前列腺癌的生物学标志物、治疗靶点及其用途的制作方法
本申请是申请日为2011年9月16日、名称为《前列腺癌的生物学标志物、治疗靶点及其用途》的中国专利申请201180073445.7的分案申请。
技术领域
本发明涉及癌症领域,特别是前列腺癌。同时,本发明涉及使用下一代测序技术,以寻找用于诊断、预后和治疗反应预测的生物学标志物和有效治疗前列腺癌的药物靶点,特别是用于前列腺癌的生物学标志物。本发明中,特别使用了RNA-Seq技术,即转录组测序技术分析前列腺癌组织和癌旁正常组织的转录组,揭示中国人前列腺癌完整的转录图谱。
背景技术:
在发达国家,前列腺癌仍是发病率最高的肿瘤,同时在男性癌症相关死亡中排第二位。全世界前列腺癌的发病率在不断上升,但在不同国家和种族中,其发病率差异很大。发病率最高的是西方国家,如美国;发病率最低的是东亚国家,如中国,这种差异可能部分是由不同种族的基因差异引起的。此外,前列腺癌是一种异质性疾病。每一个肿瘤在肿瘤进化以及生物学行为(如肿瘤休眠,局部生长,远处扩散,对治疗的反应以及复发等)上差异很大。因此,组织病理学分级分期以及Gleason评分相同、治疗方案相同的病人,其临床结局以及肿瘤进展史可能截然不同。有的病人其肿瘤处于休眠状态、局限于前列腺,可以生存10年以上,而其他病人却在诊断后2-3年死于肿瘤的远处转移。种种证据表明,前列腺癌临床行为的异质性是在肿瘤进展过程中由其内在的分子机制差异引起的。
在过去的十余年间,DNA和RNA芯片技术在分析生物学机制上应用广泛。其帮助我们对前列腺癌的发病机制有了新的了解,为我们找到用于诊断、预后和治疗反应预测的生物学标志物提供了基础。虽然目前为止,类似乳腺癌的OncotypeDx和MammoPrint的用于前列腺癌基因组预后检测极少,但一些被发现的前列腺癌分子学改变正在被应用于临床实践。Taylor等(Taylor BS,et al.(2010)Integrative genomic profiling of human prostate cancer.Cancer Cell 18(1):11-22.)通过对前列腺癌的综合基因组分析发现,某些基因拷贝数的变化可能区分进展性肿瘤和休眠性肿瘤,该发现意义重大。然而,我们仍迫切需要新的生物学标志物以更准确地检出前列腺癌并改进对肿瘤进展性及治疗结局的预测能力。
需要指出的是,虽然以基因芯片为基础的研究对我们对人类肿瘤发生发展的理解做出了重大贡献,但该技术有很大的局限性,如不能检测基因组结构的变化和碱基突变。
技术实现要素:
在过去几年中,下一代测序技术(Next Generation Sequencing,NGS)的飞速发展克服了上述不足。NGS使我们能以前所未有的高分辨率和高通量分析整个肿瘤基因组及转录组。NGS的数据能从多个角度分析基因组,如突变,转录,结构变异和转录后调节(如甲基化)。此外,NGS技术的不断改进使得科学家能够对主要的肿瘤类型的基因组进行测序。
目前,几乎所有针对前列腺癌基因组和转录组水平变化的研究都是在白人中进行,黄种人的研究极少。在本研究中,我们用RNA-Seq技术,即转录组测序技术分析了14对前列腺癌组织和癌旁正常组织的转录组。我们将所有的转录产物类型进行分析,揭示中国人前列腺癌完整的转录图谱。我们找到了很多异构体包括:外显子跳跃、内含子保留、5’和3’端选择性剪切、基因融合、点突变、长链非编码RNA,这些都可能在前列腺癌的发生和发展中起作用。我们的研究阐明了前列腺癌基因组变化的复杂图谱,证实了前列腺癌的异质性,推进了我们对中国人前列腺癌的认识。
1.前列腺癌新型融合基因的发现和验证
(1).对上海长海医院14对前列腺癌和癌旁组织中进行RNA-Seq(即转录组测序技术),发现USP9Y-TTTY15、CTAGE5-KHDRBS3、RAD50-PDLIM4、SDK1-AMACR共4个文献未报道高频融合基因及其它数十个融合基因,参见如下表1。
表1.前列腺癌新型融合基因
(2).我们在54对前列腺癌和癌旁组织中对这些融合基因进行了验证。我们设计了基因融合特异性的PCR引物。PCR和琼脂电泳后,所有RT-PCR扩增片段割胶回收(Qiagen QIAquick Gel Extraction kit)并行Sanger测序。我们发现验证的4个新型融合基因在癌组织中特异表达、频率较高(结果见图2-4)。这些融合基因之前未被报道过,但其在本研究中频率较高提示其在中国人前列腺癌的发生中起重要作用,这些可望在后续的研究中得到阐明。
(3).临床应用前景:在癌组织中表达,癌旁和正常组织中不表达的融合基因,是高度特异性的前列腺癌标记物,在血液、尿液中通过real time PCR检测,前列腺穿刺组织和术后组织通过FISH检测融合基因存在情况,用于前列腺癌病人的早期诊断、分子分型和判断病人预后,同时融合基因可作为靶向治疗的靶点。
