镍在富锂正极材料中用于抑制在充电循环期间从正极材料的气体放出和增大正极材料的电荷容量的用途的制作方法
本发明涉及一组电活性正极化合物。更具体地,本发明涉及一组含镍的富锂的高容量正极化合物的用途。
常规的锂离子电池(battery)因用于制成正极(positiveelectrode,cathode)的材料的容量而在性能方面受限。包含镍锰钴氧化物的共混物的正极材料的富锂共混物在安全性和能量密度之间存在折衷。认识到,电荷储存在这样的正极材料内的过渡金属阳离子中。已经建议,如果电荷可储存在阴离子(例如氧)上,则能显著地提升正极材料的容量和由此的能量密度,减少对这样大量的过渡金属重离子的需要。然而,提供如下材料的挑战依旧存在:其可依赖于阴离子和阳离子两者的氧化还原化学性来储存电荷,并且经受住充电/放电循环,而不损害该材料的安全性或造成会使该材料分解的不期望的氧化还原反应。
在第一方面中,本发明涉及镍在如下通式的正极材料中用于抑制在充电循环期间的气体放出的用途:
在该用途的一种具体实施方式中,x大于0.06且等于或小于0.4;y等于或大于0且等于或小于0.4;和,z等于或大于0且等于或小于0.05。
在第二方面中,本发明涉及镍在如下通式的正极材料中用于增大所述材料的电荷容量(chargecapacity)的用途:
在该用途的一种具体实施方式中,x大于0.06且等于或小于0.4;y等于或大于0且等于或小于0.4;和,z等于或大于0且等于或小于0.05。
已经发现,可通过如下实现容量改善的化合物:减少过量的锂量,并且增加镍的量。因此,可使用镍来增大nmc-型正极材料的容量。如上定义的特定化合物因过渡金属镍的氧化程度并且还因晶格内氧根离子(oxideion)的氧化而展现在容量方面的大幅增加。在不希望局限于理论的情况下,认识到,特定量的镍替换(取代)实现更大的氧的氧化还原活性,并且因此改善该材料的电化学容量。
另外,当与现有技术的过渡金属替换的nmc富锂材料相比时,本发明的化合物在电化学循环期间展现改善的稳定性。分子氧的放出(evolution)普遍存在于如下的富锂材料第三行过渡金属氧化物(li1+xm1-xo2,其中m为ti、v、cr、mn、fe、co、ni、cu或zn):其中锂已经被过渡金属离子的一些所交换。这些材料通常依赖于氧的氧化还原来改善它们的电荷容量性质。均质材料可遭受分子氧在循环期间因氧化物阴离子的氧化还原而从晶体结构逃逸。进而,这使该材料的容量和可用寿命缩减。然而,本发明材料具有改善的容量,其历经很多循环保持不变。
所述气体可为分子氧和/或二氧化碳。认识到,当由锂离子的移出(removal)导致的电荷不平衡因电子从氧阴离子移出而平衡时,所得的氧阴离子是不稳定的,这导致在充电循环期间不期望的氧化还原反应和分子氧气的放出。二氧化碳还可由于从晶格逃逸的氧与电解质溶剂(例如碳酸亚丙酯)的反应而产生。在不希望局限于理论的情况下,认识到,所述材料中相对于锂含量特定的镍含量避免晶格内的欠键合(under-bonding),使得各氧阴离子仍键合到~3个阳离子。应对该问题的潜在方案可能为将单元电池(电池,cell)的正极层或部分封装在不透气膜中。然而,这会向单元电池增添寄生质量,从而使所得电池的能量密度降低。然而,本发明的化学途径使用特定量的过渡金属调节晶格结构,减少来自所述材料的氧气的产生,而不需要向正极材料或所得的电池单元(batterycell)增添层。
