基于三氯代氮杂并苯稠环芳烃的氮掺杂多孔碳材料的制备方法及其应用与流程
本发明涉及杂原子掺杂多孔材料聚合技术领域,尤其是涉及一种基于三氯代氮杂并苯稠环芳烃的氮掺杂多孔碳材料的制备方法及其应用。
背景技术:
随着世界经济迅猛发展,能源需求量不断增大,传统的化石能源已经越难以满足人类对日益增长的能源需求,而环境污染引发的健康问题越发突出,人们对于绿色环保能源设备的研究急为迫切。
燃料电池具有转换效率高、对环境友好等优点,目前商业化的氧还原电催化剂pt/c因成本过高、易失活而限制其在工业化应用的开展。因此,寻找廉价的、高效的氧还原电催化剂对燃料电池的开发显得尤为重要。近年来,有机多孔聚合物催化、电子器件、能源存储与转化等领域极具应用潜力,引起科学家的广泛关注。最新的研究表明有机多孔碳材料在氧还原领域表现出优异的性能。相比于其他前驱体,以稠环芳烃聚合构建的共轭多孔聚合物前驱体制备的多孔碳材料具有高比表面、结构可控等优点。
因此,在燃料电池电催化氧化原应用方面,如何制备优异的氧化原催化活性和良好的稳定性的碳材料是亟待解决的问题。
技术实现要素:
本发明的目的就是为了克服上述现有技术存在的缺陷,而提供一种基于三氯代氮杂并苯稠环芳烃的氮掺杂多孔碳材料的制备方法及其应用,以制备优异的氧化原催化活性和良好的稳定性的碳材料。
本发明,以c3对称的三氯代氮杂并苯稠环芳烃为聚合单体,制备共轭多孔聚合物,在高温煅烧和氨气活化后的氮掺杂多孔碳材料(hpnc@n)有较高的比表面积。在电催化氧化原应用方面,hpnc@n表现出优异的氧化原催化活性和良好的稳定性。
本发明的目标可以通过以下技术方案来实现:
基于三氯代氮杂并苯稠环芳烃的氮掺杂多孔碳材料的制备方法,以均三溴苯为起始原料,通过buchwald-hartwig偶联、水解和脱水芳构化反应制备c3对称的三氯代氮杂并苯稠环芳烃,然后通过脱氯-碳碳偶联反应制备含氮共轭多孔聚合物,最后通过高温煅烧和氨气活化,得到氮掺杂多孔碳材料。
具体包括以下步骤:
(1)1,3,5-三(2-氨基苯甲酸甲酯)苯的合成:将1,3,5-三溴苯、邻氨基苯甲酸甲酯和碳酸铯溶解于甲苯,经冷冻-抽真空-融化循环后,在氮气下加入pd(oac)2和三(叔丁基)膦,并在氮气下回流反应70-80小时,之后冷却至室温、过滤、洗涤、萃取分离,有机相经干燥、纯化后得到产物3(nh-och3);
(2)1,3,5-三(2-羧基苯基)苯的合成:将步骤(1)得到的3(nh-och3)和naoh溶解在丙酮和水的混合物,溶液在氮气下回流搅拌15-20小时后,冷却、萃取分离,hcl酸化、过滤、干燥得到固体产物3(nh-oh);
(3)6,12,18-三氯-5,11,17-三氮杂萘并苯的制备:向干燥后的3(nh-oh)中加入pocl3,将溶液在氮气下搅拌并回流20-25小时,冷却后将溶液倒入冰nh4oh溶液中,过滤、洗涤、干燥得到产物3(ncl);
(4)含氮共轭多孔聚合物的制备:将得到的产物3(ncl)用重结晶的方法获得3(ncl)晶体,将干燥的3(ncl)晶体置于管式炉中加热进行脱氯-碳碳偶联反应,最后降至室温,得到含氮共轭多孔聚合物pnc;