2.发现差异性表达的长链非编码RNA
前列腺癌中长链非编码RNA的转录图谱。越来越多的证据表明长链非编码RNA在细胞生物学许多方面中起作用,提示其在疾病的病因学,包括肿瘤发生机制中起作用。到目前为止,之前的研究都未涉足肿瘤中长链非编码RNA的整体转录水平改变。因此,我们首先在前列腺癌组织及其配对癌旁正常组织中分析了长链非编码RNA的整体转录谱,发现每个标本中平均有1599个已知长链非编码RNA表达。接下来,我们在前列腺癌组织和配对癌旁正常组织比较了长链非编码RNA的表达水平,发现平均有406个长链非编码RNA在二者间有差异性表达(倍数改变>=2,假阳性率,False positive Rate,FDR<=0.001),其中137个长链非编码RNA在50%的前列腺癌中都呈现一致的上调或下调。
因为大多数长链非编码RNA被发现与转录调节有关,我们研究了长链非编码RNA表达量的变化对前列腺癌基因表达的影响。我们分析了每个长链非编码RNA与所有基因表达量的相关性。使用绝对相关系数大于0.85、假发现率小于0.01为界值,我们发现与长链非编码RNA高度相关的基因。非常有趣的是,有23个长链非编码RNA与全基因组中数百个基因显著相关,而其他大多数基因仅与几个基因相关,或者根本就不相关。这提示长链非编码RNA可能有转录调节以外的功能,比如在转录后水平的调节。出人意料的是,除了两个长链非编码RNA外,几乎所有的长链非编码RNA与基因表达呈正相关,提示这些长链非编码RNA可能促进基因的表达。
为了研究长链非编码RNA与前列腺癌的关系,我们选择了4个长链非编码RNA(两个已知:DD3和MALAT1;两个新发现:FR257520和FR348383),并用qRT-PCR在两组前列腺标本中检测它们的表达量。第一组是40对前列腺癌组织及其配对癌旁正常组织,第二组是15个正常人前列腺组织和15个前列腺癌组织。qRT-PCR和RNA-seq结果有很强的相关性。与RNA-Seq结果一致,在大多数前列腺癌标本中PCA3、MALAT1和FR348383过表达,而FR257520表达量降低。PCA3过表达的结果与之前认为其可能成为新的诊断标志物的研究类似,但我们首次发现MALAT1、FR257520和FR348383在前列腺癌中表达与正常前列腺有明显差异。
临床应用前景:在血液、尿液中通过real time PCR检测长链非编码RNA存在情况,用于前列腺癌病人的早期诊断、分子分型,同时可作为靶向治疗的靶点,判断病人预后。我们的研究结果表明137个长链非编码RNA可以作为生物标志物,具体参见表2。
表2.137个长链非编码RNA
3、单核苷酸多态性和点突变的检测
我们使用SOAPsnp(Li RQ,Li YR,Fang XD,Yang HM,Wang J,et al.(2009)SNP detection for massively parallel whole-genome resequencing.Genome Research 19:1124-1132.)检测单核苷酸多态性。Sanger测序验证突变。我们通过以下步骤减少单核苷酸多态性检测的假阳性率,包括删除一致性质量低于20的SNP、位于剪接供体位点5bp以内的SNP以及读数支持不超过2个的SNP。为了找到新的SNP,我们进一步在已报道的六大SNP数据库进行筛选(YH,1000genomes,Yoruba,Korean,Watson and NCBI dbSNP)。
前列腺癌突变谱。我们在前列腺癌组织中平均找到1725个点突变。然而,只有一小部分(平均1.5%)位于基因的编码区。有趣的是,有的点突变位于长链非编码RNA。绝大多数突变(91.7%)是T:A到C:G的突变。对该发现的一个合理的解释是,这种点突变发生在RNA编辑的时候,RNA编辑通过将腺嘌呤核苷改变为次黄嘌呤核苷,后者翻译时被读作鸟嘌呤核苷,从而导致特定的RNA核苷酸的改变。
在290个基因的编码区中共找到309个点突变。其中115个为沉默突变、181个错义突变、13个为无义突变。这些突变都未在多于一个肿瘤组织中发现,提示在这些前列腺癌样本中没有热点突变。然而,我们发现有3个样本有位于UTP14C基因不同位置的突变,有两个样本有位于4个基因(CBARA1,FRG1,NAMPT和ZNF195)不同位置的突变。我们用基因组PCR、RT-PCR和Sanger测序证实了30个突变。其中27个在基因组水平证实,29个在cDNA水平证实。
