采用分子印迹固相萃取法分离纯化发酵液中吡咯喹啉醌的方法与流程
本发明涉及一种采用分子印迹固相萃取法分离纯化发酵液中吡咯喹啉醌的方法,属于生物化工技术领域。
背景技术:
吡咯喹啉醌(PQQ)是一种水溶性醌类化合物,广泛存在于食物和动物中,是葡萄糖脱氢酶和乙醇脱氢酶的辅酶。由于PQQ具有很重要的生理功能,因此PQQ也被认为是一种新的B族维生素。研究表明,PQQ能够阻止淀粉样蛋白的形成,抑制C-端缩短的变异型突触核蛋白的细胞毒性,因此PQQ可以用于神经性和精神性失调症的治疗。另外,PQQ也可以有效阻止氧化应激引起的神经退行性病变。简单来说,PQQ与神经系统相关的功能有如下四种:(1)抗氧化,清除自由基;(2)影响呼吸链功能,维护线粒体能量代谢;(3)刺激神经生长因子的分泌,修复和促进神经生长;(4)延缓α-synuclein蛋白的沉积,防止神经细胞纤维化。因此,有关研究人员认为PQQ对帕金森病和老年性痴呆等多种神经变性性疾病具有潜在的治疗价值。所以,寻找简单易行的获得PQQ的方法,进行产业化生产具有重大的意义。
目前常用的PQQ合成方法主要是化学法合成,但是化学合成PQQ步骤多,产率低,异构体和副产品的去除需要多步纯化,并且需要用到多种有毒试剂,污染环境。因此,研究如何利用非化学方法获得PQQ更具有产业化意义。PQQ广泛存在于革兰阴性菌中,但合成量却有所不同,有些菌只产生痕量PQQ,供正常的生理代谢需求,如恶臭假单胞菌;还有些菌却能产生过量的PQQ,并分泌到胞外。迄今发现的能产生过量PQQ的野生菌包括氧化葡萄糖杆菌属(Gluconobacter)、无色杆菌属(Achromobacter)、甲烷单胞菌属(Methanomonas)、甲基菌属(Methy-lobacillus)、甲基单胞菌属(Methylomonas)、嗜甲基菌属(Methy-lophilus)及黄色杆菌属(Xanthobacter)等。目前,已经有采用微生物发酵的方法生产PQQ的相关研究,所使用的原始菌种PQQ产量在0.07-7mg/L,发酵时间为2-5天左右,但是这些研究中并没有提供简单易行的从微生物发酵液中分离纯化PQQ,使PQQ的制备实现工业化规模生产的方法。
分子印迹聚合物(MIPs)是一种对目标分子具有特定吸附能力的聚合物。自1972年Wulff等成功制备出MIPs以来,印迹技术研究发展迅速。目前MIPs已在色谱固定相、固相萃取、传感器、膜分离、酶催化等领域得到广泛应用。将MIPs作为固相萃取填料,可以增强萃取的选择性和吸附容量,具有很好的应用前景。
技术实现要素:
本发明所要解决的技术问题是提供一种采用分子印迹固相萃取法分离纯化发酵液中吡咯喹啉醌的方法,该方法能够简单、高效、大规模分离纯化吡咯喹啉醌,更适于吡咯喹啉醌的产业化发展。
为了实现上述目的,本发明所采用的技术方案是:
采用分子印迹固相萃取法分离纯化发酵液中吡咯喹啉醌的方法,包括以下步骤:
(1)制备:利用吡咯喹啉醌生产菌制备含有吡咯喹啉醌的发酵液;