(5)氮掺杂多孔碳材料的制备:将含氮共轭多孔聚合物pnc置于管式炉中,先在氩气保护下,加热至800-1000℃,碳化时间为1-3小时,然后通入氨气活化,最后在氮气氛围下降至室温,得到氮掺杂多孔碳材料hpnc@n。
优选地,步骤(1)中,1,3,5-三溴苯、邻氨基苯甲酸甲酯与碳酸铯的摩尔比为1:3.5:5。
优选地,步骤(1)中,需进行至少三次冷冻-抽真空-融化的循环,纯化采用硅胶柱色谱法,其中,乙酸乙酯与石油醚的体积比为2:1。
优选地,步骤(2)中,3(nh-och3)和naoh的摩尔比为1:12。
优选地,步骤(2)中,丙酮和水的体积比为1:1;萃取分离时加入的混合物是ch2cl2和水(v:v=1:1)的混合物,hcl酸化是将水相的ph值调至1。
优选地,步骤(3)中,pocl3需用氢化钙除水并蒸馏提纯。
优选地,步骤(4)中,pnc的制备工艺为:在氮气保护下,以10℃min-1的速率加热至400℃,反应时间为3小时。
优选地,步骤(5)中,在氩气氛围下的升温速率为10℃min-1,氨气活化时间为15分钟。
所述氮掺杂多孔碳材料用于燃料电池的电催化氧化。
将氮掺杂多孔碳材料加入到质量分数为0.05%的nafion溶液中超声2小时得到分散均匀的材料浆料,取12μl的浆料滴在玻碳电极表面,自然晾干后即可进行电化学氧还原进行测试。
本发明中,浆料由10mg新型氮掺杂多孔碳材料加入1ml质量分数为0.05%的nafion溶液制得。
本发明利用脱氯-碳碳偶联反应制备了基于三氯代氮杂并苯稠环芳烃的含氮共轭多孔聚合物,并对其进行高温煅烧和氨气活化处理,使其具有更高的比表面积和大量的催化活性位点,在氧化原方面具有良好的性能和优异的稳定性。与现有技术相比,本发明所制备的新型氮掺杂多孔碳材料具有高比表面、低骨架密度和良好的化学稳定性等优点,在氧化原领域有着较好的性能。
附图说明
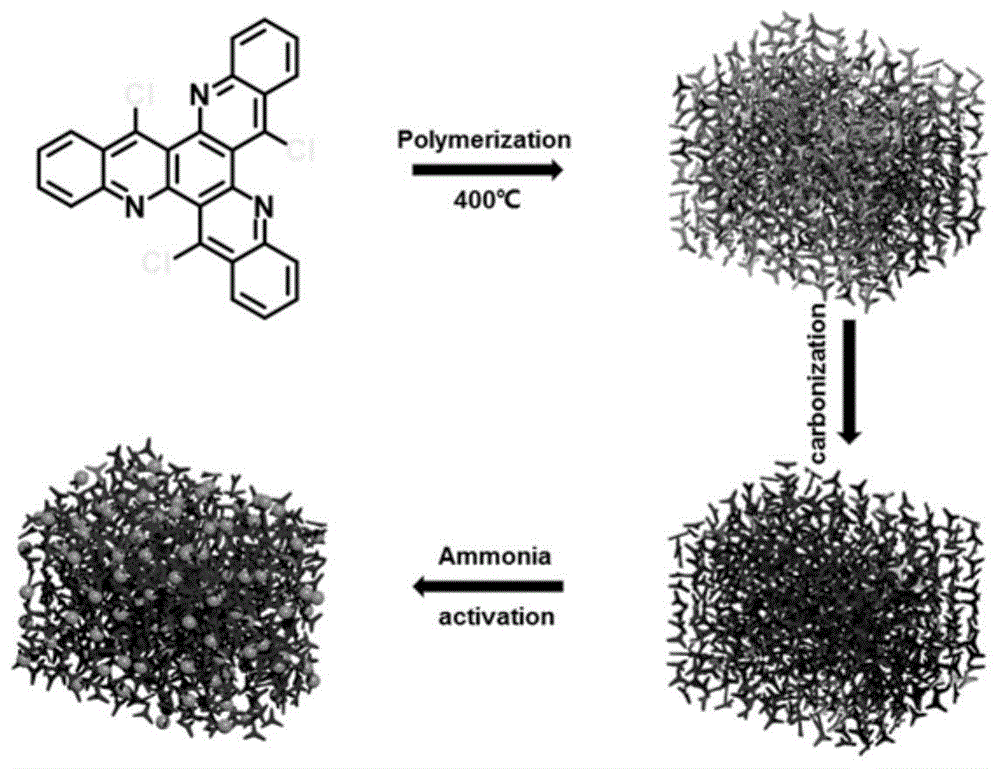
图1是氮掺杂多孔碳材料的制备路线图;
图2是1,3,5-三(2-氨基苯甲酸甲酯)苯3(nh-och3)的核磁共振氢谱图(1hnmr);
图3是1,3,5-三(2-羧基苯基)苯3(nh-oh)的核磁共振氢谱图(1hnmr);
图4是6,12,18-三氯-5,11,17-三氮杂三奈3(ncl)的核磁共振氢谱图(1hnmr);
图5是3(ncl)晶体和pnc的在氮气氛围中的tga曲线;
图6是3(ncl)晶体和pnc的红外光谱图;
图7是hpnc@n-800、hpnc@n-900和hpnc@n-1000的拉曼图谱;
图8是hpnc@n-800、hpnc@n-900和hpnc@n-1000的氮气吸脱附等温线;
图9是hpnc@n-800、hpnc@n-900和hpnc@n-1000在氮气和氧气下的cv曲线;
图10是hpnc@n-800、hpnc@n-900和hpnc@n-1000的rrde曲线;
图11是hpnc@n-900不同转速下的rde线性扫描伏安曲线;
图12是hpnc@n-900的稳定性和耐甲醇性能;
图13是hpnc-800、hpnc-900和hpnc-1000的氮气吸脱附等温线;