我们还找到183有突变的基因,但大多数都是低频率突变。这与Taylor等(Taylor BS,et al.(2010)Integrative genomic profiling of human prostate cancer.Cancer Cell 18(1):11-22.)报道的138个基因结果一致。在30个基因进行突变验证发现RNA-Seq发现突变的准确性分别为96.7%(cDNA水平)和90%(基因组水平)。1个样本有KLK3基因突变。令人吃惊的是,所有样本都没有P53和PTEN突变,而这两个基因是COSMIC数据库中与前列腺癌相关度最高的基因。虽然大多数突变的基因之前未在前列腺癌中被报道过,其中118个在其它肿瘤中被发现过,提示这些基因的突变可能也导致前列腺癌。
临床应用前景:从前列腺穿刺组织或手术后组织中提取DNA后行PCR后送测序检测SNP和点突变存在情况,用于前列腺癌病人分子分型和药物治疗靶标,判断病人预后。本发明提供的183个基因的194个突变参见表3,其中优选的30个基因突变如表8所示
表3.前列腺癌特异性基因突变
4.选择性剪切的检测
选择性剪切(alternative splicing,AS)是真核细胞中的普遍现象,它能使基因转录出不同的mRNA产物,进而可能翻译出不同的蛋白异构体。
(1).我们使用SpliceMap来寻找剪切位点,然后运用不同方法检测不同类型的选择性剪切包括外显子跳跃、内含子保留以及选择性5’和3’剪切位点。首先我们找到28个标本转录组中所有的选择性剪切。然后我们找到仅存在于癌组织样本而其配对癌旁组织没有的选择性剪切。我们找到了数千个选择性剪切,通过非冗余读序筛出一组高度可靠地差异性剪切。在超过一半的前列腺癌样本中发现有KLK3(也叫PSA)基因的内含子保留,这可能产生一种新的蛋白序列。选择性剪切的转录产物和蛋白都可能作为前列腺癌诊断的新生物学标记物。在一部分前列腺癌样本中发现有AMACR基因的外显子跳跃。这两种选择性剪切方式都用RT-PCR在测序组得到了验证。我们同时在另外40对样本中用RT-PCR进行了验证,发现绝大多数癌组织样本中有PSA内含子保留,而癌旁组织中几乎没有。PSA是为数不多的几个常规用于诊断的生物学标志物。然而,目前以PSA为基础的筛查手段准确度有限。我们新发现的PSA内含子保留可能有助于改进PSA的敏感性和特异性。40个癌组织样本中仅9个有AMACR基因外显子跳跃。
(2).临床应用前景:在血液、尿液中通过real time PCR或者ELISA检测选择性剪切的存在情况,用于前列腺癌病人的早期诊断、分子分型,同时可作为靶向治疗的靶点,判断病人预后。
表4.选择性剪切体,包括3'剪切位点变异,5'剪切位点变异,外显子跳跃和内含子保留四种方式。
3'剪切位点变异
5'剪切位点变异
外显子跳跃
内含子保留
为了理解前列腺癌中上述分子遗传学改变,我们把与基因融合、点突变、差异性表达、肿瘤特异性差异性剪切相关的肿瘤与Taylor描述的调节异常的信号通路相对比。依据文献资料,我们把肿瘤中过表达的基因以及已知的癌基因定义为激活基因,把肿瘤中表达下调的基因以及已知的抑癌基因定义为失活基因。我们计算了每个激活基因、失活基因在14个标本中的频率。如果肿瘤标本在信号通路中有一个或多个基因有点突变、基因融合、差异性表达或肿瘤特异的选择性剪切,我们就认为肿瘤在该信号通路发生了改变。我们发现有3个很常见的信号通路(AR、Ras-PI3K-AKT和RB)在前列腺癌中发生了变化。
与其它很多肿瘤一样,前列腺癌是一种遗传性疾病,是由一系列基因改变的累积引起的。因此,更详细的基因特征分析将有助于更好地理解这些疾病并促进研发新的个体化的靶向治疗。此外,不同种族特别是白人和黄种人之间前列腺癌发病率和临床预后差异很大。然而,虽然白人的前列腺癌基因谱被研究得很深入,黄种人中的相关研究极少。本研究中,我们通过14对癌组织及配对癌旁正常组织进行RNA-Seq研究了上述两个问题。这同时也是首次同时揭示前列腺癌转录组的多个方面,包括基因融合、选择性剪切、病毒转录片段和长链非编码RNA的表达以及体细胞突变。通过对上述方面的研究,我们发现不同前列腺癌病人转录组有很大的异质性。对这些不同的基因改变的综合分析发现与中国人前列腺癌发生相关的信号通路与白人类似。这些发现为研究中国人前列腺癌的发病机制提供了新的可能,同时提供了治疗前列腺癌的可能方式。
附图说明
图1.系统肿瘤转录组分析流程图。
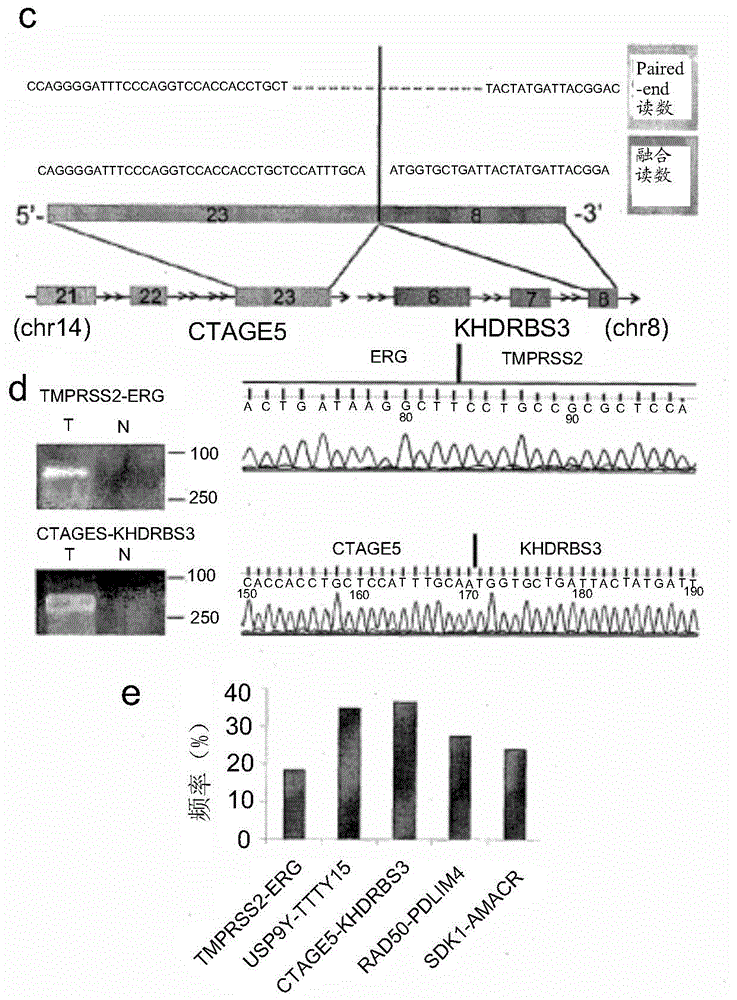
图2.融合基因示意图。其中图2c是CTAGE5-khdrbs3融合基因示意图,ctage5的第23个外显子与khdrbs3第8个外显子融合在一起;图2d是Tmprss2-erg融合基因示意图,Tmprss2第1个外显子与ERG第4个外显子融合在一起;图2e是5个融合基因的发生频率。
图3.融合基因示意图。其中图3a是USP9Y-TTTY15融合示意图,USP9Y的第3个外显子和TTTY15的第4个外显子融合在一起;图3b是USP9Y-TTTY15的RT-PCR结果。
图4.融合基因示意图。其中图4a RAD50-PDLIM4融合基因RT-PCR和sanger测序结果;图4b是SDK1-AMACR融合基因RT-PCR和sanger测序结果。