在一种具体实施方式中,y和z两者均等于0;和,x等于或大于0.3且等于或小于0.4。因此,正极材料可选自li1.333ni0.3mn0.5667o2或li1.066ni0.4mn0.533o2之一。
在一种具体实施方式中,z等于0;x等于0.2;和,y等于或大于0.15且等于或小于0.2。因此,正极材料可选自li1.1333co0.2ni0.2mn0.4667o2或li1.15co0.15ni0.2mn0.5o2之一。
在一种具体实施方式中,x等于0.2;z等于0.05;和,y等于或大于0.1且等于或小于0.15。因此,正极材料可选自li1.15ni0.2co0.1al0.05mn0.5o2或li1.1333ni0.2co0.15al0.05mn0.4667o2之一。
这些具体化合物已经展现低的气体放出和改善的电荷容量以及良好的经过若干循环的稳定性。
所述化合物可被限定为具有层状结构。典型地,层状结构已经显示出具有最高的能量密度。当以层状形式时,所述材料可进一步使用如下通式进行定义:(1-a-b-c)li2mno3·alicoo2·blini0.5mn0.5o2·clialo2,使得a、b和c等同于以上各通式的值。
为了使本发明可更容易地被理解,现将通过示例的方式参考附图描述本发明的实施方式,在图中:
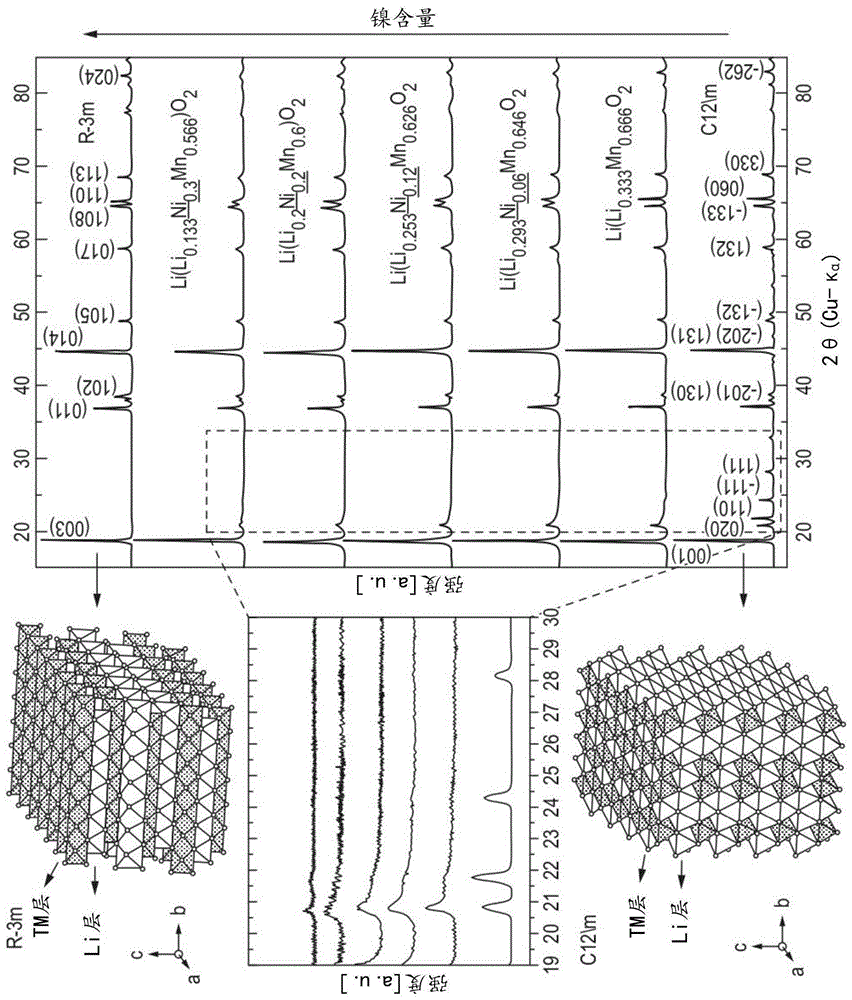
图1显示根据实施例1a的合成材料相较于c12/m和r-3m对称晶格的经计算的图案(分别在该图的底部和顶部处显示)的粉末x-射线衍射图案;
图2显示根据实施例1b的合成材料的粉末x-射线衍射图案;
图3显示根据实施例1c的合成材料的粉末x-射线衍射图案;
图4显示根据实施例1a的合成材料的第一次循环的恒电流负荷曲线;
图5显示根据实施例1b的合成材料的第一次循环的恒电流负荷曲线;
图6显示根据实施例1c的合成材料的第一次循环的恒电流负荷曲线;
图7显示经镍掺杂的li2mno2材料的oems分析;和
图8显示根据实施例1c的材料之一的oems分析。
现将参考以下实施例对本发明进行说明。
实施例1–经镍替换的富锂材料的合成
1a)使用甲醛-间苯二酚溶胶凝胶合成途径合成如下的具有通式
将按化学计量的量的ch3cooli·2h2o(98.0%,sigmaaldrich(rtm))、(ch3coo)2mn·4h2o(>99.0%,sigmaaldrich(rtm))和(ch3coo)2ni·4h2o(99.0%sigmaaldrich(rtm))溶解在50ml水中,其中0.25mmol的ch3cooli·2h2o(99.0%,sigmaaldrich(rtm))对应于相对于0.01摩尔所合成的材料5摩尔%的锂。同时,将0.1mol的间苯二酚(99.0%,sigmaaldrich(rtm))溶解在0.15mol甲醛(36.5%w/w水溶液,fluka(rtm)中。所有试剂在它们各自的溶剂中一旦完全溶解,就将这两种溶液混合并将混合物剧烈搅拌一小时。随后,将包含5%摩尔过量的锂的所得溶液在油浴中在80℃下加热直至形成均匀的白色凝胶。