(2)萃取分离:采用分子印迹固相萃取法对含有吡咯喹啉醌的发酵液进行萃取,将含有吡咯喹啉醌的发酵液上样于装填有分子印迹聚合物的分子印迹萃取柱内,然后依次用乙醇和乙醇-有机酸混合液洗脱,收集乙醇-有机酸混合洗脱液,减压浓缩,得到吡咯喹啉醌粗品;
(3)纯化:将吡咯喹啉醌粗品用超纯水溶解,调节pH至3-4,加入乙醇,搅拌后静置,得到吡咯喹啉醌。
所述的分子印迹聚合物是以二氧化硅为基材,吡咯喹啉醌为模板分子,甲基丙烯酸为功能单体,乙二醇二甲基丙烯酸酯为交联剂,采用热聚合技术制备而成。
所述的分子印迹聚合物的制备方法为:将100-300mg吡咯喹啉醌、0.1-0.5mL甲基丙烯酸,加入到50-200mL乙腈溶液中,室温机械搅拌6-20h;然后加入100-800mg二氧化硅微球,超声波分散;再加入0.6-1.0mL乙二醇二甲基丙烯酸酯和50-150mg偶氮二异丁腈,通入氮气5min,密封,加热至50-70℃,机械搅拌反应8-20h,分离,得到聚合材料;将聚合材料用乙醇洗涤5-10次后,50℃真空干燥8-16h;用体积比6-10:1乙醇/乙酸混合溶液反复洗脱干燥后的聚合材料,除去聚合材料中的模板分子吡咯喹啉醌,直至洗脱液中检测不到模板分子吡咯喹啉醌,最后用蒸馏水洗涤洗脱后的聚合材料至中性,50℃真空干燥12h,得到分子印迹聚合物,备用。
所述的二氧化硅微球的粒径为100-200目。
所述的分子印迹聚合物与含有吡咯喹啉醌的发酵液的固液比1:100-500(g/ml)。
含有吡咯喹啉醌的发酵液中吡咯喹啉醌的量以不超过分子印迹聚合物的最大吸附量为宜。优选的,所述的含有吡咯喹啉醌的发酵液中吡咯喹啉醌的浓度为100-200mg/L。
所述的乙醇-有机酸混合液中乙醇和有机酸的体积比为8-10:1-2;所述的有机酸为甲酸、乙酸或三氟乙酸中的任意一种。
所述的减压浓缩的真空度为﹣0.9MPa,转速为150-180转/秒。
步骤(3)中吡咯喹啉醌粗品用超纯水溶解至终浓度为9-12g/L。
步骤(3)中超纯水与乙醇的体积比为3-5:1。
步骤(3)中调节pH所用试剂为盐酸。
步骤(3)中搅拌温度为20-25℃,搅拌时间为5-6h,静置时间为12-24h。
所述的吡咯喹啉醌生产菌包括扭脱甲基杆菌(Methylobacterium extorquens),肺炎克雷伯氏菌(Klebsiella Pneumonia),氧化葡萄糖酸杆菌(Gluconobacter oxidans),绿脓杆菌(Pseudomonas aeruginosa),链霉菌(Streptomyces rochei)。
本发明分子印迹固相萃取分离吡咯喹啉醌的方法的原理见图1。
有益效果
1、本发明以二氧化硅为基材,吡咯喹啉醌为模板分子,甲基丙烯酸为功能单体,乙二醇二甲基丙烯酸酯为交联剂,采用热聚合技术制备得到吡咯喹啉醌分子印迹聚合物。该吡咯喹啉醌分子印迹聚合物具有较好的特异、选择吸附目标物吡咯喹啉醌的性能,最大吸附容量为可达10mg/mg(分子印迹聚合物)。以该聚合物为固相萃取填料对目标产物吡咯喹啉醌进行固相萃取,实现了从发酵液中高效分离纯化吡咯喹啉醌的目的。