图5.长链非编码的差异表达。其中图5c是长链非编码RNA DD3MALAT1FR0257520 FR0348383在40对癌和癌旁组织中的差异表达;图5d是长链非编码RNA:DD3、MALAT1、FR0257520和FR0348383在前列腺癌和良性前列腺增生组织中的差异表达。
具体实施方式
下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限定本发明的范围。
除非另有定义,否则本文中所使用的科学和技术术语具有本领域技术人员通常理解的含义。为了更好的理解本发明,特别提供了下列术语的定义。
发现融合基因、长链非编码RNA、突变、选择性剪切的共同步骤:收集前列腺癌病人样本—>癌组织及癌旁组织行冰冻切片后由病理学家检查保证质量—>制备cDNA文库—>RNA-Seq—>将测序结果在基因组和转录组定位—>将基因和长链非编码RNA表达水平标准化后找到差异表达的长链非编码RNA、选择性剪切以及肿瘤特异性的突变、融合基因。
本发明一方面提供了用于前列腺癌的生物学标志物,包括如表1所示的融合基因、表2所示的长链非编码RNA、表3所示的基因突变、表4所示的选择性剪切中的一种或多种。
本发明所述的生物学标志物,其进一步可用作前列腺癌的早期诊断标志物、药物治疗有效性判断标志物或患者预后标志物。
在本发明的具体实施方式中,所述的生物学标志物中,所述融合基因包括表6的83个融合基因中的一种或多种,优选的包括表6中下划线所示的35个融合基因中的一种或多种。
在本发明的具体实施方式中,所述的生物学标志物中,所述融合基因包括USP9Y-TTTY15、CTAGE5-KHDRBS3、RAD50-PDLIM4、SDK1-AMACR中的一种或多种,优选地融合基因USP9Y-TTTY15、CTAGE5-KHDRBS3、RAD50-PDLIM4、SDK1-AMACR用表5所述的引物进行扩增。
在本发明的具体实施方式中,所述的生物学标志物中,所述长链非编码RNA包括DD3、MALAT1、FR0257520、FR0348383中的一种或多种,优选地所述长链非编码RNA:DD3、MALAT1、FR0257520、FR0348383用表7所述的引物进行扩增。
在本发明的具体实施方式中,所述的生物学标志物中,所述基因突变包括如表8所示的30个基因突变中的一种或多种,优选地表8所示的30个基因突变用表9所述的引物进行扩增。
在本发明的具体实施方式中,所述的生物学标志物中,所述选择性剪切包括PSA或AMACR,优选地选择性剪切PSA或AMACR用表10所述的引物进行扩增。
本发明另一方提供了所述的生物学标志物在作为诊断前列腺癌的试剂或者治疗前列腺癌的药物的靶点中的用途,特别是用作前列腺癌的早期诊断标志物、药物治疗有效性判断标志物或患者预后标志物的用途。
本发明另一方面进一步提供了用于扩增所述的生物学标志物的引物或所述生物学标志物的探针在制备用于为诊断前列腺癌的试剂中的用途。其中,所述引物可用于特异性扩增所述生物学标志物,所述探针特异性与所述生物学标志物结合,从而指示所述生物学标志物的存在。
在本发明的具体实施方式中,提供用于扩增所述的生物学标志物的引物,其中所述引物优选地包括表5所述的引物,其用于融合基因USP9Y-TTTY15、CTAGE5-KHDRBS3、RAD50-PDLIM4、SDK1-AMACR;表7所示的引物,其用于扩增长链非编码RNA:DD3、MALAT1、FR0257520、FR0348383;表9所示的引物,其用于扩增表8所示的30个基因突变;表10所示的引物,其用于扩增选择性剪切PSA或AMACR。
在本发明的具体实施方式中,提供了表5所述的引物在制备诊断前列腺癌的试剂中的用途。
在本发明的具体实施方式中,提供了表7所示的引物在制备诊断前列腺癌的试剂中的用途。
在本发明的具体实施方式中,提供了表9所示的引物在制备诊断前列腺癌的试剂中的用途。
在本发明的具体实施方式中,提供了表10所示的引物在制备诊断前列腺癌的试剂中的用途。
实施例
实施例1.差异基因表达分析
1.收集前列腺癌病人样本
病人和样本。
14对用于RNA-Seq的前列腺癌组织和癌旁正常组织取自上海长海医院。54对用于基因融合验证的样本:23对来自上海长海医院、17对来自江苏省立医院、14对来此中山大学第三附属医院。一组40对用于选择性剪切、长链非编码RNA验证的前列腺癌和癌旁组织取自上海长海医院。另一组用于长链非编码RNA验证的15个肿瘤样本和15个BPH(良性前列腺增生)样本分别取自江苏省立医院和上海长海医院。RNA-Seq的规程以及其后续试验得到了3家医院伦理委员会的批准。所有病人都填写了书面知情同意书,授权我们使用他们的样本。
2.癌组织及癌旁组织行冰冻切片后由病理学家检查保证质量