图14是hpnc-800、hpnc-900和hpnc-1000在氮气和氧气下的cv曲线;
图15是hpnc-800、hpnc-900和hpnc-1000的rrde曲线;
图16是hpnc-800、hpnc-900和hpnc-1000的拉曼图谱。
具体实施方式
下面结合附图和具体实施例对本发明进行详细说明。
本发明实施例中,1,3,5-三溴苯,邻氨基苯甲酸甲酯,碳酸铯,醋酸钯等直接购买于aladdin。
对中间产物、最终产物的结构进行表征,其测试方法如下:
对中间产物的组成表征采用brukerav500型核磁共振仪测定1,3,5-三(2-氨基苯甲酸甲酯)苯3(nh-och3),1,3,5-三(2-羧基苯基)苯3(nh-oh),6,12,18-三氯-5,11,17-三氮杂萘并苯3(ncl)的核磁共振氢谱(1hnmr),cdcl3作溶剂。
采用pyris1热重分析仪,nicolet6700傅里叶变换红外光谱仪,senterrar200-l拉曼光谱仪对小分子3(ncl),含氮共轭多孔聚合物pnc及氮掺杂多孔碳材料hpnc@n进行结构表征。
采用afmsrce2759型进行电化学工作站对氮掺杂多孔碳材料进行电催化氧还原测试。
实施例1
一种基于c3对称的三氯代氮杂并苯稠环芳烃用于氮掺杂多孔碳材料(hpnc@n-900)的制备和应用,制备路线如图1所示,具体内容如下:
(1)1,3,5-三(2-氨基苯甲酸甲酯)苯(3(nh-och3))的合成:将1,3,5-三溴苯(1.0当量),邻氨基苯甲酸甲酯(3.5当量),碳酸铯(5.0当量)的混合物溶解于甲苯中。在三次冷冻-抽真空-融化循环后,在氮气下加入pd(oac)2(0.06当量)和三(叔丁基)膦(0.18当量),然后将混合物在氮气下回流搅拌72小时。冷却至室温后,过滤,滤液用饱和nh4cl(1m)洗涤两次,分离出有机相,用na2so4干燥,过滤并减压浓缩,粗产物利用硅胶柱色谱法(乙酸乙酯:石油醚=2:1,v/v)纯化,得到3(nh-och3),为浅黄色固体粉末,收率为87%。
3(nh-och3)的核磁氢谱见图2。结构数据如下,1hnmr(400mhz,cdcl3):δ=9.45(s,3h;nh),7.96(d,j=7.8hz,3h;ar-h),7.36(m,6h;ar-h),6.84(s,3h;ar-h),6.75(m,3h;ar-h),3.90(s,3h;och3)。
(2)1,3,5-三(2-羧基苯基)苯(3(nh-oh))的合成:将3(nh-och3(1.0当量)和naoh(12.0当量)溶解在丙酮和水的混合物(v:v=1:1)中。将溶液在氮气下回流搅拌16小时。冷却至室温后,加入ch2cl2和水(v:v=1:1)的混合物,萃取分离,水相通过加入hcl溶液使溶液ph值达到1。酸化过程中产生大量的沉淀,过滤得到的沉淀,在100℃下真空干燥,得到白色固体,收率为95%。
3(nh-oh)的核磁氢谱见图3。结构数据如下,1hnmr(400mhz,dmso-d6):δ=9.6(s,3h;nh),7.89(dd,j=1.5hz,7.8hz,3h;ar-h),7.42(m,6h;ar-h),6.81(m,3h;ar-h),6.75(s,3h;ar-h)。