2、吡咯喹啉醌分子印迹固相萃取是一种新的萃取分离体系,本发明采用吡咯喹啉醌分子为模板分子,形成具有吡咯喹啉醌分子印迹的聚合物,该聚合物可以选择性吸附吡咯喹啉醌,而不吸附其他水溶性杂质,从而高效、高选择性地分离纯化吡咯喹啉醌。实验表明,本发明分离得到的吡咯喹啉醌纯度可达99%以上,回收率可达90%以上。
3、本发明的方法工艺操作简单、成本低廉,便于大规模工业化生产,对促进吡咯喹啉醌的产业化发展具有重要意义。
附图说明
以下结合附图对本发明的具体实施方式作进一步详细说明。
图1为本发明分子印迹固相萃取分离吡咯喹啉醌的方法的原理图。
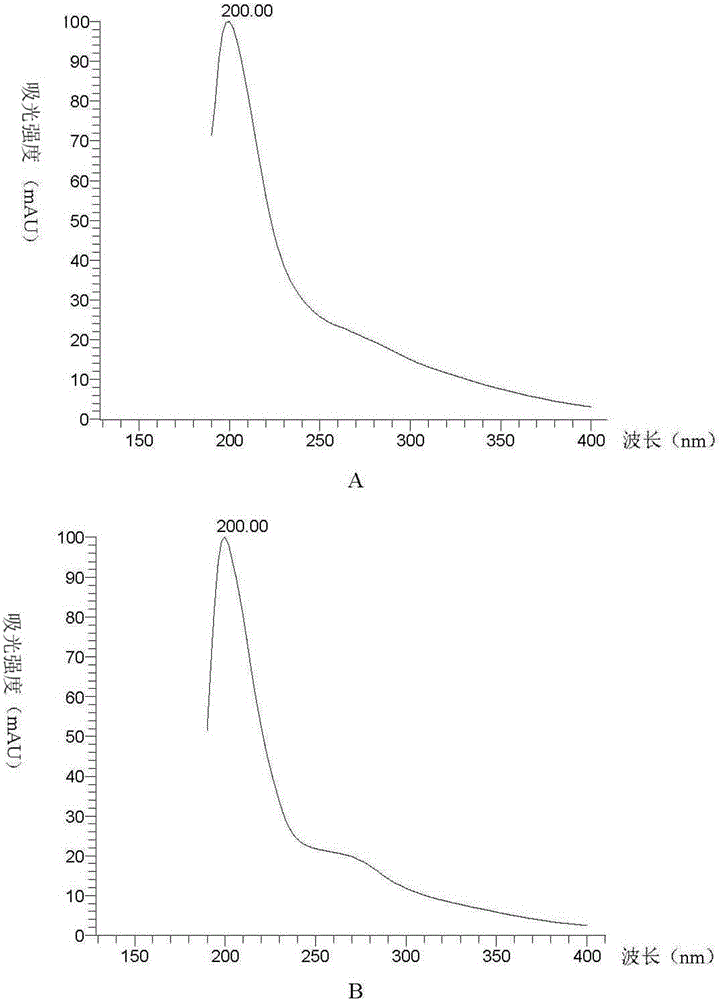
图2为本发明PQQ产物的紫外光谱图,其中,A为PQQ标准品,B为本发明PQQ产物。
图3为本发明PQQ产物的质谱图。
图4为本发明萃取洗脱液在230nm下HPLC色谱图。
图5为本发明PQQ产物在230nm下HPLC色谱图。
具体实施方式
以下结合实施例对本发明的具体实施方式作进一步详细说明。
本发明所用的培养基配制方法如下:
发酵培养基:每升培养基中含有40g山梨醇、20g酵母提取物、5g(NH4)2SO4、2g KH2PO4、5g MgSO4·H2O。
富集培养基:每升培养基中含有80g山梨醇、40g酵母提取物、10g(NH4)2SO4、4g KH2PO4、10g MgSO4·H2O。
本发明减压浓缩所用的设备为旋转蒸发器。
实施例1
一种采用分子印迹固相萃取法分离纯化发酵液中吡咯喹啉醌的方法,包括以下步骤:
(1)制备:利用吡咯喹啉醌生产菌制备含有吡咯喹啉醌的发酵液,具体方法为:将氧化葡萄糖酸杆菌按照5%的接种量,接种于发酵培养基中,在28℃条件下震荡培养3d,得到种子液;然后将种子液按照10%的接种量,接种于富集培养基,在28℃条件下震荡培养5d,培养液5℃、9000r/min离心15min,收集上清液,得到吡咯喹啉醌浓度为100-200mg/L的富含吡咯喹啉醌的发酵液;
(2)萃取分离:采用分子印迹固相萃取法对含有吡咯喹啉醌的发酵液进行萃取,将含有吡咯喹啉醌的发酵液上样于装填有分子印迹聚合物的分子印迹萃取柱内,分子印迹聚合物与含有吡咯喹啉醌的发酵液的固液比为1:100(g/ml),然后依次用10倍柱体积的乙醇和10倍柱体积的乙醇-甲酸混合液洗脱,收集乙醇-甲酸混合洗脱液,减压浓缩,得到吡咯喹啉醌粗品;减压浓缩的真空度为﹣0.9MPa,转速为170转/秒;
所述的乙醇-甲酸混合液中乙醇和甲酸的体积比为8:1;