病理检查
癌组织和癌旁正常组织冰冻切片进行HE染色(苏木精-伊红染色)后由本研究的病理学家检查以保证所选组织癌组织密度超过80%,同时癌旁正常组织中没有癌组织。所有病理样本被另一个病理学家复查。如果出现结论不一致的情况,两位病理学家共同探讨以决定结论。
3.制备cDNA文库和RNA-Seq
寡聚脱氧胸苷磁珠用于从总RNA中分离多聚A mRNA。用片段化缓冲液将纯化mRNA片段化。将这些短片段作为模板,用随机六聚体引物来合成第一段cDNA链。第二段cDNA链用缓冲液、dNTPs、RNase H和DNA多聚酶I合成。短双链cDNA片段用QIAQuick PCR extraction kit(vendor)纯化并用EB缓冲液洗脱以修复末端并加上“A”。接着,短片段被连接到Illumina sequencing adaptors上。目的片段大小的DNA被割胶纯化用于PCR扩增。用Illumina HiSeqTM 2000对扩增文库进行测序。
cDNA文库构建使用Illumina公司提供的mRNA-Seq 8-Sample Prep Kit(货号为:RS-100-0801)进行,其具体操作流程为:寡聚脱氧胸苷磁珠用于从总RNA中分离多聚A mRNA。用片段化缓冲液将纯化mRNA片段化。将这些短片段作为模板,用随机六聚体引物来合成第一段cDNA链。第二段cDNA链用缓冲液、dNTPs、RNase H和DNA多聚酶I合成。短双链cDNA片段用QIAQuick PCR extraction kit(Qiagen)纯化并用EB缓冲液洗脱以修复末端并加上“A”。接着,短片段被连接到Illumina sequencing adaptors上。目的片段大小的DNA被割胶纯化用于PCR扩增。通过使用Agilent 2100Bioanalyzer生物分析仪和Stepone plus荧光定量PCR仪对cDNA文库进行质量检测后(合格标准为:PCR扩增产物大小为322±20bp,其中插入短片段大小为200±20bp,文库摩尔浓度不低于1.3nM),使用用Illumina HiSeqTM 2000对扩增文库进行测序。
4.数据分析
原始读数筛选
将测序仪生成的图像通过配套的测序仪控制软件进行base calling处理。原始序列储存为fastq格式。分析数据前删除脏读数。我们用三个标准删除脏读数:
1)删除脏读数;
2)删除“N”碱基超过2%的读数;
3)删除有50%以上QA≤15碱基的低质量读数。
所有以下分析都基于整理后的读数。
将读数在人类基因组和转录组上定位。
我们使用的基因组和转录组的参考序列是从UCSC网站下载(hg18version)。我们使用SOAP2(Short Oligonucleotide Analysis Package(SOAP)aligner(SOAP2);Li R,Yu C,Li Y,Lam TW,Yiu SM,et al.(2009)SOAP2:an improved ultrafast tool for short read alignment.Bioinformatics 25:1966-1967)方法将整理后的读数分别与基因组和转录组进行对比。每个读数的不匹配数不能超过3个。
基因和长链非编码RNA表达水平的标准化。
能被定位到特定基因的读数用于计算表达水平。基因表达水平是每百万读段中来自于某基因每千碱基长度的读段数。公式如下:
C是所选基因读数的拷贝数;N是所有读数基因的拷贝数;L是所选基因外显子的总长度。对于有超过一个选择性转录产物的基因,最长的转录产物用于计算RPKM。RPKM法能够消除不同基因长度和序列差异对基因表达计算的影响。因此,RPKM之可以直接用于比较样本间基因的表达差异。
我们用相同方法计算非编码RNA表达水平。
5.差异表达基因分析
参考“数字基因表达谱的显著性”(例如Audic S&Claverie JM(1997)The significance of digital gene expression profiles.Genome Res 7(10):986-995),我们用假发现率<=0.001和倍数改变>=2作为标准找到了在14对前列腺癌组织和配对癌旁正常组织中差异表达的基因。每个样本生成平均66,432,064个读数和5.98Gb大小的测序的核苷酸。通过SOAP2技术,我们把84.4%的读数定位到人类基因组(UCSC hg18 version)。通过对比癌组织和配对癌旁正常组织的转录组序列,我们在每个前列腺癌标本中找到了一些基因融合、差异性表达的长链非编码RNA、选择性剪切和差异性表达的基因。此外,我们发现平均每个癌组织样本有1725个点突变。这些结果揭示前列腺癌中存在着很大的异质性,同时信号通路及分子机制在前列腺癌的发生中起作用。
实施例2.前列腺癌新型融合基因的发现和验证