(3)6,12,18-三氯-5,11,17-三氮杂萘并苯(3(ncl))的合成:将3(nh-oh)(1.0当量)在110℃下真空干燥4小时,加入pocl3。将混合物在氮气下搅拌并回流24小时。冷却至室温后,将溶液缓慢倒入冰nh4oh溶液中。过滤得到的沉淀物用大量ch2cl2洗涤,并在40℃下真空干燥得到棕色固体,收率为72%。
3(ncl)的核磁氢谱见图4。结构数据如下,1hnmr(400mhz,cdcl3):8.67(d,j=8.6hz,3h;ar-h),8.31(d,j=8.6hz,3h;ar-h),7.89(t,j=8.3hz,3h;ar-h),7.76(t,j=8.1hz,3h;ar-h)。
(4)含氮共轭多孔聚合物(pnc)的制备:用重结晶的方法获得3(ncl)晶体,将干燥的3(ncl)晶体均匀的铺在陶瓷舟中,并置于管式炉中,在氮气气保护下,以10℃min-1的速率加热至400℃,反应3小时,最后降至室温,得到黑色的固体粉末,收率为62%。
3(ncl)晶体和pnc在氮气氛围下的热重曲线如图5所示,当温度升至400℃左右时,3(ncl)晶体有明显的质量下降过程,质量下降约35%,说明在此温度区间内,3(ncl)分子中的氯(cl)元素开始脱落。随着温度的升高,脱氯交联之后的聚合物具有良好的热稳定性。为了进一步认证这一点,我们另外单独测试了pnc的热稳定性,从曲线可以看出pnc在1000℃时质量仍能保持90%左右,这点可能归因于3(ncl)的刚性骨架和高度交联的共轭多孔聚合物。3(ncl)和pnc的红外光谱如图6所示,3(ncl)中存在1569cm-2处为吡啶环上c-n键的伸缩振动峰,744cm-2处为c-cl键的伸缩振动峰;而pnc中,c-cl键的强度明显减弱,说明3(ncl)在400℃下发生聚合反应,成功转化为高度交联的含氮共轭多孔聚合物pnc。
(5)氮掺杂多孔碳材料(hpnc@n-900)的制备:将预交联后的pnc均匀的铺在陶瓷舟中,并置于管式炉中,在氩气保护下,以10℃min-1的速率加热至900℃,并保持2小时,然后通氨气活化时间为15分钟,最后降至室温,得到黑色的固体粉末。
氮掺杂多孔碳的拉曼光谱如图7所示,hpnc@n-900在1334以及1590cm-1附近都有两个明显的衍射峰,分别对应材料的d峰和g峰。d峰代表碳边缘的不对称缺陷,g峰对应于材料的e2g伸缩振动,我们通常用id/ig的值来表征材料的缺陷和有序度,比值越大说明材料的石墨化程度越低,缺陷程度越大。hpnc@n-900的id/ig值为1.06,说明高温热解可以实现高石墨化程度的多孔碳材料。从图8可以得知,hpnc@n-900的bet比表面积达到了763.9m2g-1。
(6)hpnc@n-900的氧还原性能测试:以0.1m的koh作为电解液,采用传统的三电极体系,ag/agcl为参比电极,铂丝为对电极,负载了hpnc@n-900的玻碳电极为工作电极。工作电极的制备过程如下:称取10mg的hpnc@n-900加入到1ml配好的质量分数为0.05%的nafion溶液超声2小时,得到分散均匀的材料浆料,取12μl配好的浆料滴在玻碳电极表面,自然晾干后即可进行氧还原性能测试,扫描速度为100mvs-1,电压范围为0-1v。