所述的分子印迹聚合物的制备方法为:将100mg吡咯喹啉醌、0.1mL甲基丙烯酸,加入到50mL乙腈溶液中,室温机械搅拌6h;然后加入100mg二氧化硅微球,超声波分散;再加入0.6mL乙二醇二甲基丙烯酸酯和50mg偶氮二异丁腈,通入氮气5min,密封,加热至50℃,机械搅拌反应20h,分离,得到聚合材料;将聚合材料用乙醇洗涤5次后,50℃真空干燥8h;用体积比6:1乙醇/乙酸混合溶液反复洗脱干燥后的聚合材料,除去聚合材料中的模板分子吡咯喹啉醌,直至洗脱液中检测不到模板分子吡咯喹啉醌,最后用蒸馏水洗涤洗脱后的聚合材料至中性,50℃真空下干燥12h,得到分子印迹聚合物,备用;
所述的二氧化硅微球的粒径为100-200目;
(3)纯化:将吡咯喹啉醌粗品用超纯水溶解至吡咯喹啉醌终浓度为9g/L,加入盐酸调节pH至3,加入乙醇,超纯水与乙醇的体积比为3:1,在20℃条件下搅拌5h后静置24h,得到吡咯喹啉醌的晶体。
实施例2
一种采用分子印迹固相萃取法分离纯化发酵液中吡咯喹啉醌的方法,包括以下步骤:
(1)制备:利用吡咯喹啉醌生产菌制备含有吡咯喹啉醌的发酵液,具体方法为:将氧化葡萄糖酸杆菌按照5%的接种量,接种于发酵培养基中,在28℃条件下震荡培养3d,得到种子液;然后将种子液按照10%的接种量,接种于富集培养基,在28℃条件下震荡培养5d,培养液5℃、9000r/min离心15min,收集上清液,得到吡咯喹啉醌浓度为100-200mg/L的富含吡咯喹啉醌的发酵液;
(2)萃取分离:采用分子印迹固相萃取法对含有吡咯喹啉醌的发酵液进行萃取,将含有吡咯喹啉醌的发酵液上样于装填有分子印迹聚合物的分子印迹萃取柱内,分子印迹聚合物与含有吡咯喹啉醌的发酵液的固液比1:200(g/ml),然后依次用10倍柱体积的乙醇和10倍柱体积的乙醇-乙酸混合液洗脱,收集乙醇-乙酸混合洗脱液,减压浓缩,得到吡咯喹啉醌粗品;减压浓缩的真空度为﹣0.9MPa,转速为160转/秒;
所述的乙醇-乙酸混合液中乙醇和乙酸的体积比为10:2;
所述的分子印迹聚合物的制备方法为:将200mg吡咯喹啉醌、0.3mL甲基丙烯酸,加入到100mL乙腈溶液中,室温机械搅拌10h;然后加入300mg二氧化硅微球,超声波分散;再加入0.8mL乙二醇二甲基丙烯酸酯和80mg偶氮二异丁腈,通入氮气5min,密封,加热至60℃,机械搅拌反应10h,分离,得到聚合材料;将聚合材料用乙醇洗涤8次后,50℃真空干燥10h;用体积比8:1乙醇/乙酸混合溶液反复洗脱干燥后的聚合材料,除去聚合材料中的模板分子吡咯喹啉醌,直至洗脱液中检测不到模板分子吡咯喹啉醌,最后用蒸馏水洗涤洗脱后的聚合材料至中性,50℃真空下干燥12h,得到分子印迹聚合物,备用;
所述的二氧化硅微球的粒径为100-200目;
(3)纯化:将吡咯喹啉醌粗品用超纯水溶解至吡咯喹啉醌终浓度为10g/L,加入盐酸调节pH至4,加入乙醇,超纯水与乙醇的体积比为3:1,在21℃条件下搅拌6h后静置20h,得到吡咯喹啉醌的晶体。
实施例3
一种采用分子印迹固相萃取法分离纯化发酵液中吡咯喹啉醌的方法,包括以下步骤:
