二次酸浸法制备纳米氧化镁的方法与流程
本发明涉及纳米材料工艺的技术领域,尤其涉及二次酸浸法制备纳米氧化镁的方法。
背景技术:
目前,纳米mgo最常用的制备方法有固相法、液相法、气相法。
固相法的优点是设备和工艺简单,操作方便安全,成本低,反应条件容易控制。但容易引入杂质,纯度低,颗粒不均匀,形状难以控制,易团聚。制备纳米氧化镁的固相法主要有机械粉碎法,机械粉碎法通常用于固体物质直接制备纳米粉体材料,即通过机械力将氧化镁粉末进一步细化,但机械粉碎法难以得到粒径小于100nm的纳米粒子,且粉碎过程还易混入杂质,粒子形状难以控制,难以达到工业应用的要求。
液相法是目前广泛采用的制备纳米金属氧化物粉体的方法,已用于制备纳米氧化镁的液相法有:直接沉淀法、溶胶—凝胶法等。
直接沉淀法是在金属盐溶液中加入沉淀剂,使生成的沉淀从溶液中析出,将阴离子从沉淀中除去,再经热分解制得纳米氧化物。一般用于做沉淀剂的有氨水、氢氧化钠、碳酸氨、碳酸钠等。直接沉淀法操作简便易行,对设备、技术要求不高,不易引入杂质,产品纯度高,有良好的化学计量性,制备成本较低;但产品粒度较大,粒度分布较宽。
溶胶-凝胶法是以有机或无机盐为原料,在有机介质(如乙醇等)中进行水解、缩聚反应,使溶液经溶胶-凝胶化过程得到凝胶,凝胶经干燥、煅烧得到产品。如:将明胶溶解在热的氯化镁水溶液,冷却形成凝胶后加入到过量的草酸铵溶液中浸泡,离心分离,用离子水洗干净后用乙醇分散于110℃干燥,最后于500℃煅烧2h得到7nm左右的纳米氧化镁产品。又如:将硝酸镁加入到熔融的硬脂酸中,加热、搅拌,20min后得到浅黄色透明溶胶,然后自然冷却至室温得到凝胶,再将凝胶置于马弗炉中,在400℃以上的温度下煅烧一定时间,得到白色纳米mgo微粒。再如:以mg(oc2h5)2、乙醇为原料,以草酸为催化剂,制得30nm的mgo粉体。该法得到的纳米粉体粒度分布窄、分散性好、纯度高,并且煅烧温度低、反应易控制、副反应少、工艺操作简单,但原料成本较高。
气相法分为物理气相沉积法(pvd)和化学气相沉积法(cvd)两种。物理气相沉积法是利用电弧、高频或等离子体高温热源将氧化物加热使之气化,然后聚成粒子。化学气相沉积法是利用挥发性金属化合物或金属单质蒸汽通过化学反应生成所需化合物。根据反应的类型,化学气相沉积法又可分为气相氧化法、气相热解法、气相水解法等。气相法优点是产品纯度高、分散性好、粒度分布窄,可以连续生产,生产能力大,粒度可以控制。但产品的收集还存在问题,设备昂贵,反应温度高,能耗大。
技术实现要素:
本发明的目的是为了克服上述现有技术的缺点,提供二次酸浸法制备纳米氧化镁的方法,该二次酸浸法制备纳米氧化镁的方法以白云石为原料经二次酸浸、氨水沉淀法,可制得结晶良好的纳米mg(oh)2并通过在不同温度下煅烧得到不同形态的纳米氢氧化镁,该方法的镁离子最大提取率为94.64%,mgo纯度大于99.73%,钙的含量不超过0.16%,其他铁、锰、锌、铜、铝等元素总含量不超过0.05%。所得产品具有均匀而致密、粒度小、分布窄、团聚少、分散性良好等优点。
