步骤:
步骤Ⅰ、预冷冻:冷冻温度-60℃,冷冻时间2小时;
步骤Ⅱ、预冷冻后升温至25℃,保持1.5小时;
步骤Ⅲ、将步骤Ⅱ得到的晶体加入真空冷冻干燥机中,设置冷阱温度为-80℃,真空度为50pa,冷冻干燥时间36h;
步骤二、将固液混合物过滤、洗涤和干燥得到复合物球形晶体。
实施例25:
步骤一、取质量比为2:1:2:1:1的CL-20、高氯酸铵、奥克托今、立痛定和对硝基苯酚有机小分子加入到装有超声波装置的超临界反应装置中,然后加入氯苯、水和表面活性剂十六烷基吡啶,在体系密封后通入二氧化碳至40MPa,并设定体系温度在50℃;在施加超声的超临界二氧化碳体系中搅拌混合1小时,然后卸去二氧化碳压力,温度为5℃,搅拌0.5小时,然后再次注入二氧化碳至压力为80MPa,在施加超声的超临界二氧化碳体系中搅拌2小时,卸压,采用电泳法让晶体析出,得到固液混合物;所述搅拌的速度为800rpm;所述超声的频率为50kHz,超声波功率密度为2000W/L,超声波采用间歇辐照,间歇辐照时的间歇时间为6s/10s(超声连续辐照时间/超声间歇时间);所述冷冻干燥包括以下步骤:
步骤二、将固液混合物过滤、洗涤和干燥得到复合物球形晶体。
实施例26:
步骤一、取质量比为3:1的奥克托今和昔多芬有机小分子加入到装有超声波装置的超临界反应装置中,然后加入乙酸乙酯、水和表面活性剂十六烷基吡啶,在体系密封后通入二氧化碳至45MPa,并设定体系温度在40℃;在施加超声的超临界二氧化碳体系中搅拌混合1小时,然后卸去二氧化碳压力,温度为0℃,搅拌0.5小时,然后再次注入二氧化碳至压力为70MPa,在施加超声的超临界二氧化碳体系中搅拌2小时,卸压,采用冷冻干燥法让晶体析出,得到固液混合物;所述搅拌的速度为800rpm;所述超声的频率为50kHz,超声波功率密度为2000W/L,超声波采用连续辐照;所述冷冻干燥包括以下步骤:
步骤Ⅰ、预冷冻:冷冻温度-70℃,冷冻时间1小时;
步骤Ⅱ、预冷冻后升温至25℃,保持2小时;
步骤Ⅲ、将步骤Ⅱ得到的晶体加入真空冷冻干燥机中,设置冷阱温度为-95℃,真空度为60pa,冷冻干燥时间12h;
步骤二、将固液混合物过滤、洗涤和干燥得到复合物球形晶体。
实施例27:
步骤一、取0.6g六硝基六氮杂异伍兹烷(CL-20)加入到盛有6mL乙酸乙酯的反应瓶中,再加入18mL蒸馏水,然后放在搅拌器中在40℃下以1000rpm进行搅拌,在搅拌过程中加入3×10-4g表面活性剂明胶,待乳液分散均匀后,以1200W的功率进行超声乳化60min,得到均匀分散的乳液;采用加入非溶剂乙醇萃取的方法让晶体析出,得到固液混合物;所述非溶剂乙醇的用量为15倍的溶质质量;
步骤二、将固液混合物过滤、洗涤和干燥得到球形晶体;图1示出了a)CL-20球形晶体的扫描电镜示意图;从图1中可以看出,CL-20球形晶体的粒径分布均匀,颗粒表面光滑致密,该结构极大地降低了炸药的感度,同时增加了流散性和装填密度,为高能炸药的应用展现出了良好的应用前景。
实施例28:
步骤一、取0.4g 2,4,6-三硝基甲苯(TNT)加入到盛有5mL乙酸乙酯的反应瓶中,再加入20mL蒸馏水,然后放在搅拌器中在40℃下以800rpm进行搅拌,在搅拌过程中加入3×10-4g表面活性剂聚乙烯吡咯烷酮,待乳液分散均匀后,以1200W的功率进行超声乳化60min,得到均匀分散的乳液;采用加入非溶剂乙醇萃取的方法让晶体析出,得到固液混合物;所述非溶剂乙醇的用量为15倍的溶质质量;