(1)制备:利用吡咯喹啉醌生产菌制备含有吡咯喹啉醌的发酵液,具体方法为:将氧化葡萄糖酸杆菌按照5%的接种量,接种于发酵培养基中,在28℃条件下震荡培养3d,得到种子液;然后将种子液按照10%的接种量,接种于富集培养基,在28℃条件下震荡培养5d,培养液5℃、9000r/min离心15min,收集上清液,得到吡咯喹啉醌浓度为100-200mg/L的富含吡咯喹啉醌的发酵液;
(2)萃取分离:采用分子印迹固相萃取法对含有吡咯喹啉醌的发酵液进行萃取,将含有吡咯喹啉醌的发酵液上样于装填有分子印迹聚合物的分子印迹萃取柱内,分子印迹聚合物与含有吡咯喹啉醌的发酵液的固液比1:400(g/ml),然后依次用10倍柱体积的乙醇和10倍柱体积的乙醇-三氟乙酸混合液洗脱,收集乙醇-三氟乙酸混合洗脱液,减压浓缩,得到吡咯喹啉醌粗品;减压浓缩的真空度为﹣0.9MPa,转速为180转/秒;
所述的乙醇-三氟乙酸混合液中乙醇和三氟乙酸的体积比为9:2;
所述的分子印迹聚合物的制备方法为:将250mg吡咯喹啉醌、0.4mL甲基丙烯酸,加入到150mL乙腈溶液中,室温机械搅拌15h;然后加入500mg二氧化硅微球,超声波分散;再加入0.9mL乙二醇二甲基丙烯酸酯和100mg偶氮二异丁腈,通入氮气5min,密封,加热至60℃,机械搅拌反应15h,分离,得到聚合材料;将聚合材料用乙醇洗涤10次后,50℃真空干燥12h;用体积比9:1乙醇/乙酸混合溶液反复洗脱干燥后的聚合材料,除去聚合材料中的模板分子吡咯喹啉醌,直至洗脱液中检测不到模板分子吡咯喹啉醌,最后用蒸馏水洗涤洗脱后的聚合材料至中性,50℃真空下干燥12h,得到分子印迹聚合物,备用;
所述的二氧化硅微球的粒径为100-200目;
(3)纯化:将吡咯喹啉醌粗品用超纯水溶解至吡咯喹啉醌终浓度为11g/L,加入盐酸调节pH至3.2,加入乙醇,超纯水与乙醇的体积比为4:1,在23℃条件下搅拌5h后静置16h,得到吡咯喹啉醌的晶体。
实施例4
一种采用分子印迹固相萃取法分离纯化发酵液中吡咯喹啉醌的方法,包括以下步骤:
(1)制备:利用吡咯喹啉醌生产菌制备含有吡咯喹啉醌的发酵液,具体方法为:将氧化葡萄糖酸杆菌按照5%的接种量,接种于发酵培养基中,在28℃条件下震荡培养3d,得到种子液;然后将种子液按照10%的接种量,接种于富集培养基,在28℃条件下震荡培养5d,培养液5℃、9000r/min离心15min,收集上清液,得到吡咯喹啉醌浓度为100-200mg/L的富含吡咯喹啉醌的发酵液;
(2)萃取分离:采用分子印迹固相萃取法对含有吡咯喹啉醌的发酵液进行萃取,将含有吡咯喹啉醌的发酵液上样于装填有分子印迹聚合物的分子印迹萃取柱内,分子印迹聚合物与含有吡咯喹啉醌的发酵液的固液比1:500(g/ml),然后依次用10倍柱体积的乙醇和10倍柱体积的乙醇-甲酸混合液洗脱,收集乙醇-甲酸混合洗脱液,减压浓缩,得到吡咯喹啉醌粗品;减压浓缩的真空度为﹣0.9MPa,转速为150转/秒;
所述的乙醇-甲酸混合液中乙醇和甲酸的体积比为9:1;