图9a和图9b分别为hpnc@n材料在n2和o2条件下的循环伏安曲线。hpnc@n-900在n2条件下曲线没有氧化还原峰;但在o2条件下,循环伏安曲线在-0.28v(vsrhs)有明显的还原峰,说明hpnc@n-900存在氧还原活性。从图10的旋转盘环电极(rrde)的测试结果可知,hpnc@n-900的起始电位与半波电位分别为0.89和0.75v(vs.rhe),极限电流密度最高能达到5.5macm-2。旋转圆盘电极(rde)测试是在10mvs-1的扫描速度,旋转速度在225、400、625、900、1225、1600rpm下进行。如图11所示,hpnc@n-900的rde曲线随着转速的增大(225rpm-2025rpm),电流密度逐渐增大,这说明hpnc@n-900在高转速下能保持良好的氧还原催化活性。图12表明hpnc@n-900在氧还原测试中具有良好的稳定性,在循环10小时后,性能仍保持着93.1%的稳定性,高于商业化pt/c(81.2%);同时,hpnc@n-900在测试过程中具有极好的耐甲醇性能。
实施例2
步骤(1)~(4)同实施例1。
步骤(5)氮掺杂多孔碳材料(hpnc@n-800)的制备:将预交联后的pnc均匀的铺在陶瓷舟中,并置于管式炉中,在氩气保护下,以10℃min-1的速率加热至800℃,并保持2小时,然后通氨气活化时间为15分钟,最后降至室温,得到黑色的固体粉末。
hpnc@n-800的拉曼光谱见图7,hpnc@n-800的id/ig值为1.07。图8可知hpnc@n-800的bet比表面积为345.2m2g-1,相比于hpnc@n-900,煅烧温度的降低,降低了hpnc@n-800的石墨化程度;同时,多孔碳材料内部的孔隙也减少,从而降低了比表面积。
图9中,hpnc@n-800在n2条件下曲线没有氧化还原峰;但在o2条件下曲线在-0.28v(vsrhs)有明显的还原峰,说明hpnc@n-800存在氧还原活性。从图10的旋转盘环电极(rrde)的测试结果可知,hpnc@n-800的起始电位与半波电位分别为0.79和0.7v(vs.rhe),极限电流密度最高能达到4macm-2。相比于hpnc@n-800,hpnc@n-900具有较好的氧还原性能,这是由于较高的煅烧温度使得多孔碳材料具有较好的导电性和高的比表面积。
实施例3
步骤(1)~(4)同实施例1。
步骤(5)氮掺杂多孔碳材料(hpnc@n-1000)的制备:将预交联后的pnc均匀的铺在陶瓷舟中,并置于管式炉中,在氩气保护下,以10℃min-1的速率加热至1000℃,并保持2小时,然后通氨气活化时间为15分钟,最后降至室温,得到黑色的固体粉末。
hpnc@n-1000的拉曼光谱见图7,hpnc@n-800的id/ig值为1.05。图8可知hpnc@n-1000的bet比表面积为249.8m2g-1相比于hpnc@n-900,煅烧温度的升高,虽然提高多孔碳的石墨化程度和导电性,但聚合物内部孔道的坍塌降低了材料的比表面积。