本发明解决其技术问题所采用的技术方案是:二次酸浸法制备纳米氧化镁的方法,包括如下步骤:
(1)以1050℃煅烧白云石2h,加水以70℃下消化40min;然后用盐酸第一次进行酸浸,盐酸用量与钙离子的摩尔比为2:1;
(2)用硫酸进行第二次酸浸,硫酸用量与镁离子的摩尔比为4:1;
(3)用15%浓度氨水沉淀溶液的沉淀物,然后用15%浓度的乙醇洗涤,氨水用量为与镁离子的摩尔比为4:1;
(4)把沉淀物干燥后在550℃或850℃温度下煅烧2h得到纳米氧化镁。
进一步的,产物中纳米mgo纯度大于99.73%,钙的含量不超过0.16%,其他铁、锰、锌、铜、铝等元素总含量不超过0.05%。
进一步的,所述的步骤(4)中在550℃煅烧生成的纳米氧化镁为纳米氧化镁微片,850℃煅烧生成的纳米氧化镁是纳米氧化镁球形粒子。
综上所述,本发明的二次酸浸法制备纳米氧化镁的方法以白云石为原料经二次酸浸、氨水沉淀法,可制得结晶良好的纳米mg(oh)2并通过在不同温度下煅烧得到不同形态的纳米氢氧化镁,该方法的镁离子最大提取率为94.64%,mgo纯度大于99.73%,钙的含量不超过0.16%,其他铁、锰、锌、铜、铝等元素总含量不超过0.05%。所得产品具有均匀而致密、粒度小、分布窄、团聚少、分散性良好等优点。
附图说明
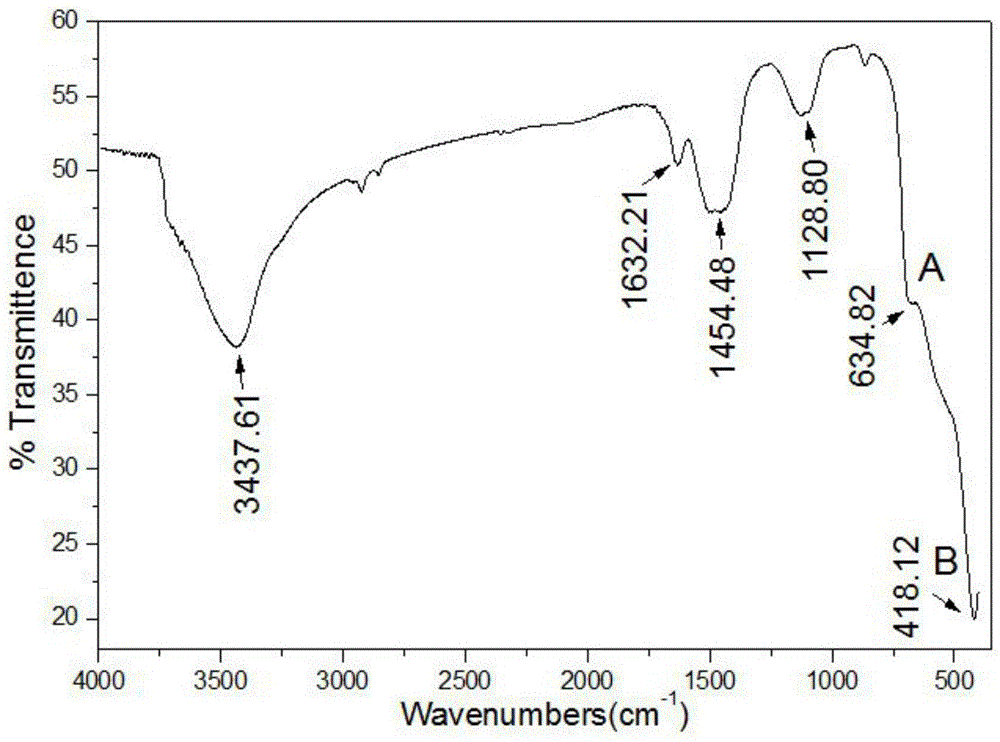
图1是前驱体mg(oh)2经650℃煅烧后mgo的红外吸收光谱图;
图2是前驱体mg(oh)2在不同温度下煅烧2h后得到纳米氧化镁的碘吸附曲线图;
图3是前驱体mg(oh)2在不同温度下煅烧2h后得到纳米氧化镁的比表面积曲线;
图4是纳米氧化镁的晶体点阵示意图;
图5是前驱体mg(oh)2在550℃煅烧1h后得到纳米氧化镁的sem图。
具体实施方式
实施例1
本实施例1所描述的二次酸浸法制备纳米氧化镁的方法,包括如下步骤:
(1)以1050℃煅烧白云石2h,加水以70℃下消化40min;然后用盐酸第一次进行酸浸,盐酸用量与钙离子的摩尔比为2:1;
(2)用硫酸进行第二次酸浸,硫酸用量与镁离子的摩尔比为4:1;
(3)用15%浓度氨水沉淀溶液的沉淀物,然后用15%浓度的乙醇洗涤,氨水用量为与镁离子的摩尔比为4:1;
(4)把沉淀物干燥后在550℃或850℃温度下煅烧2h得到纳米氧化镁。
在本实施例中,产物中纳米mgo纯度大于99.73%,钙的含量不超过0.16%,其他铁、锰、锌、铜、铝等元素总含量不超过0.05%。
如图1所示,在400~800cm-1波数范围内,吸收谱窄而尖锐,说明经650℃煅烧处理后的mgo结晶较完整。在3437.61cm-1及1454.48cm-1处的吸收峰分别归属为水分子的o-h的伸缩以及弯曲振动,表明mgo中有水分存在;在1632.21cm-1及1128.80cm-1处的吸收峰分别归属为co2的伸缩及弯曲振动,说明mgo表面吸附了co2气体。