最后,将凝胶在90℃干燥过夜并且然后在500℃以15小时和在800℃以20小时进行热处理。
1b)对于掺杂有镍的含钴的正极材料,使用甲醛-间苯二酚溶胶凝胶合成途径合成通式为
将按化学计量的量的ch3cooli·2h2o(98.0%,sigmaaldrich(rtm))、(ch3coo)2mn·4h2o(>99.0%,sigmaaldrich(rtm))(ch3coo)2ni·4h2o(99.0%sigmaaldrich(rtm)和(ch3coo)2co·4h2o(99.0%sigmaaldrich(rtm))溶解在50ml水中,其中0.25mmol的ch3cooli·2h2o(99.0%,sigmaaldrich(rtm))对应于相对于0.01摩尔所合成的材料5摩尔%的锂。同时,将0.1mol的间苯二酚(99.0%,sigmaaldrich(rtm))溶解在0.15mol甲醛(36.5%w/w水溶液,fluka(rtm))中。所有试剂在它们各自的溶剂中一旦完全溶解,就将这两种溶液混合并将混合物剧烈搅拌一小时。随后,将包含5%摩尔过量的锂的所得溶液在油浴中在80℃下加热直至形成均匀的白色凝胶。
最后,将凝胶在90℃干燥过夜并且然后在500℃以15小时和在800℃以20小时进行热处理。
1c)对于掺杂有镍的含钴-铝的正极材料,使用甲醛-间苯二酚溶胶凝胶合成途径合成通式为
为了获得0.01mol的最终产物,计算所有试剂的比率。
将按化学计量的量的ch3cooli·2h2o(98.0%,sigmaaldrich(rtm))、(ch3coo)2mn·4h2o(>99.0%,sigmaaldrich(rtm))、(ch3coo)2co·4h2o(99.0%sigmaaldrich(rtm))、al2(so4)3·4h2o(sigmaaldrich(rtm))和(ch3coo)2ni·4h2o(99.0%sigmaaldrich(rtm))溶解在50ml水中,其中0.25mmol的ch3cooli·2h2o(99.0%,sigmaaldrich(rtm))对应于相对于0.01摩尔所合成的材料5摩尔%的锂。同时,将0.1mol的间苯二酚(99.0%,sigmaaldrich(rtm))溶解在0.15mol甲醛(36.5%w/w水溶液,fluka(rtm))中。所有试剂在它们各自的溶剂中一旦完全溶解,就将这两种溶液混合并将混合物剧烈搅拌一小时。随后,将包含5%摩尔过量的锂的所得溶液在油浴中在80℃下加热直至形成均匀的白色凝胶。
最后,将凝胶在90℃干燥过夜并且然后在500℃以15小时和在800℃以20小时进行热处理。
实施例2–经镍替换的富锂材料的结构分析和表征
通过如下考察根据实施例1a-c的材料:采用装备有9kwcu旋转阳极的rigaku(rtm)smartlab实施粉末x-射线衍射(pxrd);和,通过brukeravanceiii400wd核磁共振仪对所述材料收集mas-nmr谱图。