图9中,hpnc@n-1000在n2条件下曲线没有氧化还原峰;但在o2条件下曲线在-0.28v(vsrhs)有明显的还原峰,说明hpnc@n-1000存在氧还原活性。从图10的旋转盘环电极(rrde)的测试结果可知,hpnc@n-1000的起始电位与半波电位分别为0.79和0.7v(vs.rhe),极限电流密度最高能达到4.8macm-2。相比于hpnc@n-1000,hpnc@n-900具有较好的氧还原性能,这是由于多孔碳材料的丰富的缺陷和高的比表面积可以实现优异的氧还原性能。
实施例4
步骤(1)~(4)同实施例1。
步骤(5)氮掺杂多孔碳材料(hpnc@x,x=800,900,1000)的制备:将预交联后的pnc均匀的铺在陶瓷舟中,并置于管式炉中,在氩气保护下,以10℃min-1的速率加热至800,900或1000℃,并保持2小时,最后降至室温,得到黑色的固体粉末,分别命名为hpnc-800,hpnc-900,hpnc-1000。从图13可知,hpnc-800,hpnc-900,hpnc-1000的比表面积分别为256.7、459.6和126.3m2g-1。
(6)碳材料的氧还原性能测试:以0.1m的koh作为电解液,采用传统的三电极体系,ag/agcl为参比电极,铂丝为对电极,负载了测试材料的玻碳电极为工作电极。工作电极的制备过程如下:称取10mg的hpnc-x加入到1ml配好的质量分数为0.05%的nafion溶液超声2小时得到分散均匀的材料浆料,取12μl配好的浆料滴在玻碳电极表面,自然晾干后即可进行测试。
如图14所示,hpnc系列材料在在n2条件下曲线没有氧化还原峰;但在o2条件下,循环伏安曲线在-0.28v(vsrhs)处有明显的还原峰,说明hpnc系列材料存在氧还原活性。hpnc系列材料的旋转盘环电极(rrde)的测试结果如图15所示,相比于hpnc-800和hpnc-1000,hpnc-900具有更好的氧还原性能,起始电位与半波电位分别为0.79和0.75v(vs.rhe),极限电流密度最高能达到4macm-2,这是由于900℃煅烧可以更好的平衡多孔碳材料的比表面积和材料的催化位点含量等各项参数,从而具有最好的氧还原性能。图16是hpnc系列材料的拉曼光谱,hpnc-800,hpnc-900与hpnc-1000的id/ig值分别为1.06、1.04、1.03,结果表明hpnc@n系列多孔碳材料具有更多的缺陷结果,因此,氨气活化可以使材料的缺陷程度增大,从而提高材料的活性位点,有利于明显提高多孔碳材料的电催化氧还原性能。
上述的对实施例的描述是为便于该技术领域的普通技术人员能理解和使用发明。熟悉本领域技术的人员显然可以容易地对这些实施例做出各种修改,并把在此说明的一般原理应用到其他实施例中而不必经过创造性的劳动。因此,本发明不限于上述实施例,本领域技术人员根据本发明的揭示,不脱离本发明范畴所做出的改进和修改都应该在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!