a峰和b峰分别对应于mg-o键的伸缩振动和弯曲振动。对应本征态mgo红外吸收光谱,伸缩振动峰强度加强而弯曲振动峰强度减弱。当纯的氧化镁掺入钙、铁等之后,pauling离子半径较大的ca2+(r=0.098nm)或fe3+(r=0.088nm)等小部分取代mg2+(r=0.085nm),使晶格畸变度δ(δ=c/a-1)变大,使氧化镁立方相的不对称性增大,则晶格振动产生的电偶矩及其极化率必然升高,这就使mg-o键的伸缩振动得到增强。同时,氧化镁中部分有机物官能团分解的残留物产生的范德华力及几何空间构型对mg-o键的弯曲振动亦产生一定的削弱,这就导致红外光谱弯曲振动强度的减弱。
mgo键的伸缩振动峰出现在634.82cm-1,mgo键的弯曲振动峰出现在418.12cm-1,而本征态氧化镁mg-o键的伸缩振动峰是在550cm-1,弯曲振动峰为450cm-1。与本征态氧化镁相比较,伸缩振动吸收峰a向高波数方向移动(蓝移)而弯曲振动吸收峰b向短波数方向移动(红移),伸缩振动吸收峰a峰蓝移了约84.82cm-1,弯曲振动吸收峰b峰红移了约31.88cm-1,此种反常红外特性国内外均未见报道。低维纳米材料表面体系的特殊红外性能极大地取决于纳米尺度、纳米结构以及纳米粒子间的相互作用。纳米片层mgo由于其结构既不同于长程有序的晶体,也不同于长程无序、短程有序的非晶态,因而其光学性质表现出异于晶态和非晶态的现象。
这种纳米菱片厚度仅10-20nm左右,而面积最大可达1μm2左右,因而其表面粒子量子尺寸效应十分显著,处于片层表面态的原子、电子行为异常,表面原子配位不饱和,垂直于表面的悬键垂直伸缩振动变得活跃,也就是说与mgo纵向声子振动有关的谱带得到加强,能级间距加大,因而吸收峰向高能方向移动;同时纳米片层表面张力较大,而片层界面组元体积分数极高,导致链的振动频率升高,颗粒内部晶格畸变使氧化镁立方相的不对称性增大,链长改变,使得伸缩振动吸收峰发生蓝移。纳米氧化镁片的红外吸收带发生红移是由于纳米片层晶粒组元微结构变化大,界面原子配位数不足,失配键较多,表面原子易吸附气体分子形成吸附层,使晶体场效应变弱,导致弯曲振动移向较低的能量处而发生红移。
纳米材料普遍存在红外吸收峰的蓝移现象,但同一样品在相同条件下的氧化镁伸缩振动吸收峰红移、弯曲振动蓝移共同存在,这一现象未见报道。mgo伸缩振动峰蓝移,弯曲振动峰红移表明晶粒的吸收光谱宽频化,这是因为这种特殊的纳米片层晶粒大的比表面导致了其平均配位数的下降,不饱和键和悬键增多,片层mgo分子与单一的普通振动模不同,存在一个较宽的键振动模的分布;同时,晶格畸变产生的晶格张力导致mg-o键的松懈,这就导致了纳米菱片红外吸收带的宽化。
活性是决定氧化镁功能的重要物理化学性质,mgo活性的差异,主要来源于mgo雏晶的大小及结构不完整等因素。若结构松弛、晶格畸变、缺陷较多,则活性较高。反之,mgo晶粒较大、结构紧密,晶格完整,其活性较低。氧化镁活性可用吸碘值、比表面积、柠檬酸值(或醋酸值)、水化率等法来表示。氧化镁的比表面积或吸碘值越大,活性就越高。