图1(经镍掺杂的li2mno2)、2a和b(分别为经镍掺杂的钴组成1和2)以及3a和3b(分别为经镍掺杂的铝钴组成1和2)显示所合成的材料的粉末x-射线衍射图案。它们以在过渡层中具有一定的阳离子排序(有序性)的层状材料为特征。所有图案似乎显示与具有r-3m空间群的密堆积层状结构例如litmo2一致的主峰。在范围为20-30的2θ角度下观察到另外峰,其无法被归属为r-3m空间。该排序源于在
实施例3–经镍替换的富锂材料的电化学分析
通过用biologicvmp3和maccor4600系列的恒电位仪进行的恒电流循环对根据实施例1a-c的材料进行电化学表征。将所有样品组装到相对于金属锂的不锈钢纽扣电池中,并且在相对于li+/li为2v和4.8v之间以100次循环在50mag-1电流比率(currentrate)下循环。所使用的电解质为lp30(lipf6在1:1重量比的ec:dmc中的1m溶液)。
图4-6显示根据实施例1的各材料在第一次循环的充电和随后的放电期间的电位曲线。
图4显示根据实施例1a的各材料在第一次循环的充电和随后的放电期间的电位曲线。所有的样品均呈现不同长度的以相对于li+/li0的4.5v为中心的高电压平台,而在充电开始时存在的倾斜区域随着镍的掺杂量而在长度上逐步地增加。该区域的延长可归因于镍从ni+2向ni+4的氧化,似乎与仅对镍的氧化还原活性负责的将被提取的锂(即电荷)的量良好一致。因此,如所预期的,li2mno3没有展现任何的预平稳(平台前,pre-plateau)区域,而经x=0.3掺杂的
在第一次放电期间,所述材料均未显示可逆平台的存在,表明在锂离子从各样品的晶格中提取(充电)和插入到其中(放电)期间所遵循的热力学途径的不同。
对于根据实施例1a的所有材料,第一次循环呈现由于不可逆的高电位平台的存在引起的最低库伦效率值。库伦效率似乎在前五次循环内从第一次循环值(大约60-70%)快速地提升至高于98%的值。然而,对此,li2mno3和其中x=0.06的
图5(经镍掺杂的钴正极材料)显示根据实施例1b的材料在第一次循环的充电和随后的放电期间的电位曲线。两个样品均呈现不同长度的以相对于li+/li0的4.5v为中心的高电压平台、和在充电开始时的倾斜区域。该区域的延长可归因于镍从ni+2向ni+4和co+3向co+4的氧化,并且似乎与仅对过渡金属的氧化还原活性负责的将被提取的锂(即电荷)的量良好一致。
在第一次放电期间,所述材料均未显示可逆平台的存在,表明在锂离子从各样品的晶格中提取(充电)/插入到其中(放电)期间所遵循的热力学途径不同。
对于实施例1b的材料,第一次循环呈现由于不可逆的高电位平台的存在引起的最低库伦效率值。所述库伦效率似乎在前五次循环内从第一次循环值(大约60-80%)快速地提升至高于98%的值。
通过用biologicvmp3和maccor4600系列的恒电位仪进行的恒电流循环对根据实施例1c的材料进行电化学表征。将所有样品组装到相对于金属锂的不锈钢纽扣电池中,并且在相对于li+/li为2v和4.8v之间以100次循环在50mag-1电流比率下循环。所使用的电解质为lp30(lipf6在1:1重量比的ec:dmc中的1m溶液)。