所述的分子印迹聚合物的制备方法为:将300mg吡咯喹啉醌、0.5mL甲基丙烯酸,加入到200mL乙腈溶液中,室温机械搅拌20h;然后加入800mg二氧化硅微球,超声波分散;再加入1.0mL乙二醇二甲基丙烯酸酯和150mg偶氮二异丁腈,通入氮气5min,密封,加热至70℃,机械搅拌反应8h,分离,得到聚合材料;将聚合材料用乙醇洗涤10次后,50℃真空干燥16h;用体积比10:1乙醇/乙酸混合溶液反复洗脱干燥后的聚合材料,除去聚合材料中的模板分子吡咯喹啉醌,直至洗脱液中检测不到模板分子吡咯喹啉醌,最后用蒸馏水洗涤洗脱后的聚合材料至中性,50℃真空下干燥12h,得到分子印迹聚合物,备用;
所述的二氧化硅微球的粒径为100-200目;
(3)纯化:将吡咯喹啉醌粗品用超纯水溶解至吡咯喹啉醌终浓度为12g/L,加入盐酸调节pH至3.6,加入乙醇,超纯水与乙醇的体积比为5:1,在25℃条件下搅拌6h后静置12h,得到吡咯喹啉醌的晶体。
实验例
1、产物分析
1.1、紫外光谱分析
采用紫外光谱仪对本发明的结晶产物与PQQ标准品同时进行检测,结果见图2。结果表明,本发明的结晶产物与PQQ标准品的紫外光谱结果一致。
1.2、质谱分析
采用质谱仪对本发明的结晶产物进行检测,结果见图3。将本发明的结晶产物质谱数据与已知PQQ质谱数据对比,结果表明,本发明的结晶产物与已知PQQ质谱数据一致。
1.3、HPLC法检测
检测方法:采用Waters Symmetry 300C18液相色谱柱,以乙腈:水(乙腈和水均含2%甲酸)为流动相,流速1mL/min,梯度洗脱(30-90%,30min),检测波长230nm。
检测对象:萃取洗脱液(乙醇-有机酸混合洗脱液)、PQQ结晶产物。
结果分析:HPLC分析结果见图4、5。图4表明,萃取洗脱液中有较高含量的目标物,萃取具有较高选择性;图5表明,通过重结晶,结晶产物纯度达到99%以上。
1.4、回收率
以实施例1-4吡咯喹啉醌的发酵液为样品溶液,研究该分子印迹固相萃取对吡咯喹啉醌的分离效果,采用高效液相色谱技术检测乙醇-有机酸混合洗脱液中吡咯喹啉醌的含量,计算回收率(表1)。
表1 本发明PQQ回收率的结果分析
表1结果表明,本发明分子印迹固相萃取法分离纯化发酵液中吡咯喹啉醌的效果非常好,回收率可达90%以上。
2、分子印迹聚合物吸附容量的测定
称取于10mg分子印迹聚合物吸附管中,然后各加入10mL质量浓度分别为2、4、8、9、10、11、12g/L的PQQ标准溶液,室温下,静态吸附12h,离心,分离吸附饱和后的分子印迹聚合物与吸附后的溶液,吸附后的溶液中PQQ的质量浓度采用高效液相色谱测定(表2)。
表2 本发明分子印迹聚合物吸附容量的结果分析(质量浓度单位:g/L)
表2结果表明,本发明制备的分子印迹聚合物具有较好的特异、选择吸附目标物吡咯喹啉醌的性能,最大吸附容量为可达10mg/mg(分子印迹聚合物)。
- 还没有人留言评论。精彩留言会获得点赞!