在我们将短RNA读数与参考基因组比较时发现,有的序列要分成两段才能和基因组相配对。这类读数需满足以下条件:
a)较短片段长度不短于8bp;
b)注意不管内含子在什么位置(从5’到3’,正链或负链)
对两段的对位分析,我们允许不超过一个的不匹配和无空位对位。
RT-PCR和测序验证基因融合。我们在转录水平对RNA-Seq得到的基因融合进行验证。我们设计了基因融合特异性的PCR引物。PCR和琼脂电泳后,所有RT-PCR扩增片段割胶回收(Qiagen QIAquick Gel Extraction kit)并行Sanger测序。用这种方法我们验证了5个融合基因,分别是TMPRSS2-ERG,USP9Y-TTTY15,SDK1-AMACR,CTAGE5-KHDRBS3,RAD50-PDLIM4,其中除TMPRSS2-ERG外的其他4个融合基因是本发明人新发现的。
4个新发现的融合基因是:
>39a fwd chrY 155 39b fwd chrY
USP9Y-TTTY15
GATAACTACATAAAGAGACAAAAAAAAGAAAAAAGAGCAAAGATCTGTGCTGTGTCAAGTATGACAGCCATCACTCATGGCTCTCCAGTAGGAGGGAACGACAGCCAGGGCCAGGTTCTTGATGGCCAGTCTCAGCATCTCTTCCAACAGAACCAGgaatcaaacttgacgtatggagccaagaaagcccttggaaaaactggcctcatattttgtgtacacagtccctgtacagggtttctgacctgtg
>31a fwd chr7 12131b rev chr5
SDK1-AMACR
ACCTTCCTGGTGCCCCATCCAACCTGGTCATTTCCAACATCAGCCCTCGCTCCGCCACCCTTCAGTTCCGGCCAGGCTATGACGGGAAAACGTCCATCTCCAGGTGGATTGTTGAGGGGCAGgtgtcatggagaaactccagctgggcccagagattctgcagcgggaaaatccaaggcttatttatgccaggctgagtggatttggccagtcaggaagcttctgccggttagctggccacgatatcaactatttggctttgtcag
>2a site:235 ID:4253 fwd_chr14<=>fwd_chr8 ID:10656
CTAGE5-KHDRBS3
AATTTAAATGTGCCTGATTCATCTCTCCCTGCTGAAAATGAAGCCACTGGCCCTGGCTTTGTTCCTCCACCTCTTGCTCCAATCAGAGGTCCATTGTTTCCAGTGGATGCAAGAGGCCCATTCTTGAGAAGAGGACCTCCTTTCCCCCCACCTCCTCCAGGAGCCATGTTTGGAGCTTCTCGAGATTATTTTCCACCAGGGGATTTCCCAGGTCCACCACCTGCTCCATTTGCAAtggtgctgattactatgattacggacatggactcagtgaggagacttatgattcctacg
>44a fwd chr5 113 44b fwd chr5 10111(RAD50) 8572(PDLIM4)
CAAAAAGAAACTGAACTTAATAAAGTAATAGCTCAACTAAGTGAATGCGAGAAACACAAAGAAAAGATAAATGAAGATATGAGACTCATGAGACAAGATATTGATACACAGAAGgtccatgctggcagcaaggctgcattggctgccctgtgcccaggagacctgatccaggccatcaatggtgagagcacagagctcatgacacacctggaggcacagaaccgcatcaagggctgccacgatcacctcacactgtctgtgagcag
其中大写字母表示第一个基因的序列,小写字母表示第二个基因的序列。
对于这5个融合基因的扩增引物如下表5。
表5.5个融合基因的扩增引物
PCR条件是:95℃10秒;60℃30秒;72℃90秒;38-43个循环。
使用PCR纯化试剂盒PCR Cleanup Kit 50-prep(AXYGEN,Cat No.AP-PCR-50,Lot No.KB10101204-G)进行PCR产物纯化,对PCR产物进行2%琼脂糖凝胶电泳,使用胶回收试剂盒DNA Gel Extraction Kit 50-prep(AXYGEN,Cat No.AP-GX-50,Lot No.KE10101204-G)进行胶回收。
有融合基因的电泳图片,分别参加见图2d(TMPRSS2-ERG和CTAGE5-KHDRBS3),图3a和b(USP9Y-TTTY15)和图4a(RAD50-PDLIM4),图4b(SDK1-AMACR)。