如图2所示,把前驱体纳米氢氧化镁在550℃下煅烧2h,所获纳米氧化镁的吸碘值分别为312.5mgi2/gmgo和60.4mgi2/gmgo。表明前驱体氢氧化镁的晶形和形貌是决定氧化镁活性的首要指标,制备高活性氧化镁必须使用晶粒小、分散度高、具有一定活性形貌的氢氧化镁。由图2可以看出,随着煅烧温度的增加,氧化镁的吸碘值先增大,后减小,在850℃有一突变。
如图3所示,煅烧温度对氧化镁活性有较大影响。随着煅烧温度的增加,氧化镁的比表面积先增大,后减小,且在850℃有一突变,这与氧化镁的碘吸附曲线变化规律一致。
前驱体于500℃煅烧两小时,所形成的氧化镁为纳米微片组成的空间格子,而此格子具有较大的孔隙结构,因而极易吸附气体,故其碘吸附值和比表面积较大;前驱体550℃煅烧两小时后的产物比表面积最大,其原因在于550℃煅烧下的产物,片层厚度最小,微片面积最大,其表面原子畸变度大,表面缺陷较多,而且微片表面有大量细微的孔隙结构,表面大量的细微孔隙对吸附气体分子极其有利,且纳米微片本身交错生长形成的空间格子同样容易吸附气体,故其比表面积最大,活性最高;随着煅烧温度的升高,氧化镁的碘吸附值和比表面积随之下降,但在850℃有一个拐点,其碘吸附值和比表面积相当大,原因在于850℃的煅烧温度下,氢氧化镁的纳米微片结构不再被保留,形成的氧化镁不再是微片结构,而是细小的球形粒子,粒子尺寸为15nm左右,因此具有较大的比表面积;850℃之后,随着煅烧温度的升高,所形成的氧化镁比表面积急剧下降,原因在于,高温度煅烧下形成的氧化镁开始大量烧结团聚,引起粒子尺寸的急剧膨胀、变大,反应在吸附值上,即为比表面积的下降。
综上所述,可见前驱体于550℃或850℃左右煅烧2h可得活性较大的纳米氧化镁产品,550℃煅烧生成的为纳米氧化镁微片,而850℃煅烧生成的是纳米氧化镁球形粒子。
前驱体mg(oh)2的焙烧有3个阶段:(1)热分解开始,此时已分解的mg和o处于分解前的晶格点阵上;(2)已分解的mg和o从原始晶格点阵上移动形成新的mgo晶核,构成多空隙的具有前驱体残余骨架的结构;(3)已分解的mg和o在mgo晶核上长大,构成完整的晶格点阵,形成产物,热分解完毕。氧化镁为面心立方结构晶体,面心立方具有最紧密堆积结构,如图4所示,纳米氧化镁的晶格畸变在图4中并未显示,图4中的大球为o离子,小球为mg离子。
如图5所示,可以很清晰的看出来,mg(oh)2分解中间产物仍然继承了mg(oh)2的微片结构,并且微片面积有所增大,生长较为均匀,其外延辐射状生长的特点也同样被保留了下来。
以上所述,仅是本发明的较佳实施例而已,并非对本发明的技术方案作任何形式上的限制。凡是依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明的技术方案的范围内。
- 还没有人留言评论。精彩留言会获得点赞!