图6显示根据实施例1c的各材料在第一次循环的充电和随后的放电期间的电位曲线。两个样品均呈现不同长度的以相对于li+/li0的4.5v为中心的高电压平台、和在充电开始时的倾斜区域。该区域的延长可归因于镍从ni+2向ni+4和co+3向co+4的氧化,并且似乎与仅对过渡金属的氧化还原活性负责的将被提取的锂(即电荷)的量良好一致。
在第一次放电期间,所述材料均未显示可逆平台的存在,表明在锂离子从各样品的晶格中提取(充电)/插入到其中(放电)期间所遵循的热力学途径不同。
对于根据实施例1c的两种材料,第一次循环呈现由于不可逆的高电位平台的存在引起的最低库伦效率值。所述库伦效率似乎在前五次循环内从第一次循环值(大约60-80%)快速地提升至高于98%的值。
实施例4–在经镍替换的富锂材料的第一次循环期间的气体放出
将根据实施例1a的材料的一个圆片(pellet)组装到专门机械加工成用于实施现场原位电化学质谱(operandoelectrochemicalmassspectrometry,oems)测量的swagelok(rtm)测试单元电池中。通过thermo-fisher四极质谱仪进行oems实验中涉及的质谱测量。为了获得对在第一次循环期间观察的额外容量的起因的洞察,对一组材料进行oems。
图7(a)、(b)和(c)分别显示经镍掺杂的x=0.2、0.3和0.4的
co2和o2为对于所有样品检测到的唯一气态物种并且明显的趋势从图4中显现,其中随掺杂剂镍的量增加,所释放的气体量逐步地减少。
在所有情形中首先检测到co2,其在高电位平台(大约4.5v,相对于li+/li0)区域开始处达到峰值并逐步地减少直至充电结束。
co2量随镍含量的增大而减少,但从未消除。另一方面,对于本发明的材料,分子氧似乎以在充电结束方向上达到其最大值的类尖峰方式释放。在其中x=0.4的高ni替换的情形中,已经表明,几乎完全抑制o2并且所检测的co2量骤降(图7(c))。该结果暗示,镍在通过减少失氧过程而使氧化物结构在高电位下稳定化中起着重要作用。
将组成1li1.1333co0.15al0.05ni0.2mn0.4667o2(来自实施例1c的组成)的一个圆片组装到专门机械加工成用于实施现场原位电化学质谱(oems)测量的swagelok(rtm)测试单元电池中。通过thermo-fisher四极质谱仪进行该oems实验中涉及的质谱测量。为了获得对在第一次循环期间观察的额外容量的起因的洞察,对一组材料进行oems。
图8分别显示经镍掺杂的li1.1333co0.15al0.05ni0.2mn0.4667o2的oems分析。该图显示各材料在前两次循环期间的恒电流曲线(上图)、氧气痕量和二氧化碳痕量(下图)。对于所有材料,以0.7ml/min的通量速率使用氩气作为载气,并且使电极相对于金属锂以15mag-1的比率在相对于li+/li0为2v和4.8v之间循环。所使用的电解质为lipf6在碳酸亚丙酯中的1m溶液。
co2为对于所有样品检测到的唯一气态物种并且随掺杂剂镍的量增加,所释放的气体量逐步地减少。co2在高电位平台(大约4.5v,相对于li+/li0)区域开始处达到峰值并逐步地减少直至充电结束。
- 还没有人留言评论。精彩留言会获得点赞!