步骤二、将固液混合物过滤、洗涤和干燥得到球形晶体;图1示出了b)TNT球形晶体的扫描电镜示意图;从图1中可以看出,TNT球形晶体的粒径分布均匀,颗粒表面光滑致密,该结构极大地降低了炸药的感度,同时增加了流散性和装填密度,为高能炸药的应用展现出了良好的应用前景。
实施例29:
步骤一、取黑索今加入到盛有环氧丙烷的反应器中,再加入水,然后放在搅拌器中在50℃下以1500rpm进行搅拌,在搅拌过程中加入表面活性剂聚乙烯吡咯烷酮,待乳液分散均匀后,以800W的功率进行超声乳化360min,得到均匀分散的乳液;采用冷冻干燥让晶体析出;得到固液混合物;所述环氧丙烷与水的质量比为1:12;所述环氧丙烷和水的总质量为40倍的黑索今的质量;所述表面活性剂的用量为0.005倍的黑索今的质量;所述冷冻干燥包括以下步骤:
步骤Ⅰ、预冷冻:冷冻温度-60℃,冷冻时间2小时;
步骤Ⅱ、预冷冻后升温至25℃,保持1.5小时;
步骤Ⅲ、将步骤Ⅱ得到的晶体加入真空冷冻干燥机中,设置冷阱温度为-80℃,真空度为50pa,冷冻干燥时间12h;
步骤二、将固液混合物过滤、洗涤和干燥得到黑索今球形晶体。
实施例30:
步骤一、取呋喃苯胺酸有机小分子加入到装有超声波装置的超临界反应装置中,然后加入乙酸乙酯、水和表面活性剂十六烷基吡啶,在体系密封后通入二氧化碳至30MPa,并设定体系温度在50℃;在施加超声的超临界二氧化碳体系中搅拌混合1小时,然后卸去二氧化碳压力,温度为0℃,搅拌0.5小时,然后再次注入二氧化碳至压力为80MPa,在施加超声的超临界二氧化碳体系中搅拌2小时,卸压,得到均匀分散的乳液,采用冷冻干燥法让晶体析出,得到固液混合物;所述搅拌的速度为800rpm;所述超声的频率为50kHz,超声波功率密度为2000W/L,超声波采用连续辐照;所述冷冻干燥包括以下步骤:
步骤Ⅰ、预冷冻:冷冻温度-60℃,冷冻时间1小时;
步骤Ⅱ、预冷冻后升温至25℃,保持2小时;
步骤Ⅲ、将步骤Ⅱ得到的晶体加入真空冷冻干燥机中,设置冷阱温度为-90℃,真空度为60pa,冷冻干燥时间36h;
步骤二、将固液混合物过滤、洗涤和干燥得到呋喃苯胺酸球形晶体。
本发明中将有机小分子的复合、超细化、球形化三者结合起来,对于含能材料来说可以大幅度降低感度、提高流散性和装填密度,同时降低其在含能材料的3D打印或PBX炸药浆料的粘度;对于药物来说可以改变其熔点、溶解度,提高药物的稳定性和生物利用度,同时有利于将其压成药片;对于导电有机晶体、非线性光学晶体、染料、照相原料色素及农用化学品来说,可以扩展其研究及应用领域,并且在本发明中,采用在超临界反应装置中进行物料的乳化,通过该方法,得到的晶体的粒径分布更加均匀,颗粒表面更加光滑致密,并且晶体结构极大地降低了炸药的感度,同时增加了流散性和装填密度,为高能炸药的应用展现出了良好的应用前景。
尽管本发明的实施方案已公开如上,但其并不仅仅限于说明书和实施方式中所列运用,它完全可以被适用于各种适合本发明的领域,对于熟悉本领域的人员而言,可容易地实现另外的修改,因此在不背离权利要求及等同范围所限定的一般概念下,本发明并不限于特定的细节和这里示出与描述的图例。
- 还没有人留言评论。精彩留言会获得点赞!