筛选高频的基因融合。用RT-PCR验证了基因融合以后,我们在另外54对样本中验证了每个(以上4个)融合基因。首先提取所有样本的RNA并逆转录为cDNA。RT-PCR引物与上述验证引物相同。测序样本的cDNA作为阳性对照。
前列腺癌基因融合图谱。转录组测序最早被用于检测前列腺癌中的基因融合现象。使用配对末端读数,我们一共找到了84个基因融合。除了众所周知的TMPRSS2-ERG基因融合外,我们找到了83个新的基因融合,这些在之前针对白人的研究中都未被报道过。35个新的和1个以前熟知的基因融合仅见于前列腺癌组织而未见于配对癌旁正常组织中(见下划线部分的融合基因),另外有融合基因在癌旁正常组织表达(见黑体加粗部分),具体生物学意义暂时不明,还有如下4个融合基因在癌和癌旁均有。
只在癌中表达的基因融合定义为肿瘤特异性基因融合。每个癌组织样本的基因融合数分别为1到6个不等。所述83个新基因融合如表6所示,其中的35个新的基因融合以下划线标出
表6.83个新基因融合
最常见的基因融合是TMPRSS2-ERG和USP9Y-TTTY15。二者均见于14个测序前列腺癌组织样本中的3个样本。我们通过RNA-Seq检测到另一个最常见的融合基因是位于Y染色体上的USP9Y-TTTY15。USP9Y编码一个类似于泛素特异性蛋白酶的蛋白,而TTTY15是一个非编码RNA。USP9Y基因缺失或突变与男性不育有关。然而,之前的研究都未揭示上述两种基因与肿瘤发生有关。RNA-Seq结果中,USP9Y基因的3号外显子和TTTY15基因的3号外显子融合形成的USP9Y-TTTY15频率(3/14=21.4%)与TMPRSS2-ERG相同。但是,RT-PCR发现54个前列腺癌组织中19个有USP9Y-TTTY15。该融合基因之前未被报道过,但其在本研究中频率较高提示其在中国人前列腺癌的发生中起重要作用,这些可望在后续的研究中得到阐明。有趣的是,用开放阅读框(ORF)预测工具Six-Frame Translation发现该融合基因的转录产物似乎没有开放阅读框,提示其可能是非编码RNA。我们还发现该融合可能导致USP9Y功能的缺失和一个新的非编码的融合基因转录产物。该融合基因在测序样本和验证样本中较高的出现频率提示其在前列腺癌中起重要作用。
在该54对前列腺癌样本中,我们还验证了另外3个(CTAGE5-KHDRBS3、SDK1-AMACR和RAD50-PDLIM4)基因融合,他们的频率分别是37%,20%,33.3%。
实施例3.前列腺癌长链非编码RNA的发现和验证
(1).从http://www.ncrna.org/frnadb/download下载ncRNA数据库,然后删除片段小于200nt的ncRNA、zRNA和非人类RNA并得到2981个长链非编码RNA。接下来我们用该数据库计算长链非编码RNA的表达水平。配对癌和癌旁标本的长链非编码RNA差异性表达的标准为:假发现率<=0.001、倍数改变>=2。选择在超过50%样本中一致上调或下调的长链非编码RNA进行监督聚类分析(使用cluster 3.0对基因和长链非编码RNA表达谱进行分层聚类分析)。进一步行长链非编码RNA和基因的相关分析。我们选择在超过50%前列腺癌样本中一致上调或下调的长链非编码RNA并分析它们与所有在前列腺癌组织中发现的基因的相关性。长链非编码RNA和基因的表达水平(RPKM)用作计算相关系数R。
(2).qRT-PCR验证长链非编码RNA(我们使用Power SYBR Green Mastermix试剂在Applied Biosystems Step One Plus做qRT-PCR。GAPDH引物用作内参。如上所述一组40对前列腺癌和癌旁组织取自上海长海医院,另一组用于15个肿瘤样本和15个BPH样本分别取自江苏省立医院和上海长海医院,用于长链非编码RNA验证。使用两步法PCR扩增标准程序:Stage1:预变性(Reps:1;95℃30秒);Stage2:PCR反应(Reps:40;95℃5秒;60℃34秒);Dissociation Stage(解离阶段)。
设计了针对4个长链非编码RNA的引物如下表7:
表7.4个长链非编码RNA的引物
所有的实验都使用两个或三个孔进行平行重复实验,结果以相对于GAPDH的平均倍数改变绘图(图5)。我们发现有137个长链非编码RNA在50%的前列腺癌中都呈现一致的上调或下调。我们分析了每个长链非编码RNA与所有基因表达量的相关性发现有23个长链非编码RNA与全基因组中数百个基因显著相关,而其他大多数基因仅与几个基因相关,或者根本就不相关。
结果分析部分
我们在40对前列腺癌和癌旁组织中、15个正常人前列腺组织和15个前列腺癌组织中验证发现,在大多数前列腺癌标本中PCA3(又称为DD3)、MALAT1和FR0348383过表达,而FR0257520表达量降低(图5)。PCA3过表达的结果与之前认为其可能成为新的诊断标志物的研究类似,但我们首次发现MALAT1过表达的频率在前列腺癌中很高。
本发明提供了137个长链非编码RNA可用于诊断、判断患者预后和药物反应,以及治疗的靶点,参见表2。
实施例4.单核苷酸多态性和点突变的发现和验证
(1).我们使用SOAPsnp检测单核苷酸多态性。该软件是用重复测序方法通过将测序序列与已知序列对比将新测序的个体的共有序列组装到基因组。通过将共有序列与参考序列相对比,可以找到单核苷酸多态性。
(2).我们用RT-PCR联合Sanger测序验证RNA-Seq筛选出的候选碱基对变异。PCR条件是:95℃10秒;60℃30秒;72℃90秒;38-43个循环。样品来自上海长海医院14对前列腺癌和癌旁组织。我们随机选择30个蛋白编码突变进行验证。其中27个仅存在于癌组织(cDNA和DNA中均有),而未见于癌旁正常组织(cDNA和DNA中均无)。2个变异仅见与癌组织cDNA,而未见于正常组织cDNA。1个变异在癌组织和癌旁正常组织中均没有。
表8.已经验证的30个突变,其中最右一列是用的模板分别是CDNA和DNA,S代表成功,F代表失败。
表9. 30个突变所使用的引物
(3).所有样本都没有P53和PTEN突变,而这两个基因是COSMIC数据库中与前列腺癌相关度最高的基因。虽然大多数突变的基因之前未在前列腺癌中被报道过,其中118个在其它肿瘤中被发现过,提示这些基因的突变可能也导致前列腺癌。
本发明提供了183个突变,这些突变可作为诊断标志物、预后判断、药物疗效判断和治疗靶点,具体参见表3。
实施例5.选择性剪切的发现和验证
我们用于检测选择性剪切的方法主要包括两步:
1)我们使用SOAPsplice 1.1将读数定位到人参考序列,然后根据连接点读数(与参考序列两个或以上的独立片段相对应的读数,这两个片段之间由内含子隔开)的对比结果找到剪切位点。我们尽量使用SOAPsplice的默认参数,对于完整比对的读数允许3个错配,对于分段比对的读数每个片段仅允许1个错配。
2)根据选择性剪切机制,我们使用剪切位点和对比结果来检测四种基本的选择性剪切,包括外显子跳跃、选择性5’剪切位点、选择性3’剪切位点以及内含子保留。
找出四种选择性剪切后,我们选出存在于癌组织而不存在于癌旁正常组织的选择性剪切。对每个癌组织标本,我们分别计算支持3种选择性剪切(外显子跳跃、选择性5’剪切位点和选择性3’剪切位点)相应连接位点的连接点读数数目以及内含子保留事件中保留下来的内含子的平均深度。因为每种选择性剪切数量巨大,我们通过取0.99百分位数来得到高可信度的选择性剪切,并通过画circos图以便揭示一些共有模式。以1T为例,其有2047个选择性3’剪切位点。支持选择性3’剪切位点的连接点读数从1到609不等,其0.99百分位数是69。因此,我们保留连接点读数≥69的选择性3’剪切位点。此外,我们还删除掉在癌旁正常组织中也有的选择性剪切。最后,我们得到一组与每个样本相对应的高度可信的癌特异选择性剪切。RT-PCR验证选择性剪切。我们从冰冻癌组织和癌旁组织中提取总RNA,然后取5μgRNA逆转录为cDNA(Qiagen QuantiTect Reverse Transcription kit)。我们在40对癌组织和癌旁正常组织中用RT-PCR对选择性剪切进行了验证。
PCR条件是:95℃10秒;60℃30秒;72℃90秒;33-36个循环。其中特别地两个基因引物如下:
表10.PSA和AMACR选择性剪切的扩增引物
选择性剪切 正向引物 反向引物
PSA CCAAGTTCATGCTGTGTGCT TGCCTAGTAACCGTGTGCTG
AMACR GGGAAAATCCAAGGCTTATTTATG AAGTCGTATAGAAAGGTGCTCCAC
发明提供了如表4所示的肿瘤特异性的选择性剪切,这些选择性剪切可以作为血液、尿液和组织的诊断标志物,也可作为判断预后、治疗效果的标志物,还可以作为肿瘤治疗的靶点。
在超过一半的前列腺癌样本中发现有KLK3(也叫PSA)基因的内含子保留,在一部分前列腺癌样本中发现有AMACR基因的外显子跳跃。这两种选择性剪切方式都用RT-PCR在测序组得到了验证。我们同时在40对样本(来自长海医院的40个样本)中用RT-PCR进行了验证,发现绝大多数癌组织样本中有PSA内含子保留,而癌旁组织中几乎没有。40个癌组织样本中仅9个有AMACR基因外显子跳跃。
尽管本发明的具体实施方式已经得到详细的描述,本领域技术人员将会理解。根据已经公开的所有教导,可以对那些细节进行各种修改和替换,这些改变均在本发明的保护范围之内。本发明的全部范围由所附权利要求及其任何等同物给出。
- 还没有人留言评论。精彩留言会获得点赞!